Abstract
The difficult separation of magnesium–lithium has always been a problem that impedes the comprehensive utilization of salt lake brine resources. In this paper, a method for the separation of magnesium and lithium based on the crystallization of magnesium sulfate at high-temperature supersaturation and a low viscosity was investigated. The microstructure of soluble solutions was analyzed, and the results showed that, in a single-salt solution, controlling the temperature can change the contact ion pair structure of MgSO4 solution, and the arrangement of SO42− and H2O in the second hydration layer changes. In the Li2SO4 solution, the hydrogen bonds between SO42− and H2O break, and the surrounding water structure changes, breaking the similarity of the microstructure of magnesium–lithium and enhancing the separation effect. In a multi-ion system, the change in water structure in the solution decreases with the increase in Cl− concentration. Controlling the temperature of salt lake brine with different magnesium–lithium mass ratios, it was found that the magnesium–lithium mass ratio in the brine could be reduced by one-third; when the magnesium–lithium mass ratio was 10:1~160:1, the loss of lithium could be controlled within 5%, but when the magnesium–lithium mass ratio was 5:1, the loss of lithium was 25.06%. The main reason for lithium loss is that Li2SO4 in the liquid phase enters the solid phase as a cluster and is entrapped during the MgSO4 crystallization process. The entire experiment shows that controlling the temperature process is more suitable for salt lake brine with a high magnesium–lithium ratio.
1. Introduction
The Qinghai magnesium-sulfate-subtype salt lake in China contains abundant mineral resources such as potassium, magnesium, and lithium [1]. However, lithium in the lake brine is usually present in trace amounts. Due to the high magnesium–lithium ratio (generally more than 40:1) in the brine [2] and their similar physicochemical properties, efficient separation is difficult to achieve [3]. Current separation methods result in significant lithium losses when applied to high-magnesium–lithium-ratio brines, posing a challenge for achieving enhanced separation and a reduction in lithium losses.
The traditional natural evaporation process directly utilizes MgCl2 as the raw material for magnesium–lithium separation [4]. However, the high concentration of MgCl2 solution leads to high viscosity, resulting in a lithium loss of 36~70% in the liquid phase [5]. Despite this, there exists a type of magnesium-sulfate-type brine in salt lake production that can achieve effective magnesium removal and enhance magnesium–lithium separation [6] due to its properties of being supersaturated and having a low viscosity under high-temperature conditions [7]. In fact, the temperature is higher, so the separation of salt components is easier; if the system is supersaturated, the separation effect will be significantly enhanced, and the separation performance will even be increased by 30% [7]. The properties of low viscosity and supersaturation at high temperatures are possessed by sulfate-type salt lake brines [8], which can provide convenient conditions for magnesium–lithium separation. However, the lithium loss mechanism and the process-enhanced separation mechanism of this process are still unclear, requiring clarification of the microstructure, hydrogen bonding, and complexation. Therefore, it is necessary to elucidate the effects of enhanced magnesium–lithium separation and the causes of lithium losses from changes in solution structure and molecular levels.
Methods for studying the solution structure include Raman spectroscopy, infrared spectroscopy [9], and X-ray diffraction. Generally, inorganic salts do not produce infrared characteristic peaks. However, in salt solutions, the water structure near ions can be altered. Water preferentially solvates cations with oxygen and hydroxyl groups solvate anions, increasing the complexity of the water structure and leading to spectral changes. Therefore, infrared spectroscopy can be used to characterize the microstructure of water in inorganic salt solutions by analyzing the shifts [10] and intensities of the O-H stretching band [11]. Cheng et al. [12] studied the O-D stretching band in K2SO4 and MgSO4 solutions using FTIR and analyzed the water structure changes induced by these salts. Deepak Ojha et al. [13] used two-dimensional infrared spectroscopy to investigate the effects of halide salts as solutes on the diffusion dynamics of the OD stretching vibrational spectra in the water medium at different temperatures. Guo et al. [14] used FTIR to demonstrate the strength of interactions between water and solvent molecules in the hydration layer by observing the frequency shift of the O-H bond. Moilanen et al. [15] observed the water structure surrounding ions in aqueous salt solutions using the FTIR technique. Borkowski et al. [16] observed changes in the water vibrational spectra in halide salt solutions using FTIR. Therefore, infrared spectroscopy can be used to analyze the changes in water structure caused by soluble salts with varying temperatures, providing support for magnesium–lithium separation during cooling processes.
Experimental infrared spectroscopy involves the absorption or scattering of specific wavelengths of infrared radiation to reveal the molecular composition and structural characteristics of a sample. On the other hand, simulated infrared spectroscopy can predict the infrared spectral features of a sample through theoretical calculations and various computational methods. It helps uncover the effects of different molecular configurations and environments on the infrared spectrum, aiding in the interpretation of experimental observations. The combination of both approaches allows for mutual validation and supplementation. In their study, Grabska et al. [17] employed a combination of two infrared spectroscopy techniques to provide a better explanation of water structure. Ben-Amotz [18] utilized complementary information from two infrared spectroscopy methods to elucidate ion polarization. Roget et al. [19], using infrared techniques in conjunction with kinetic analysis, investigated the interaction and structural evolution of water and chloride ions in a LiCl solution. Therefore, employing the mutual confirmation of two infrared spectroscopy methods can offer a more comprehensive understanding of the microscopic structural changes in brine during the temperature-enhanced magnesium–lithium separation process.
This study utilizes infrared spectroscopy and molecular dynamics simulations to investigate the microstructural changes in soluble magnesium–lithium salt solutions during temperature variations. It examines the effects of changes in magnesium and lithium salt solutions on the efficiency of magnesium–lithium separation and lithium losses in order to elucidate the mechanisms behind lithium loss and process enhancement.
2. Experimental Section
A simulated brine solution was prepared with a magnesium–lithium ratio of approximately 40:1, based on the composition of brine samples obtained after potassium extraction from the Qinghai salt lake, China. This solution was then subjected to room-temperature evaporation, after the addition of Na2SO4·10H2O, with a composition of Mg2+ 48 g/L, Na+ 50.23 g/L, Cl− 138.2 g/L, Li+ 1.236 g/L, K+ 13.86 g/L, and SO42− 77.25 g/L.
Single-salt solutions of MgSO4, Li2SO4, LiCl, and MgCl2, with a concentration of 2.5 mol/L, as well as simulated brine solutions with magnesium–lithium ratios of 160:1, 80:1, 40:1, 20:1, 10:1, and 5:1, were prepared and heated under reflux at 80 °C. The resulting MgSO4-LiCl saturated solution was then slowly cooled to around 60 °C, at which point the solution became supersaturated (i.e., the concentration of MgSO4 is empirically approximately 1.3 times its standard solubility). The temperature was then further lowered to room temperature (around 20 °C). During this cooling stage, approximately one-third of the dissolved MgSO4 crystallized and precipitated, significantly reducing the magnesium–lithium ratio of the brine. This is the most distinctive feature of this separation method.
Furthermore, because the brine was cooled from a relatively high temperature (60 °C), the viscosity of the brine was low during the cooling process, which prevented a large amount of Li+ from being trapped in MgSO4 crystals and then avoided serious lithium process losses.
The changes in the temperature and state of the brine solution were recorded. After the solid–liquid separation, the crystals were collected, and the magnesium–lithium separation was completed. FTIR spectroscopy was used to observe changes in the structure of the single-salt and brine solutions before and after cooling, over a range of wave numbers from 3800 cm−1 to 650 cm−1. The morphology and composition of the crystals were analyzed using SEM, EDS, and XRD, and the magnesium–lithium separation was confirmed via chemical analysis. The laboratory instruments used in this study included a Fourier transform infrared spectrometer (Thermo Fisher Scientific Nicolet iS50, Waltham, MA, USA), an X-ray diffractometer (Rigaku Corporation Smartlab, Tokyo, Japan), an inductively coupled plasma atomic emission spectrometer (Thermo Fisher Scientific ICAP6300, Waltham, MA, USA), a scanning electron microscope, and an energy dispersive X-ray spectroscopy system (JSM-IT500HR, JEOL, Tokyo, Japan), among others.
Different temperature conditions (20–60 °C) were established for the MgSO4-H2O, Li2SO4-H2O, LiCl-H2O, and MgCl2-H2O systems. The B3LYP functional was utilized, and all simulated systems were maintained at a constant pressure and temperature using the Nose–Hoover barostat and Nose–Hoover thermostat, with parameter settings of 1 atm and 298 K, 313 K, and 333 K. The vibrational frequencies and reaction kinetics were calculated with a convergence criterion of 10−6 eV. The following thresholds were used for geometry optimization: 1 × 105 Hartree for maximum energy change, 2 × 10−3 Hartree/A for maximum force, and 5 × 10−3 A for maximum displacement. The radial distribution function of anions (SO42−, Cl−) in the solute was analyzed, and the distribution of water molecules around the ions before and after temperature changes was compared to provide a molecular-level explanation for the control of magnesium–lithium separation during cooling. Similar methods have been used in many inorganic salt solution systems [20,21,22].
