

Ensuring the Quality of the Analytical Process in a Research Laboratory

 , , ,
, , ,
Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples, Reagents, and Solutions
2.2. Procedures
2.2.1. BCR Sequential Extraction
2.2.2. Determination of Extractable K and P in Arable Soil
2.2.3. Determination of Alkaline Reactivity of Raw Materials Intended for Geopolymerisation
2.3. Statistical Treatment of Experimental Data
2.4. Chalcogenide Glasses
2.4.1. Samples
2.4.2. Determination of Density
3. Results
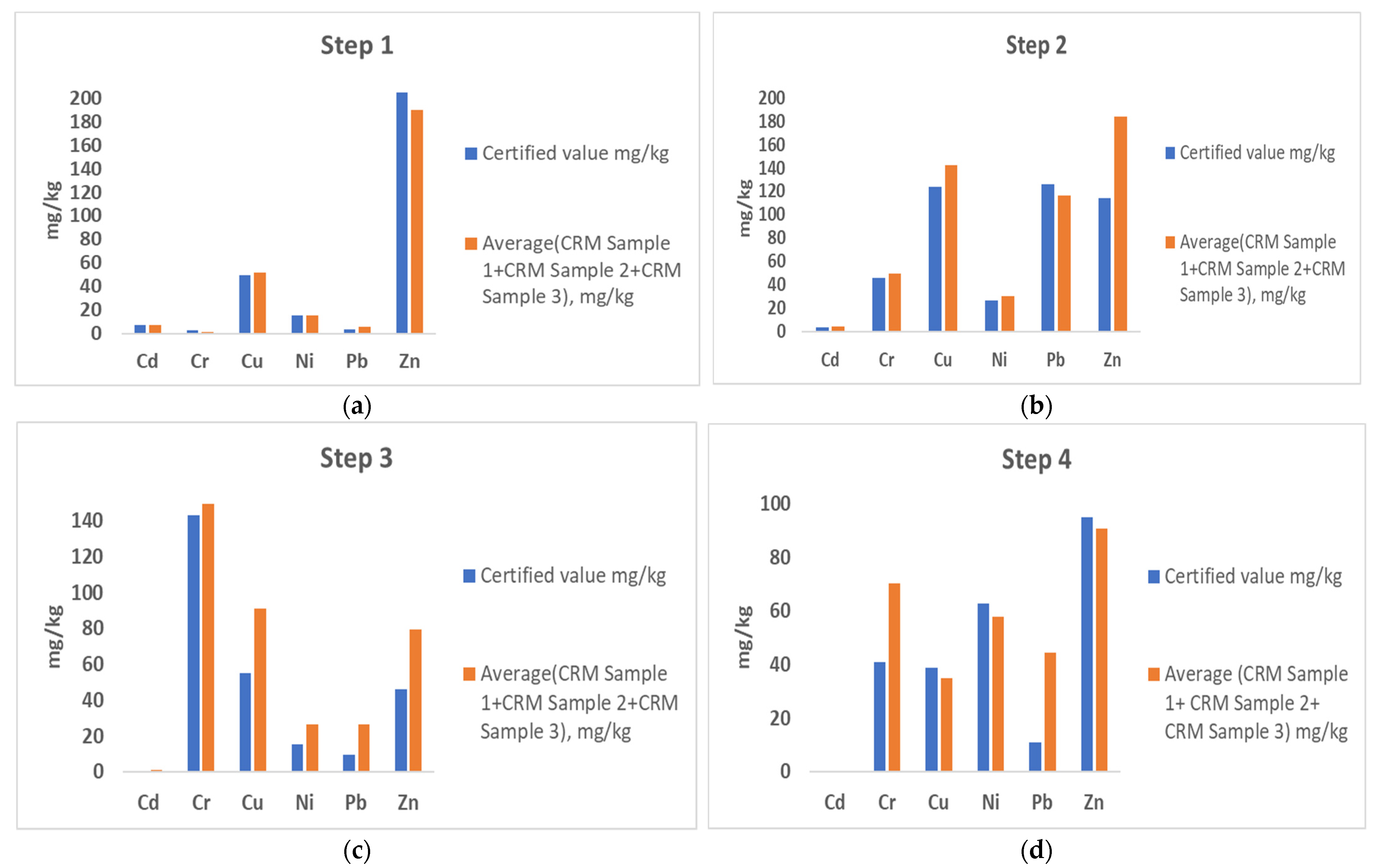
3.1. Scaling down the Standard Operating Procedure (SOP)—Sequential Extraction of Mine Tailing and Fly Ash
3.2. Standard Addition Approach as a Solution to Lack of Suitable CRM for Trueness Estimation
3.3. A New Specific Procedure—Alkaline Reactivity of Mine Tailings and Fly Ash
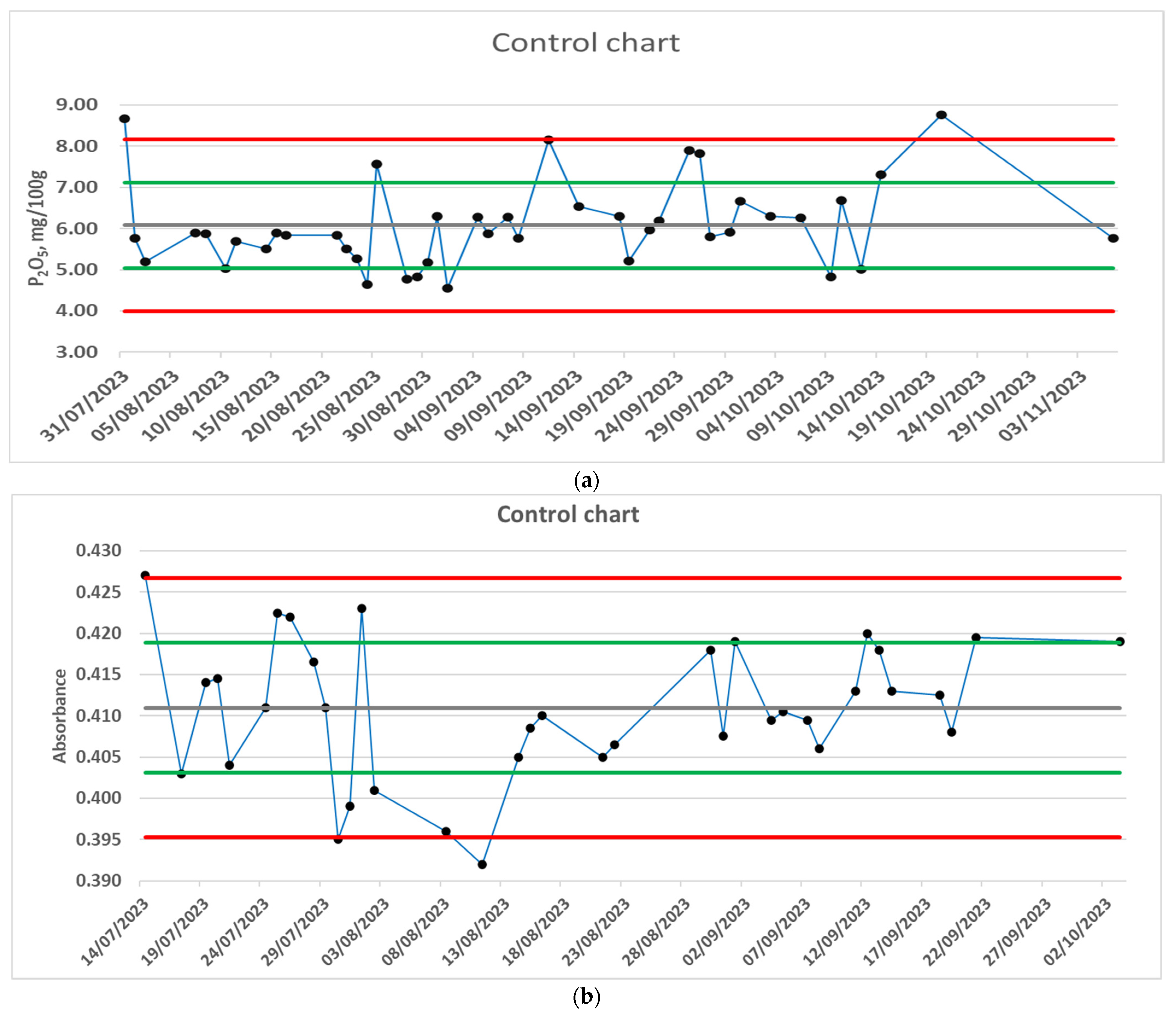
3.4. Quality Control—In-Lab Control Sample
3.5. Density of Chalcogenide Glasses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Konieczka, P.; Namiesnik, J. Quality Assurance and Quality Control in the Analytical Chemical Laboratory, 2nd ed.; CRC Press: Boca Raton, FL, USA; Taylor and Francis Group: Abingdon, UK, 2018. [Google Scholar] [CrossRef]
- Søndergaard, J.; Hansen, V.; Bach, L.; Jørgensen, C.J.; Jia, Y.; Asmund, G. Geochemical Test Work in Environmental Impact Assessments for Mining Projects in Greenland—Recommendations by DCE and GINR; Technical Report No. 132; Aarhus University, DCE—Danish Centre for Environment and Energy: Aarhus, Denmark, 2018; 46p, Available online: http://dce2.au.dk/pub/TR132.pdf (accessed on 12 December 2023).
- Matabane, D.; Codeto, T.; Mampa, R.; Ambushe, A. Sequential Extraction and Risk Assessment of Potentially Toxic Elements in River Sediments. Minerals 2021, 11, 874. [Google Scholar] [CrossRef]
- Sagagi, B.; Davidson, C.; Hursthouse, A. Adaptation of the BCR sequential extraction procedure for fractionation of potentially toxic elements in airborne particulate matter collected during routine air quality monitoring. Int. J. Environ. Anal. Chem. 2019, 101, 956–968. [Google Scholar] [CrossRef]