3. Results and Discussion
3.1. Microstructural Changes in Single-Salt Solutions at Different Temperatures
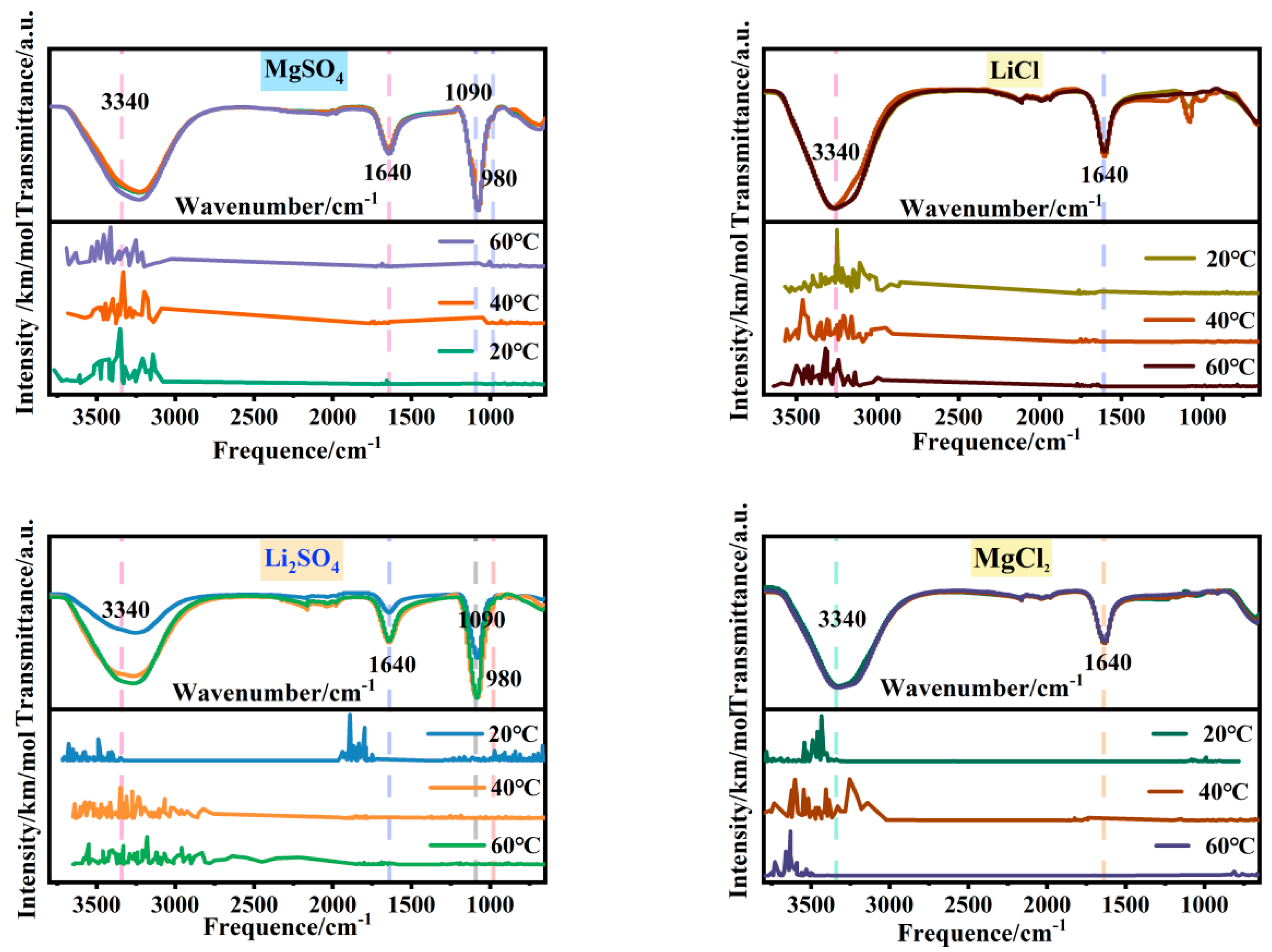
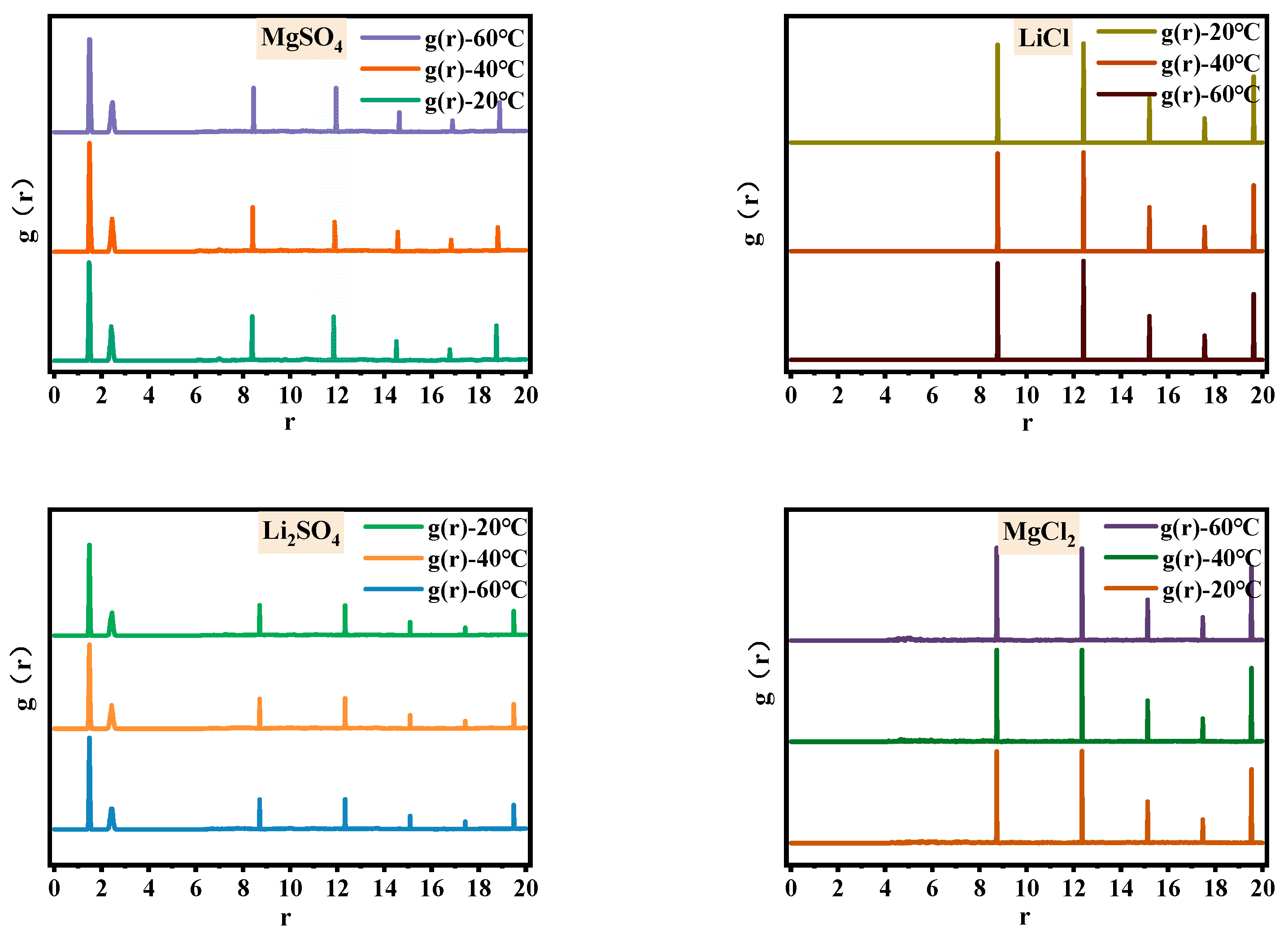
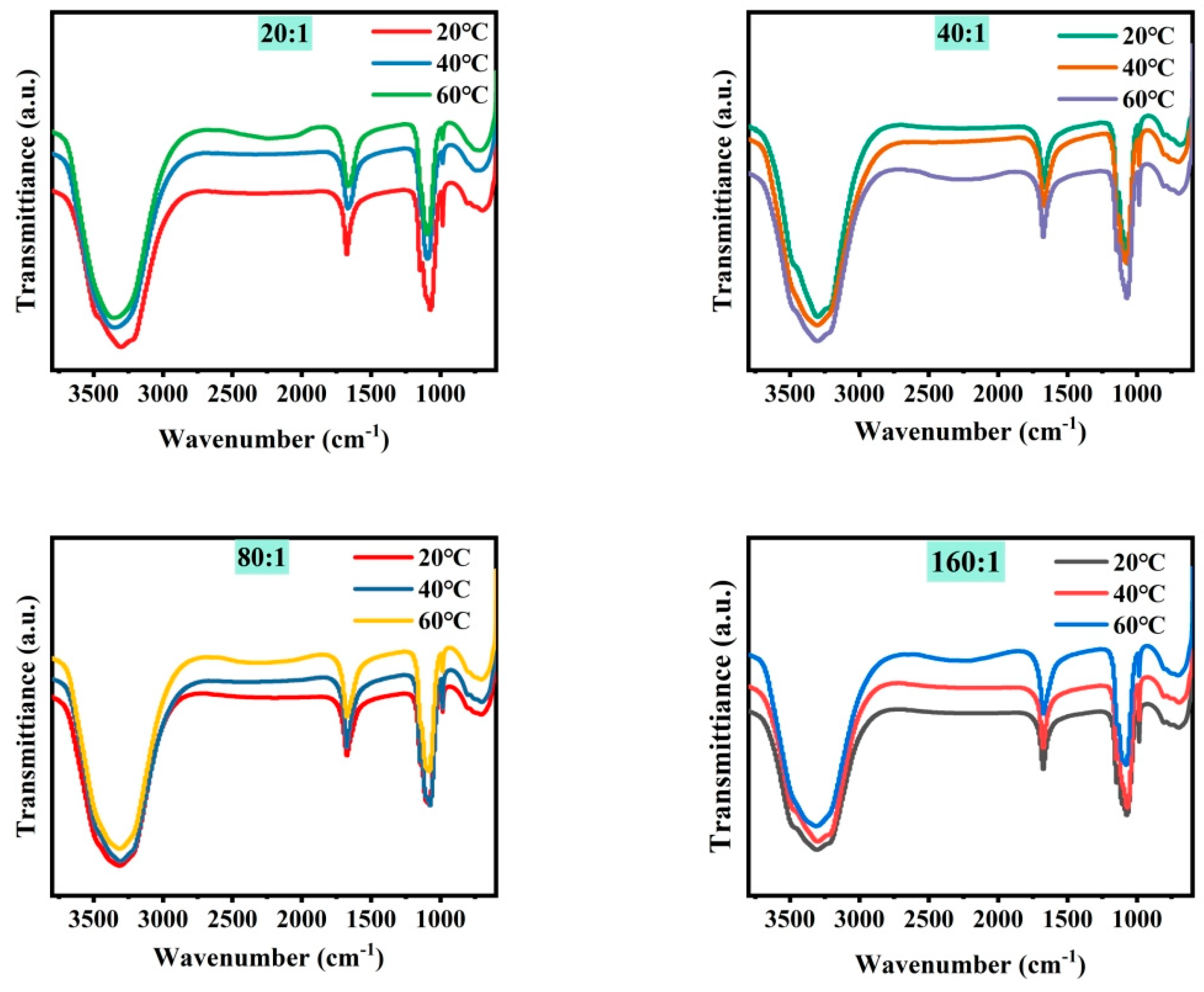
In order to address the issue of breaking the similarity between magnesium and lithium structures and enhancing their separation, this study investigates the microstructural changes in the single-salt solutions in the MgSO4-LiCl system at different temperatures. Figure 1 and Figure 2 show the Fourier transform infrared spectra and radial distribution functions of 2.5 mol/L MgSO4, Li2SO4, LiCl, and MgCl2 solutions at 20 °C, 40 °C, and 60 °C. Table 1 presents the relative peak strengths of various absorption peaks in the infrared spectra of each single-salt solution.
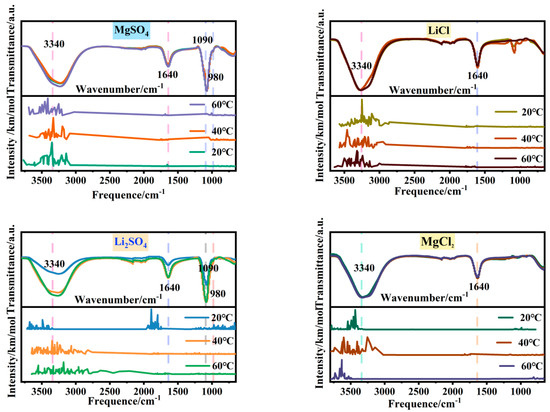
Figure 1.
FTIR spectra of magnesium sulfate, lithium sulfate, lithium chloride, and magnesium chloride solutions at different temperatures.
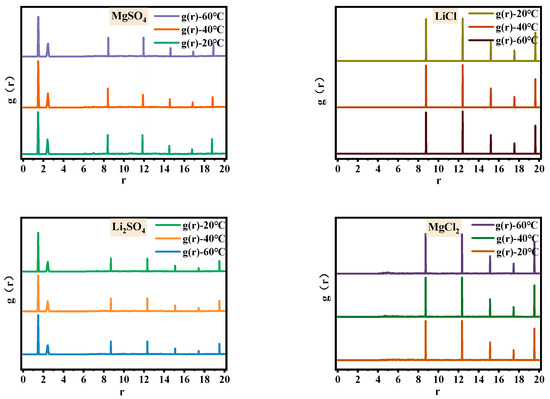
Figure 2.
Radial distribution functions of magnesium sulfate, lithium sulfate, magnesium chloride, and lithium chloride solutions at different temperatures.

Table 1.
Relative intensities of infrared spectral absorption peaks for each single-salt solution.
In Figure 1, the upper portion represents the experimental infrared spectrum, while the lower portion represents the simulated spectrum. The simulated spectrum exhibits peaks at the same positions as those observed in the experimental spectrum; the matching peak positions between simulated and experimental spectra indicate the accuracy of the solution model, thereby affirming the reliability of the radial distribution functions. From Figure 1, it can be observed that the absorption bands of O-H stretching vibrations at 3340 cm−1 in the four soluble single-salt solutions exhibit broad spectra with strong intensities, while the peak intensity and width of the O-H bending vibration absorption band at 1640 cm−1 are significantly pronounced. Both of these trends vary similarly with temperature. With increasing temperature, the O-H stretching vibration absorption band shifts to lower wavenumbers, and the peak area of Li2SO4 increases twofold at 20 °C, while the other salt solutions undergo minimal changes. In the radial distribution functions, as the temperature rises, the second hydration shell of water molecules around the anions in MgSO4 undergoes changes, while the water molecule distribution around the anions in Li2SO4, LiCl and MgCl2, and LiCl remains relatively unaffected. Therefore, it can be inferred that the increase in temperature leads to changes in the second hydration shell of MgSO4 ions and alters the number of hydrogen bonds in the Li2SO4 solution, resulting in changes in water structure. The comparable relative peak strengths of the chloride salt absorption bands are approximately 1, indicating minimal changes in their microstructure. This finding contributes to understanding the potential of altering the temperature of halide solutions to enhance the differentiation in sulfate–water structures.
In soluble salt solutions, the distinction between sulfate and chloride salts during the temperature variation lies in the presence of complex structures in sulfates, whereas chlorides exist in an ionic form. The complexation structure of sulfate solutions is manifested by two shoulder peaks related to SO42− at 1090 cm−1 and 980 cm−1 in the infrared spectra. In the radial distribution functions, two sharp peaks appear at r = 1.49 Å and r = 2.41 Å in MgSO4 and Li2SO4 solutions, providing evidence for the presence of complexation structures. The prominent structural changes in chloride salts relate to water structure. In the radial distribution functions, there are no shoulder peaks in the range of 0–8 Å, indicating the distribution of water molecules only. The peak at 1090 cm−1 corresponds to the symmetric stretching vibration of SO42−. Based on Figure 1 and Table 1, it can be observed that with increasing temperature, the relative strength of MgSO4 is approximately 1, while the peak area of Li2SO4 increases by 1.79 times at 20 °C, indicating minimal changes in the number of free SO42− ions in the MgSO4 solution and an increase in free SO42− ions in the Li2SO4 solution. The shoulder peak at 980 cm−1, attributed to the contact ion pair of SO42−, shows that the relative strengths of the absorption peaks in MgSO4 and Li2SO4 are approximately 1 with increasing temperature. However, in the radial distribution functions, the distribution of water molecules in the second hydration shell of SO42− in the MgSO4 solution undergoes changes, while the distribution of water molecules surrounding SO42− in the Li2SO4 solution remains relatively unaffected. This suggests that the microstructure of ion pairs in the MgSO4 solution undergoes changes, while the microstructure of ion pairs in the Li2SO4 solution remains unchanged. These findings widen the differentiation of magnesium–lithium solvent microstructures and provide a basis for strengthening the separation of magnesium and lithium.
Therefore, with increasing temperature, the number of water molecules in the second hydration shell surrounding the contact ion pair in the MgSO4 solution decreases. In Li2SO4 solution, the water structure changes due to variations in hydrogen bond numbers. The structures of MgCl2 and LiCl solutions remain largely unchanged, breaking the structural similarity between magnesium and lithium in solution and aiding in enhancing their separation.
3.2. Microstructure of Mixed Brine at Different Temperatures
To validate the changes in the microstructure of single-salt solutions in a cascaded temperature field, as discussed in Section 3.1, we investigated the influence of interionic temperature on the magnesium–lithium separation structure in a multi-ion system and examined the microstructural variations in brine with different magnesium–lithium mass ratios.