- Ciceri, E.; Giussani, B.; Pozzi, A.; Dossi, C.; Recchia, S. Problems in the application of the three-step BCR sequential extraction to low amounts of sediments: An alternative validate route. Talanta 2008, 76, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.A. BCR®-701: A review of 10-years of sequential extraction analyses. Anal. Chim. Acta 2010, 680, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Heltai, G.; Fekete, I.; Halász, G.; Kovács, K.; Horváth, M.; Takács, A.; Boros, N.; Győri, Z. Multi-elemental inductively coupled plasma-optical emission spectroscopic calibration problems of the sequential extraction procedure for the fractionation of the heavy metal content from aquatic sediments. Hung. J. Ind. Chem. 2015, 43, 7–13. [Google Scholar] [CrossRef]
- Kruve, A.; Rebane, R.; Kipper, K.; Oldekop, M.-L.; Evard, H.; Herodes, K.; Ravio, P.; Leito, I. Tutorial review on validation of liquid chromatography–mass spectrometry methods: Part I. Anal. Chim. Acta 2015, 870, 29–44. [Google Scholar] [CrossRef]
- Kruve, A.; Rebane, R.; Kipper, K.; Oldekop, M.-L.; Evard, H.; Herodes, K.; Ravio, P.; Leito, I. Tutorial review on validation of liquid chromatography–mass spectrometry methods: Part II. Anal. Chim. Acta 2015, 870, 8–28. [Google Scholar] [CrossRef]
- Kumkrong, P.; Dy, E.; Tyo, D.D.; Jiang, C.; Pihilligawa, I.G.; Kingston, D.; Mercier, P.H.J. Investigation of metal mobility in gold and silver mine tailings by single-step and sequential extractions. Environ. Monit. Assess. 2022, 194, 423. [Google Scholar] [CrossRef] [PubMed]
- Cappuyns, V.; Swennen, R.; Niclaes, M. Application of the BCR sequential extraction scheme to dredged pond sediments contaminated by Pb–Zn mining: A combined geochemical and mineralogical approach. J. Geochem. Explor. 2007, 93, 78–90. [Google Scholar] [CrossRef]
- Obenaus-Emler, R.; Falah, M.; Illikainen, M. Assessment of mine tailings as precursors for alkali-activated materials for on-site applications. Constr. Build. Mater. 2020, 246, 118470. [Google Scholar] [CrossRef]