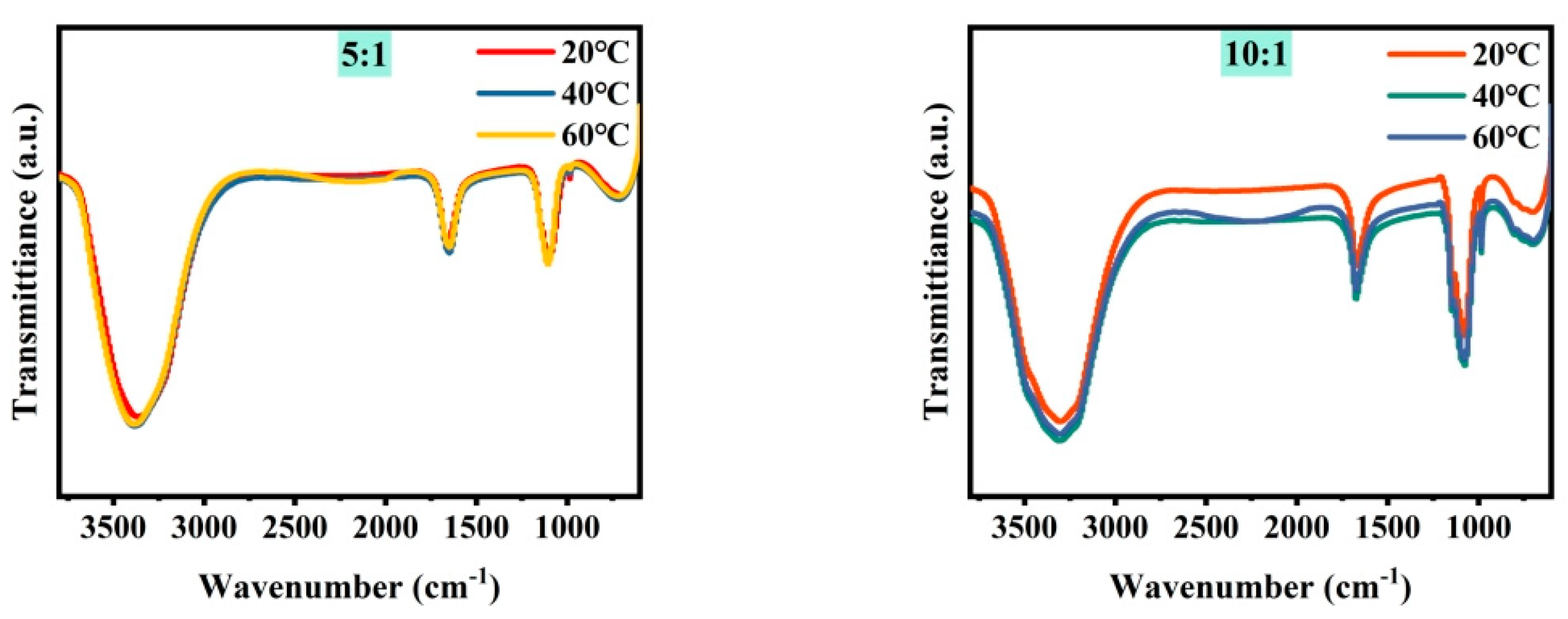
Figure 3 shows the microstructure of mixed brine with magnesium–lithium mass ratios of 5:1, 10:1, 20:1, 40:1, 80:1, and 160:1 at different temperatures. It can be seen from the combined results that with a decreasing magnesium–lithium mass ratio, the number of Cl− ions in the solution increases, and the peak intensity and area of water peaks at 3340 cm−1 and 1090 cm−1 change less during temperature variation. This is due to the easier formation of a coordination relationship between Li+ and Cl− in the solution, resulting in fewer Li2SO4 clusters. It was shown in Section 3.1 that Li2SO4 clusters play a major role in the change in water structure in the solution; therefore, the variation in the microstructure of the water structure in the solution is smaller. In a multi-ion system, as the magnesium–lithium ratio increases, the number of Cl− ions in the solution decreases, and with temperature change, the variation in water structure in the solution with hydrogen bond number becomes more significant. However, with more Cl− ions in the solution, the change in water structure with temperature is hindered, and the change in water structure around Li2SO4 clusters is suppressed, which is not conducive to the change in magnesium–lithium structure in the solution.
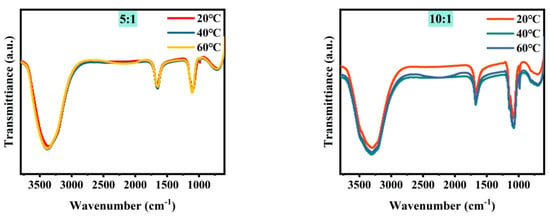
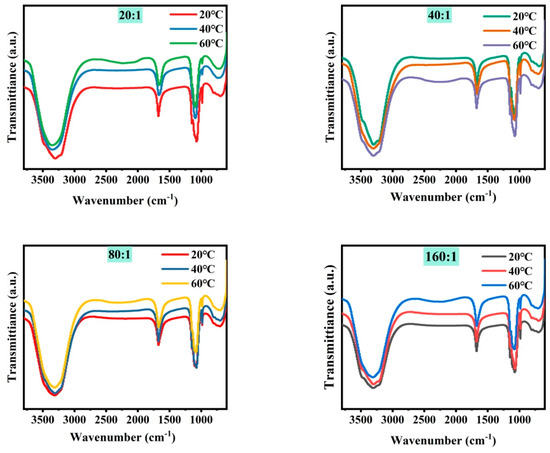
Figure 3.
FTIR spectra of simulated brine at different temperatures.
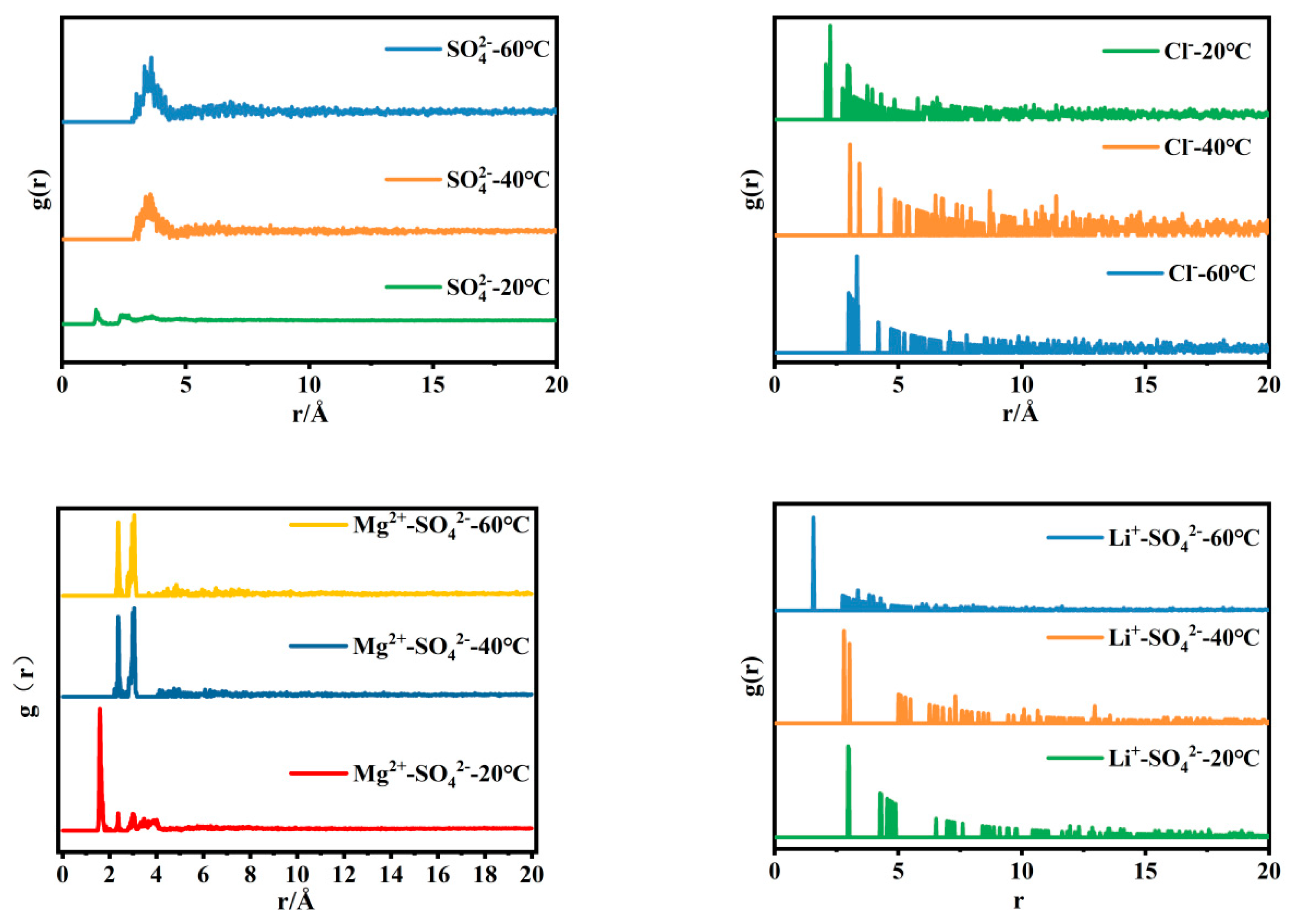
Figure 4 shows the radial distribution function of anions in simulated brine with a magnesium–lithium mass ratio of 40:1. Combined with the infrared spectra of brine, it can be seen that the peak areas of the characteristic bands of SO42− at 1090 cm−1 and 980 cm−1 decrease as the temperature increases, indicating that the free ion peak and contact ion peak of SO42− in the brine decrease with temperature increase. Correspondingly, in Figure 4, the radial distribution function of SO42− in the brine has a clear shoulder peak at r = 3.5 Å, and the stronger the shoulder peak, the stronger the interaction energy between SO42− and H2O in the solution. In the radial distribution function, the distribution of water molecules around Cl− in the brine changes less. In the radial distribution function of Mg2+-SO42−, it can be clearly seen that with temperature change, the arrangement of SO42− and H2O in the second hydration layer changes, and the ionic coordination structure undergoes a significant change. In terms of the radial distribution function of Li+-SO42−, with increasing temperature, the distance between Li+ and the hydration layer around SO42− first increases and then decreases, the hydrogen bond between SO42− and H2O breaks, and the surrounding water structure changes. Based on this, it can be inferred that the microstructure of MgSO4 ion pairs in the brine changes during the cooling process, and the distribution of water molecules around Li2SO4 clusters has a great impact. This is consistent with the microstructure changes in single-salt solutions in Section 3.1 and reinforces the magnesium–lithium separation.
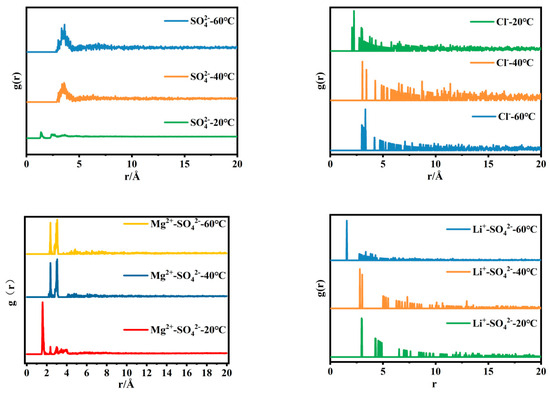
Figure 4.
Radial distribution function of SO42− and Cl− in simulated brine with a magnesium–lithium mass ratio of 40:1 at different temperatures.
Therefore, in a multi-ion system, the concentration of Cl− ions in the brine determines the change in water structure in the brine. The lower the concentration of Cl− ions, the greater the variation in the microstructure of the water with the hydrogen bond number during temperature increase, and the more significant the difference in the magnesium–lithium structure in the brine, which is more conducive to strengthening magnesium–lithium separation. This is the reason that Na2SO4 needs to be added to the raw brine to convert it into a sulfate-type solution before the brine is evaporated and separated.
3.3. Mechanism of Lithium Ion Separated from Brine Solution
The differentiation of magnesium and lithium structures by changing the temperature in multi-ion systems has been observed to enhance the separation of magnesium and lithium. During the temperature change process, the supersaturation state of brine is the key to the separation of magnesium and lithium with minimal lithium loss. However, further investigation is still required to understand the loss of lithium in the liquid phase during the cooling process. Therefore, the loss of lithium in the liquid phase is further explored by comparing isothermal evaporation and controlled cooling processes.
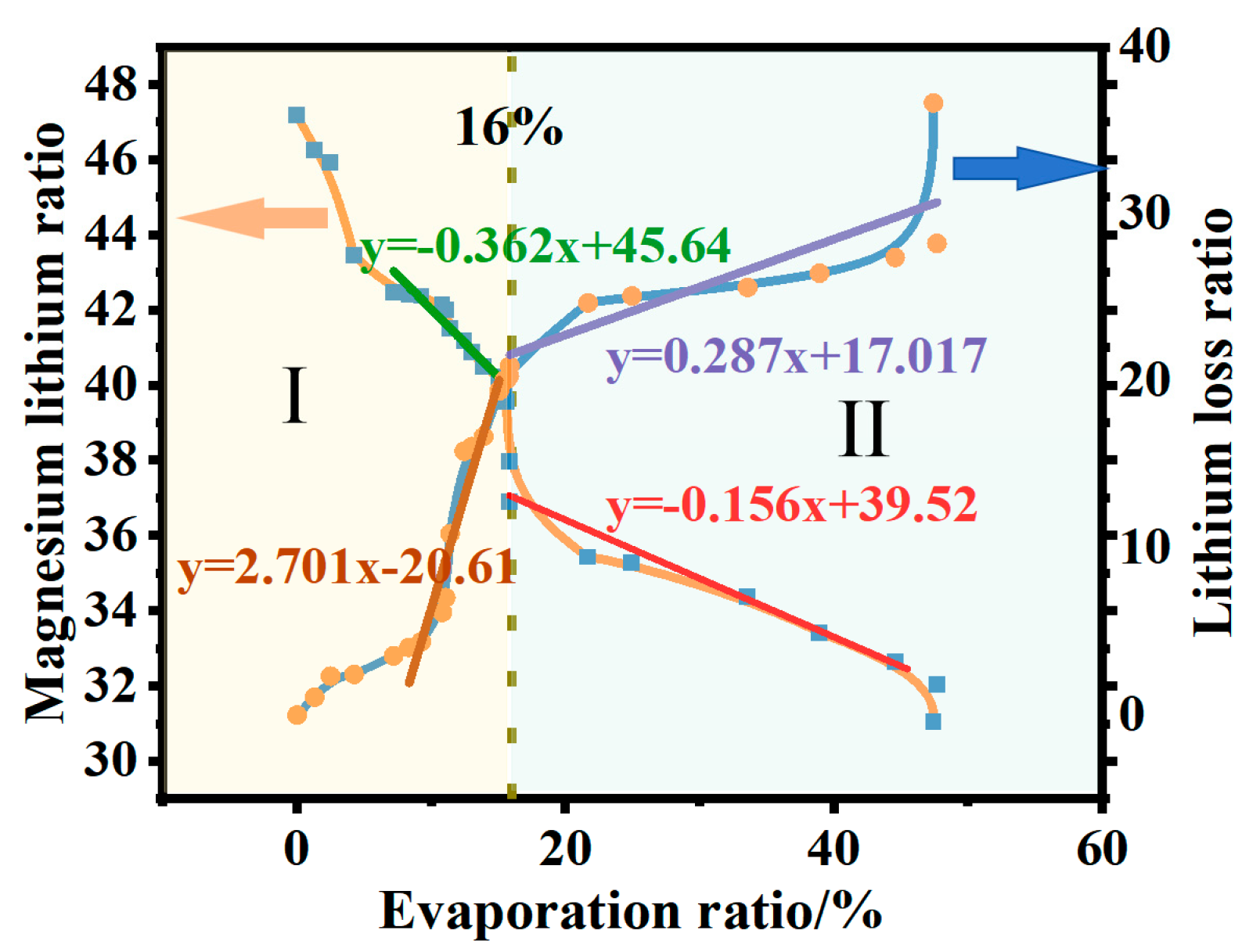
During the isothermal evaporation process of brine with a magnesium–lithium mass ratio of 40:1, variations in the concentrations of various ions were observed through ICP-OES (Thermo Fisher Scientific ICAP6300, Waltham, MA, USA) analysis as water evaporated. Figure 5 depicts the changes in the magnesium–lithium ratio and lithium loss in the liquid phase during isothermal evaporation. As brine evaporates, the magnesium–lithium ratio in the solution decreases, and the amount of lithium loss gradually increases. This could be due to the synchronous dissociation of Mg2+-Cl− complexes surrounding Mg2+ ions and the interaction of Li+-Cl− complexes, leading to partial lithium doping in magnesium crystals and resulting in lithium loss. As is shown in Figure 5, when the evaporation ratio exceeds 16%, the change in the magnesium–lithium mass ratio decreases. This is because as evaporation proceeds, the concentration of MgCl2 in brine gradually increases. The evaporation ratio of water molecules in the liquid phase is higher than that of magnesium chloride ions. MgCl2 is a structurally dispersed salt with strong water-retention capacity in the solution, resulting in increased viscosity of the brine and hindrance of water molecule evaporation [4]. This inhibition of evaporation weakens the separation of magnesium and lithium. When the evaporation ratio exceeds 50% and evaporation is stopped, the final brine magnesium–lithium ratio decreases to 23.43 with a lithium loss of 36.65%.
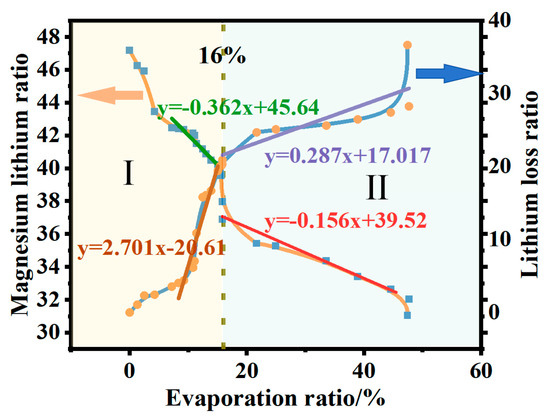
Figure 5.
Magnesium–lithium ratio and concentration changes in the liquid phase during isothermal evaporation process. (When the evaporation ratio is less than 16%, it is in Stage I, during which the lithium-magnesium mass ratio changes significantly, and the ratio of lithium loss increases. When the evaporation ratio is greater than 16%, it is in Stage II, during which the lithium-magnesium mass ratio changes minimally, and the ratio of lithium loss decreases.).