- Nikvar-Hassani, A.; Vashaghian, H.; Hodges, R.; Zhang, L. Production of green bricks from low-reactive copper mine tailings: Chemical and mechanical aspects. Constr. Build. Mater. 2022, 324, 126695. [Google Scholar] [CrossRef]
- Angelova, L.; Genova, N.; Pencheva, G.; Statkova, Y.; Yotova, V.; Surleva, A. Contribution to the Molybdenum Blue Reaction and its Application in Soil Analysis. Methods Objects Chem. Anal. 2022, 17, 59–69. [Google Scholar] [CrossRef]
- Singh, P.K.; Dwivedi, D.K. Chalcogenide glass: Fabrication techniques, properties and applications. Ferroelectrics 2017, 520, 256–273. [Google Scholar] [CrossRef]
- Kadono, K.; Kitamura, N. Recent progress in chalcogenide glasses applicable to infrared optical elements manufactured by molding technology. J. Ceram. Soc. Jpn. 2022, 130, 584–589. [Google Scholar] [CrossRef]
- Perez-Moreno, S.M.; Gazquez, M.J.; Perez-Lopez, R.; Bolivar, J.P. Validation of the BCR sequential extraction procedure for natural radionuclides. Chemosphere 2018, 198, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Rauret, G.; López- Sánchez, J.F.; Lück, D.; Yli-Halla, M.; Muntau, H.; Quevauviller, P. The certification of the extractable contents (mass fractions) of Cd, Cr, Cu, Ni, Pb and Zn in freshwater sediment following a sequential extraction procedure BCR-701. In European Commission Directorate-General for Research 2001; European Commission: Brussels, Belgium, 2001. [Google Scholar]
- Angelova, L.; Genova, N.; Stoyanova, S.; Surleva, O.; Nekov, I.-H.; Ilieva, D.; Surleva, A. Comparative study of soil test methods for determination of plant available potassium in Bulgarian arable soils. Anal. Control. 2021, 25, 182–192. [Google Scholar] [CrossRef]
- Ilieva, D.; Angelova, L.; Radoykova, T.; Surleva, A.; Chernev, G.; Vizureanu, P.; Burduhos-Nergis, D.D.; Sandu, A.V. Characterization of Bulgarian Copper Mine Tailing as a Precursor for Obtaining Geopolymers. Materials 2024, 17, 542. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, B.; Örnemark, U. (Eds.) EURACHEM Guide: The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics, 2nd ed.; 2014; Available online: https://www.eurachem.org (accessed on 20 November 2023).