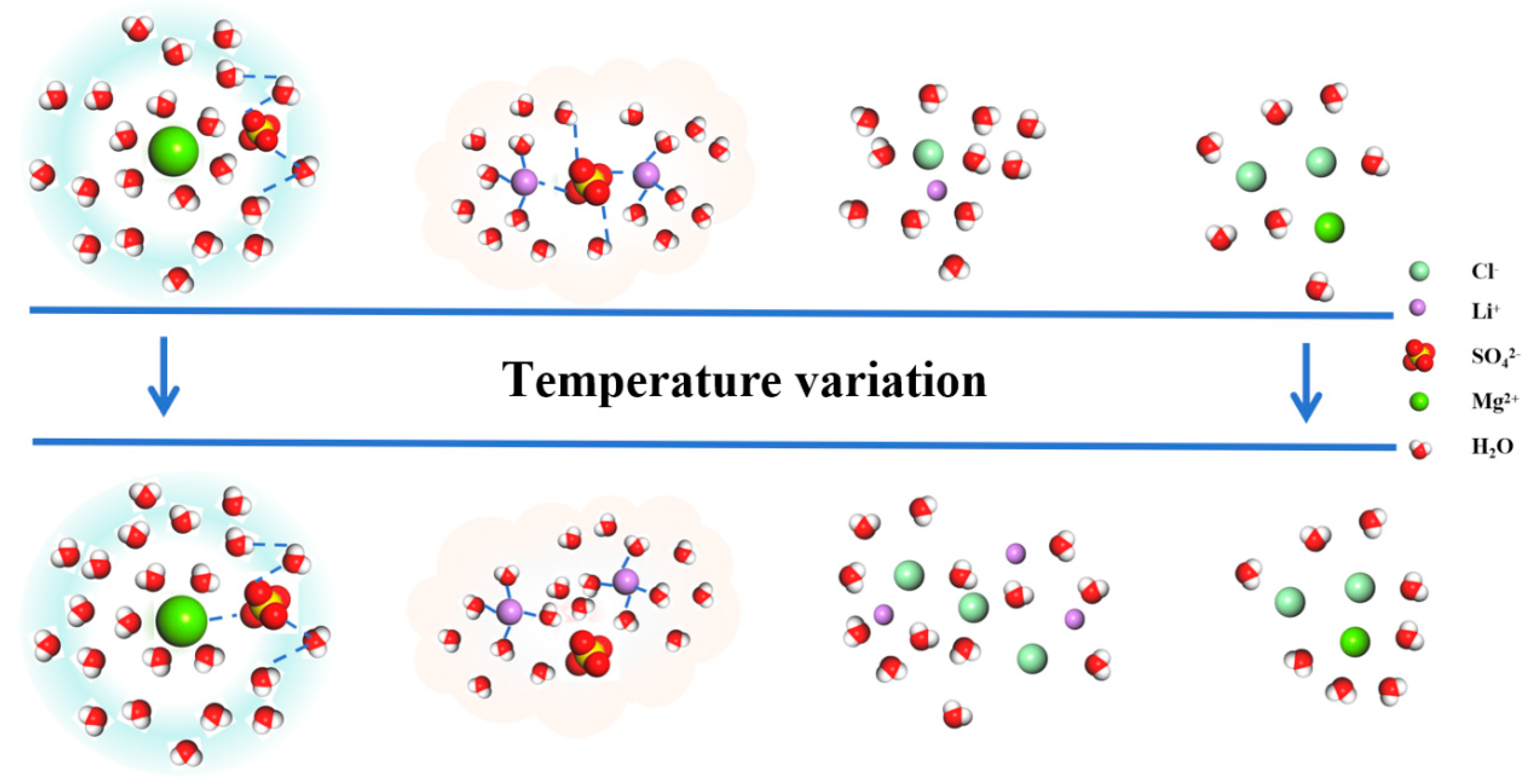
Based on the analysis of the isothermal evaporation of brine, it has been found that lithium is carried away in the form of a magnesium–lithium-complex salt (MgCl2·LiCl·6H2O) during the crystal precipitation process. As is summarized in Section 3.1 and Section 3.2, the similarity of the magnesium–lithium structure can be altered by controlling the temperature, thus allowing for improvements in the evaporation crystallization method. By analyzing the radial distribution functions between ions depicted in Figure 2, the microstructural changes between ions in the brine can be inferred. The structure of MgSO4 undergoes modifications under temperature influence, with the embedding of SO42− in the first hydration layer and increased activity of contact ions. Additionally, rearrangement occurs in the second hydration layer, promoting crystallization [20]. Similarly, the structure of Li2SO4 is affected by temperature, leading to the breaking of hydrogen bonds between water molecules and SO42− ions, resulting in changes in the surrounding water molecule structure, as illustrated in Figure 6. Consequently, through temperature control, it is possible to distinguish between the structures of MgSO4 and lithium salts in brine, thereby enhancing the separation of magnesium and lithium.
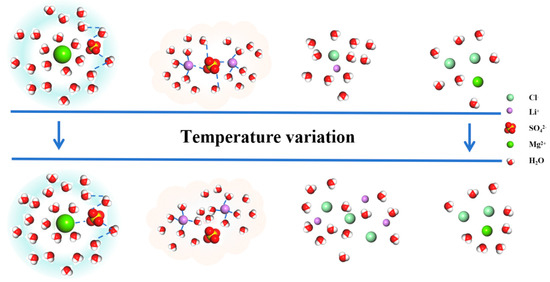
Figure 6.
Microstructural changes in various particle clusters in brine.
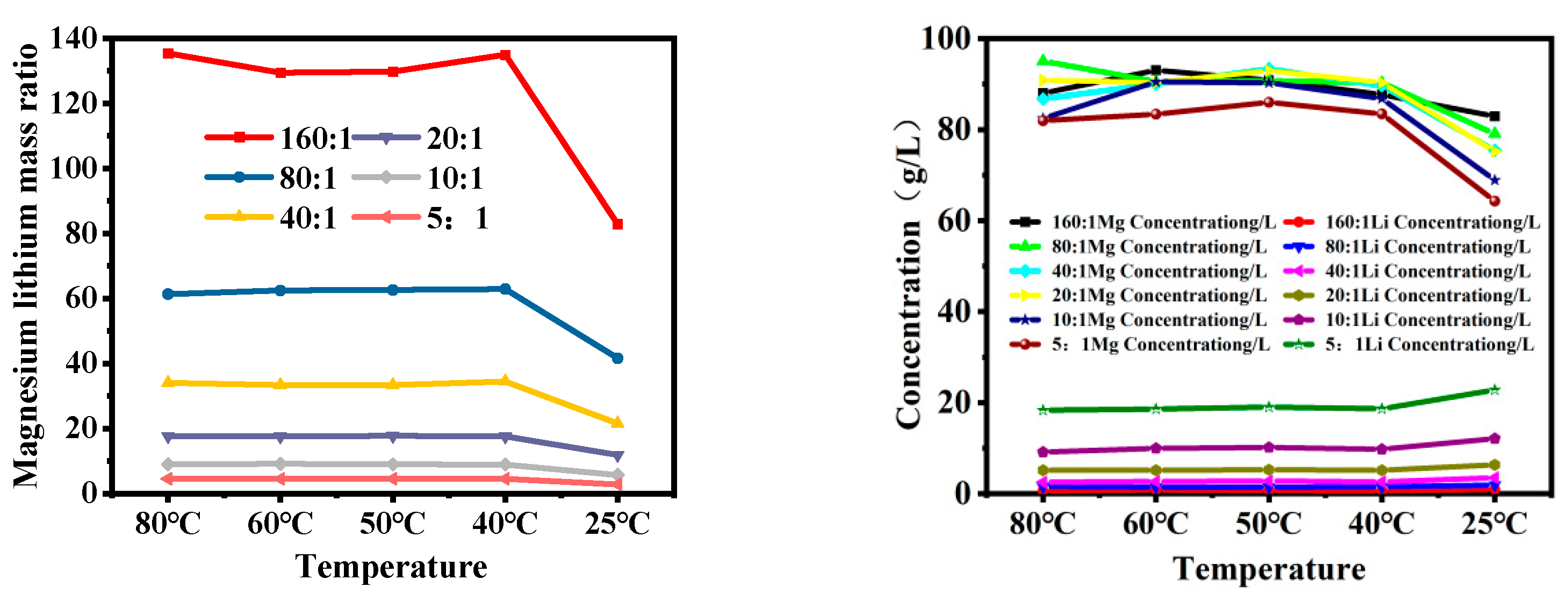
Figure 7 shows the changes in the magnesium–lithium ratio and ion concentrations of magnesium and lithium in brine with different magnesium–lithium mass ratios after cooling. It can be seen from the figure that the magnesium–lithium ratio of brine decreases by about one-third during the cooling process; when the mass ratio of magnesium to lithium is 5:1, the lithium loss is 25.06%, and when the mass ratio of magnesium to lithium is greater than 5:1, the lithium loss can be controlled within 5%. In addition, the enrichment trend of lithium in the liquid phase is smaller at 40 °C compared to 60–50 °C and 40–25 °C. This can be attributed to the fact that at 40 °C, magnesium ions have a larger charge than lithium ions, enabling them to undergo rapid movement under the influence of an electric field, forming a thicker diffusion layer and resulting in alterations in the microstructure of brine [2]. As is shown in Figure 6, LiCl exists in brine in ion form, while Li2SO4 exists in cluster form. Combined with the radial distribution function in Figure 4, the distance between hydration layers around Li2SO4 increases as the temperature decreases, promoting Li2SO4 crystallization and affecting the concentration of lithium in the solution. As a result, during the cooling process, LiCl exists in the liquid phase in ion form, while Li2SO4 clusters exist in an ion pairing structure. Changes in the hydration layers around Li2SO4 clusters due to temperature variation promote the formation of lithium sulfate crystals, causing the loss of Li+ in the liquid phase in the form of Li2SO4 clusters, thus affecting the separation of magnesium and lithium.
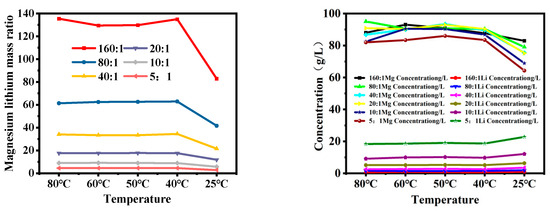
Figure 7.
Changes in magnesium concentration during cooling process with different lithium mass ratios.
3.4. Mechanism of Lithium Entrapment into Solid Phase
By referring to the radial distribution function of Li+-SO42− in Figure 4, it can be observed that as the temperature decreases, the distance between hydration layers surrounding Li+-SO42− clusters increases, thereby promoting the formation of lithium sulfate crystals. Based on this hypothesis, a chemical composition analysis was conducted on the solid phase. Table 2 presents the concentration of lithium ions in the solid phase, along with a comparison with the loss of lithium during the isothermal evaporation process, providing clear insights into the mechanism by which Li+ enters the solid phase during temperature variation.
Table 2.
Comparison of lithium content and loss in solid phase.
Through chemical composition analysis of the solid phase, the lithium content can be determined. A comparison was made between the solid phase obtained after cooling of the brine and the solid phases obtained after the cooling of MgSO4 and Li2SO4. The morphology and structure of the solid phase were studied, followed by drying treatment of the solid phase. Further clarification of the form of lithium loss was achieved through the utilization of scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDS), and X-ray diffraction (XRD) analysis.
Figure 8a,b represent the solid phase obtained by cooling crystallization of brine. Figure 8c represents the crystals obtained by cooling crystallization of a pure lithium sulfate solution, and Figure 8d represents the crystals obtained by cooling crystallization of a pure magnesium sulfate solution. A comparison reveals that the surface of MgSO4 crystals is rougher than that of Li2SO4 crystals, and according to the scale in Figure 8c,d, it can be observed that MgSO4 crystals are larger than Li2SO4 crystals. In Figure 8a, it can be observed that most of the crystals obtained after cooling are MgSO4 crystals. The MgSO4 crystals were observed to exhibit numerous small-particle crystals attached to their surfaces. The EDS analysis revealed the presence of sulfate ions without magnesium in some of these small-particle crystals. Due to the limitations of EDS in detecting lithium content, in cases where the starting materials solely consist of magnesium and lithium, the absence of magnesium and the presence of sulfur and oxygen suggest the possibility of lithium sulfate crystals. Hence, it is speculated that the change in temperature of high-magnesium–lithium-ratio brine leads to a transformation in the complex structure of MgSO4 in solution, resulting in a rearrangement of the hydration layers and promoting MgSO4 crystallization. This alteration in the water structure within the brine facilitates the generation of Li2SO4 crystals and thus, the entrapment of lithium salts in the solid phase.

Figure 8.
Solid-phase SEM and EDS results: (a,b) represent the solid phase obtained by cooling crystallization of brine; (c) represents the crystals obtained by cooling crystallization of a pure lithium sulfate solution; and (d) represents the crystals obtained by cooling crystallization of a pure magnesium sulfate solution.
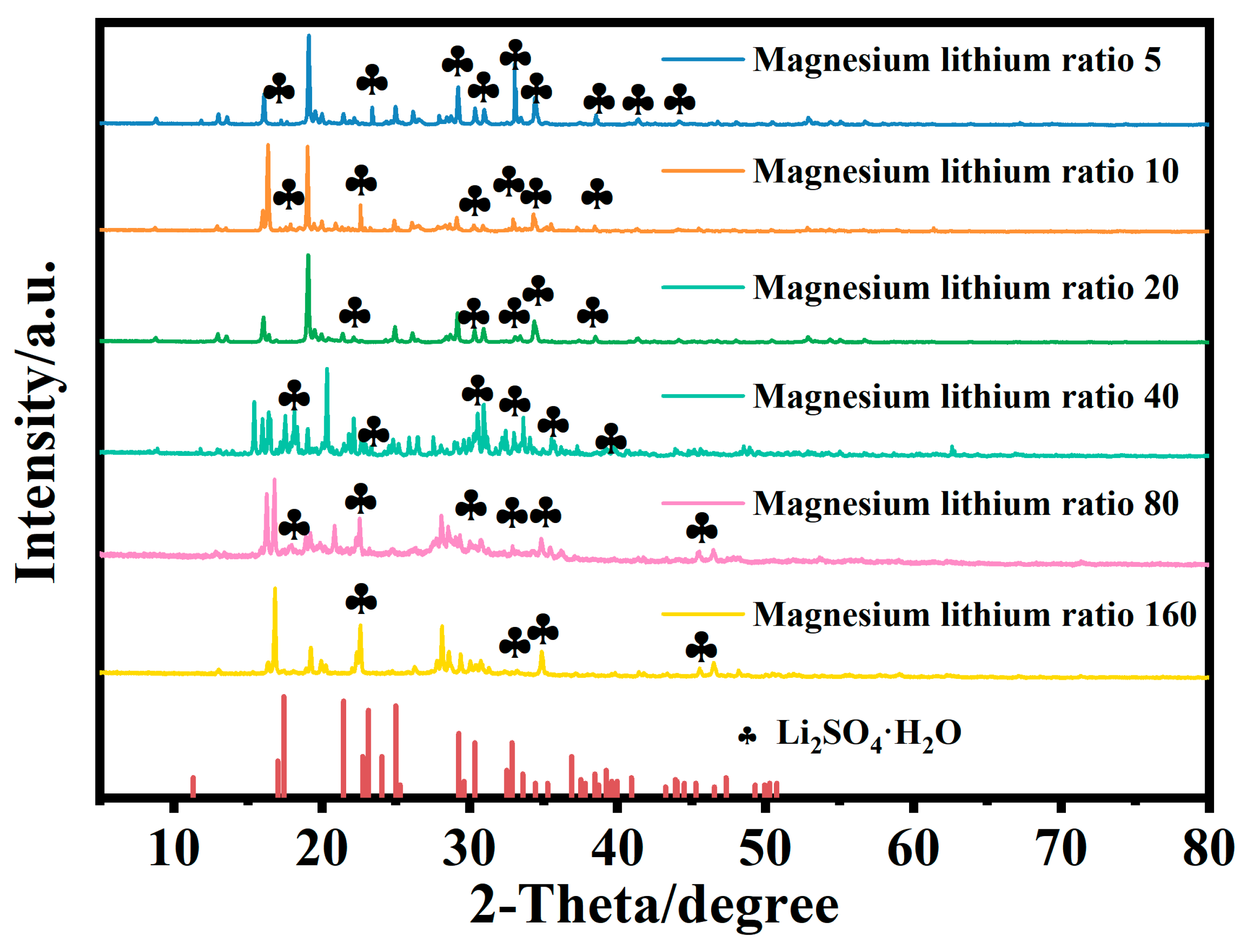
The composition of the substances was analyzed using the X-ray diffraction (XRD) method. Figure 9 depicts the XRD spectra of crystals obtained from brine with different magnesium–lithium mass ratios after controlled cooling processes.
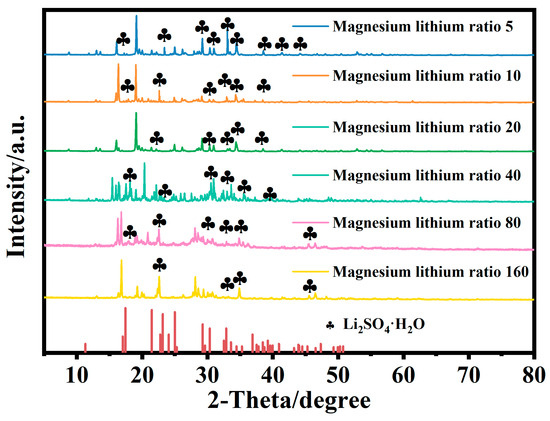
Figure 9.
XRD results of solid-phase crystallization by cooling brine with different magnesium–lithium mass ratios.