- Gupta, S.V. Practical Density Measurement and Hydrometry, 1st ed.; IoP: Bristol, UK; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar]
- Bettencourt Da Silva, R.; Bulska, E.; Godlewska-Zylkiewicz, B.; Hedrich, M.; Majcen, N.; Magnusson, B. Analytical Measurement: Measurement Uncertainty and Statistics; Joint Research Centre, Institute for Reference Materials and Measurements, European Commission: Brussels, Belgium, 2013; pp. 58–146. [Google Scholar] [CrossRef]
- Francisca, S.O.; Ramos, R.K.S.; Almeida, C.A.; Júnior, L.; Arruda, M.A.Z.; Pastore, H.O. A Straightforward Method for Determination of Al and Na in Aluminosilicates Using ICP OES. J. Braz. Chem. Soc. 2017, 28, 8. [Google Scholar] [CrossRef]
- Sivakumar, V.; Ernyei, L.; Obenauf, R.H. Matrix Effects in ICP-AES Analysis, The Application Notebook 2007; SPEX CertiPrep, Inc.: Metuchen, NJ, USA, 2007. [Google Scholar]
- Seddon, A.B. Chalcogenide glasses: A review of their preparation, properties. J. Non. Cryst. Solids 1995, 184, 44–50. [Google Scholar] [CrossRef]
- Bergin, T. An Introduction to Data Analysis: Quantitive, Qualitive and Mixed Methods, 1st ed.; SAGE Publications Ltd.: Melbourne, Australia, 2018. [Google Scholar]
- Wei, B.; Zhang, Y.; Bao, S. Preparation of geopolymers from vanadium tailings by mechanical activation. Constr. Build. Mater. 2017, 145, 236–242. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Fraction | Fe | Cu | Zn | Mn | Cd | Ni | Pb |
|---|---|---|---|---|---|---|---|
| mg/kg | |||||||
| Fraction 1 (exchangeable) | 447 | 107 | 5.5 | 20.5 | <0.25 | <0.5 | 3.8 |
| Fraction 2 (reducible) | 740 | 41.4 | 6.5 | 10.3 | <0.25 | <0.5 | 9.2 |
| Fraction 3 (oxidizable) | 8142 | 188 | 10.3 | 40.6 | <0.25 | <0.5 | 13.7 |
| F1 + F2 + F3 (total mobile) | 9329 | 337 | 22.2 | 71.4 | <0.25 | <0.5 | 26.7 |
| Fraction 4 (residual) | 10,380 | 100 | 40.2 | 102 | <0.25 | <0.5 | 17.5 |
| Total | 197,116 | 437 | 62.4 | 173 | <0.25 | <0.5 | 44.2 |