Based on the X-ray diffraction (XRD) results depicted in Figure 9, it can be observed that Li2SO4·H2O exists in the solid phases obtained during the variable-temperature crystallization process of brine with different magnesium–lithium mass ratios. Further analysis was carried out by combining scanning electron microscopy (SEM) and XRD results, which confirmed that some of the small granular crystals observed in SEM were Li2SO4 crystals. This implies that Li2SO4 crystals are entrained in MgSO4 crystals and adhere to the surface of MgSO4 in the form of small granules. Consequently, during the brine cooling process, lithium loss occurs in the form of Li2SO4·H2O crystal entrainment into MgSO4 crystals.
Therefore, during the variable-temperature enhanced magnesium–lithium separation process, the change in the MgSO4 complex structure in solution due to a decrease in temperature results in a rearrangement of the hydration layers and promotes MgSO4 crystallization. This alteration in water structure within brine increases the hydrated layer distance between Li+-SO42− clusters and enhances the generation of Li2SO4 crystals, leading to lithium loss during the magnesium–lithium separation process.
4. Conclusions
This study aims to address the issues associated with enhancing magnesium–lithium separation and reducing lithium loss through variable-temperature treatment. The microscopic structural changes, magnesium–lithium separation efficiency, and lithium loss mechanisms in high-magnesium–lithium-ratio brine solutions at different temperatures during the variable-temperature process were investigated. The following conclusions were drawn:
Through the analysis of FTIR spectra and radial distribution functions of different brine solutions, it was found that temperature can change the structure of MgSO4 solutes and the water structure of Li2SO4 in the liquid phase. The arrangement of SO42− and H2O in the second hydration layer of MgSO4 in the brine solution is changed with temperature. The hydrogen bonding between SO42− and H2O in the Li2SO4 solution is broken, resulting in changes in the surrounding water molecule structure. This change enhances the structural differences in the magnesium–lithium solution in the brine, thereby promoting magnesium–lithium separation. In a multi-ion system, the concentration of Cl− in the brine determines the changes in water structure. A lower Cl− concentration and increasing temperature result in greater changes in the water microstructure in the brine, leading to more pronounced differences in the magnesium–lithium structure and enhancing magnesium–lithium separation. In high-magnesium–lithium-ratio brine, by controlling the temperature and supersaturation state, lithium loss can be controlled within 5%. Therefore, the regulation of temperature and the supersaturation state aids in the separation of high-magnesium–lithium-ratio brine. During the cooling process, the magnesium–lithium separation is enhanced, and the cooling promotes changes in the water structure in the liquid phase, resulting in an increase in the number of hydration layers around Li+-SO42− clusters and the formation of Li2SO4 crystals. Thus, lithium is trapped in the MgSO4 crystals in the form of Li2SO4 crystals.
Author Contributions
Conceptualization, D.C. and H.C.; software, D.C.; investigation, D.A. and X.C.; data curation, D.C.; writing—original draft preparation, D.C. and D.A.; writing—review and editing, H.C. and X.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the Research Program of Qinghai Province (2023-ZJ-920M) and the Natural Science Foundation of China (U20A20149).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Liu, J.; Zhong, Y.; Ge, F.; Wang, H.; Shi, H.; Zhao, Z.; Li, L. Strategic thinking on constructing worldclass salt lake industrial base. Inorg. Chem. Ind. 2022, 54, 30–36. [Google Scholar]
- Sun, Y.; Wang, Q.; Wang, Y.; Yun, R.; Xiang, X. Recent advances in magnesium/lithium separation and lithium extraction technologies from salt lake brine. Sep. Purif. Technol. 2021, 256, 117807. [Google Scholar] [CrossRef]
- Zhou, J.; Shu, Y. Present situation and development suggestions of lithium extraction from salt lake brine in China. Ind. Min. Process. 2023, 52, 57–62. [Google Scholar]
- Bian, S.; Li, D.; Gao, D.; Peng, J.; Dong, Y.; Li, W. Hydrometallurgical processing of lithium, potassium, and boron for the comprehensive utilization of Da Qaidam lake brine via natural evaporation and freezing. Hydrometallurgy 2017, 173, 80–83. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, L.; Jiang, A.; Zhang, Y.; Zhu, X. Current situation and development suggestions of development and utilization of salt lake resources in China. Inorg. Chem. Ind. 2022, 54, 13–21. [Google Scholar]
- Shah, K.M.; Dach, E.; Winton, R.; Fan, H.; Yip, N.Y. Phase equilibria insights into amine-water-NaCl interactions in liquid-liquid biphasic systems for temperature swing solvent extraction desalination. Desalination 2023, 548, 116259. [Google Scholar] [CrossRef]
- Cheng, H.; He, Y.; Zhao, J.; Cheng, W.; Cheng, F. Pilot test and cost-based feasibility study of solar-assisted evaporation for direct preparation of high-purity magnesium sulfate hydrates from metastable Na+, Mg2+//Cl−, SO42−-H2O salt-water system. Hydrometallurgy 2019, 189, 105140. [Google Scholar] [CrossRef]
- Zhou, H.; Wu, P.; Li, W.; Wang, X.; Zhou, K.; Hao, Q. Thermodynamic modeling and phase diagram prediction of salt lake brine systems II. Aqueous Li+-Na+-K+-SO42- and its subsystems. Chin. J. Chem. Eng. 2021, 34, 134–149. [Google Scholar] [CrossRef]
- Zhao, L.J.; Zhang, Y.H.; Wei, Z.F.; Cheng, H.; Li, X.H. Magnesium sulfate aerosols studied by FTIR spectroscopy: Hygroscopic properties, supersaturated structures, and implications for seawater aerosols. J. Phys. Chem. A 2006, 110, 951–958. [Google Scholar] [CrossRef]
- Fu, J.; Schlenoff, J.B. Driving forces for oppositely charged polyion association in aqueous solutions: Enthalpic, entropic, but not electrostatic. J. Am. Chem. Soc. 2016, 138, 980–990. [Google Scholar] [CrossRef]
- Cao, Q.; Wang, X.; Miller, J.D.; Cheng, F.; Jiao, Y. Bubble attachment time and FTIR analysis of water structure in the flotation of sylvite, bischofite and carnallite. Miner. Eng. 2011, 24, 108–114. [Google Scholar] [CrossRef]
- Cheng, F.; Cao, Q.; Guan, Y.; Cheng, H.; Wang, X.; Miller, J.D. FTIR analysis of water structure and its influence on the flotation of arcanite (K2SO4) and epsomite (MgSO4·7H2O). Int. J. Miner. Process. 2013, 122, 36–42. [Google Scholar] [CrossRef]
- Ojha, D.; Chandra, A. Two-dimensional infrared spectroscopy of aqueous solutions from first principles simulations. Chem. Phys. Lett. 2020, 751, 137493. [Google Scholar] [CrossRef]
- Guo, F.; Friedman, J.M. Charge density-dependent modifications of hydration shell waters by Hofmeister ions. J. Am. Chem. Soc. 2009, 131, 11010–11018. [Google Scholar] [CrossRef] [PubMed]
- Moilanen, D.E.; Wong, D.; Rosenfeld, D.E.; Fenn, E.E.; Fayer, M.D. Ion-water hydrogen-bond switching observed with 2D IR vibrational echo chemical exchange spectroscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 375–380. [Google Scholar] [CrossRef]
- Borkowski, A.K.; Thompson, W.H. Shining (Infrared) Light on the Hofmeister Series: Driving Forces for Changes in the Water Vibrational Spectra in Alkali–Halide Salt Solutions. J. Phys. Chem. B 2022, 126, 6700–6712. [Google Scholar] [CrossRef] [PubMed]
- Beganović, A.; Beć, K.B.; Grabska, J.; Stanzl, M.T.; Brunner, M.E.; Huck, C.W. Vibrational coupling to hydration shell—Mechanism to performance enhancement of qualitative analysis in NIR spectroscopy of carbohydrates in aqueous environment. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 237, 118359. [Google Scholar] [CrossRef]
- Ben-Amotz, D. Hydration-shell vibrational spectroscopy. J. Am. Chem. Soc. 2019, 141, 10569–10580. [Google Scholar] [CrossRef]
- Roget, S.A.; Carter-Fenk, K.A.; Fayer, M.D. Water dynamics and structure of highly concentrated licl solutions investigated using ultrafast infrared spectroscopy. J. Am. Chem. Soc. 2022, 144, 4233–4243. [Google Scholar] [CrossRef]
- Cai, H.Q.; Li, H.J.; Wang, M.; Wang, C.C.; Yi, H.B. Microscopic insight into the ion aggregation characteristics in aqueous MgCl2 and MgCl2–LiCl solutions: Implications for Mg2+/Li+ separation. J. Mol. Liq. 2019, 273, 374–382. [Google Scholar] [CrossRef]
- Wang, C.C.; Wang, M.; Cai, H.Q.; Zhang, Q.W.; Li, Y.Y.; Yi, H.B. Insight into the roles of two typical ion clusters and their second hydration shells: Implication for the nucleation mechanism in MgSO4 aqueous solution. J. Mol. Liq. 2019, 278, 33–42. [Google Scholar] [CrossRef]
- Li, H.J.; Yi, H.B.; Xu, J.J. High-order Cu (II) chloro-complexes in LiCl brines: Insights from density function theory and molecular dynamics. Geochim. Et Cosmochim. Acta 2015, 165, 1–13. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).