| Fraction | Mine Tailing | CRM Certified | CRM Experimental |
|---|---|---|---|
| F1 | Fe > Cu > Mn > Zn > Pb | Zn > Cu > Ni > Cd > Pb > Cr | Zn > Cu > Ni > Cd > Pb > Cr |
| F2 | Fe > Cu > Mn > Pb > Zn | Cu > Pb > Zn > Cr > Ni > Cd | Zn > Cu > Pb > Cr > Ni > Cd |
| F3 | Fe > Cu > Mn > Pb > Zn | Cr > Cu > Zn > Ni > Pb > Cd | Cr > Cu > Zn > Ni > Pb > Cd |
| F4 | Fe > Mn > Cu > Zn > Pb | Zn > Cr > Ni > Cu > Pb > Cd | Zn > Cr > Pb, Ni > Cu > Cd |
| Sample | Standard Addition | Recovery, % | ubias, mg/100 g |
|---|---|---|---|
| dry soil | K | 87 | 0.03 |
| dry soil | P | 17 | n.a. |
| extracted solution | P | 96 | 0.01 |
| Standard Series | Calibration Curve Concentration Range 0–1 mg/L | Calibration Curve Concentration Range 0–10 mg/L |
|---|---|---|
| external standard | y = 1.106 × x + 10,644 | y = 1.106 × x − 107,298 |
| matrix-matched calibration | y = 1.106 × x + 15,700 | y = 1.106 × x + 36,475 |
| System | Methods | Value (kg/m3) |
|---|---|---|
| Pycnometer | ||
| Hydrostatic balance | ||
| Pycnometer | ||
| Hydrostatic balance |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Surleva, A.; Angelova, L.; Ilieva, D.; Ivanova, V.; Surleva, O.; Chavdarova, K. Ensuring the Quality of the Analytical Process in a Research Laboratory. Appl. Sci. 2024, 14, 3281. https://doi.org/10.3390/app14083281
Surleva A, Angelova L, Ilieva D, Ivanova V, Surleva O, Chavdarova K. Ensuring the Quality of the Analytical Process in a Research Laboratory. Applied Sciences. 2024; 14(8):3281. https://doi.org/10.3390/app14083281
Chicago/Turabian StyleSurleva, Andriana, Lyudmila Angelova, Darya Ilieva, Vladislava Ivanova, Olya Surleva, and Katrin Chavdarova. 2024. "Ensuring the Quality of the Analytical Process in a Research Laboratory" Applied Sciences 14, no. 8: 3281. https://doi.org/10.3390/app14083281
APA StyleSurleva, A., Angelova, L., Ilieva, D., Ivanova, V., Surleva, O., & Chavdarova, K. (2024). Ensuring the Quality of the Analytical Process in a Research Laboratory. Applied Sciences, 14(8), 3281. https://doi.org/10.3390/app14083281