Comparison of Water Sampling between Environmental DNA Metabarcoding and Conventional Microscopic Identification: A Case Study in Gwangyang Bay, South Korea




Abstract
:1. Introduction
2. Materials and Methods
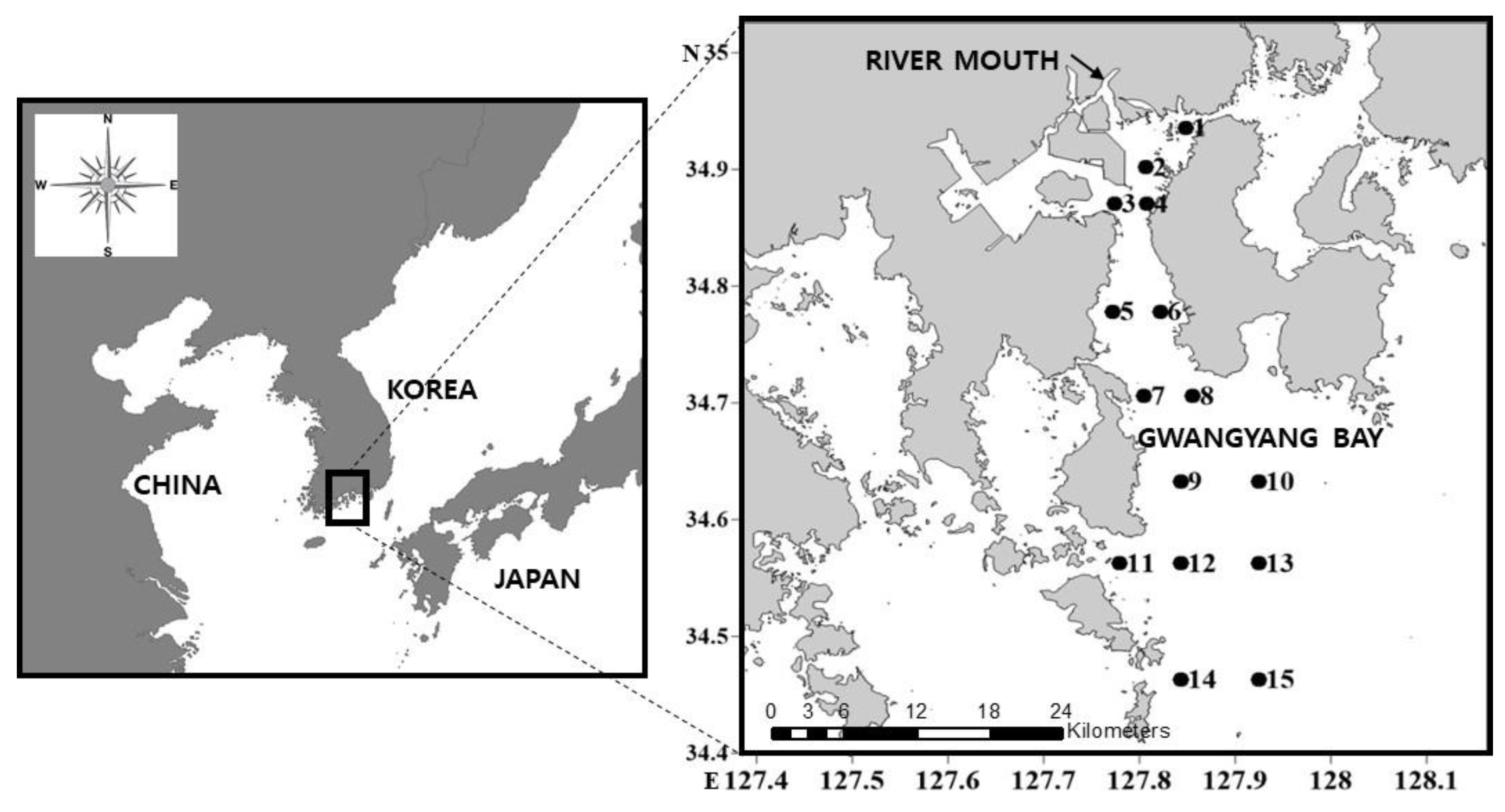
2.1. Description of the Study Site
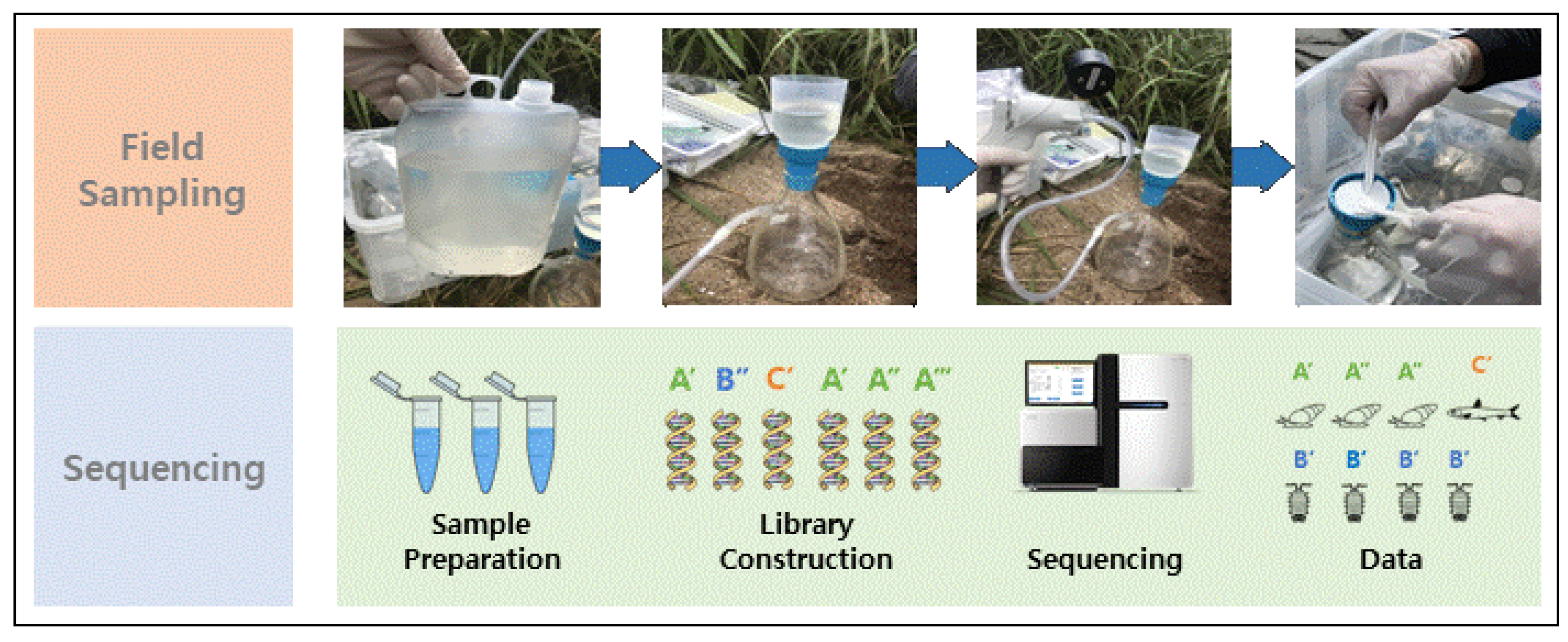
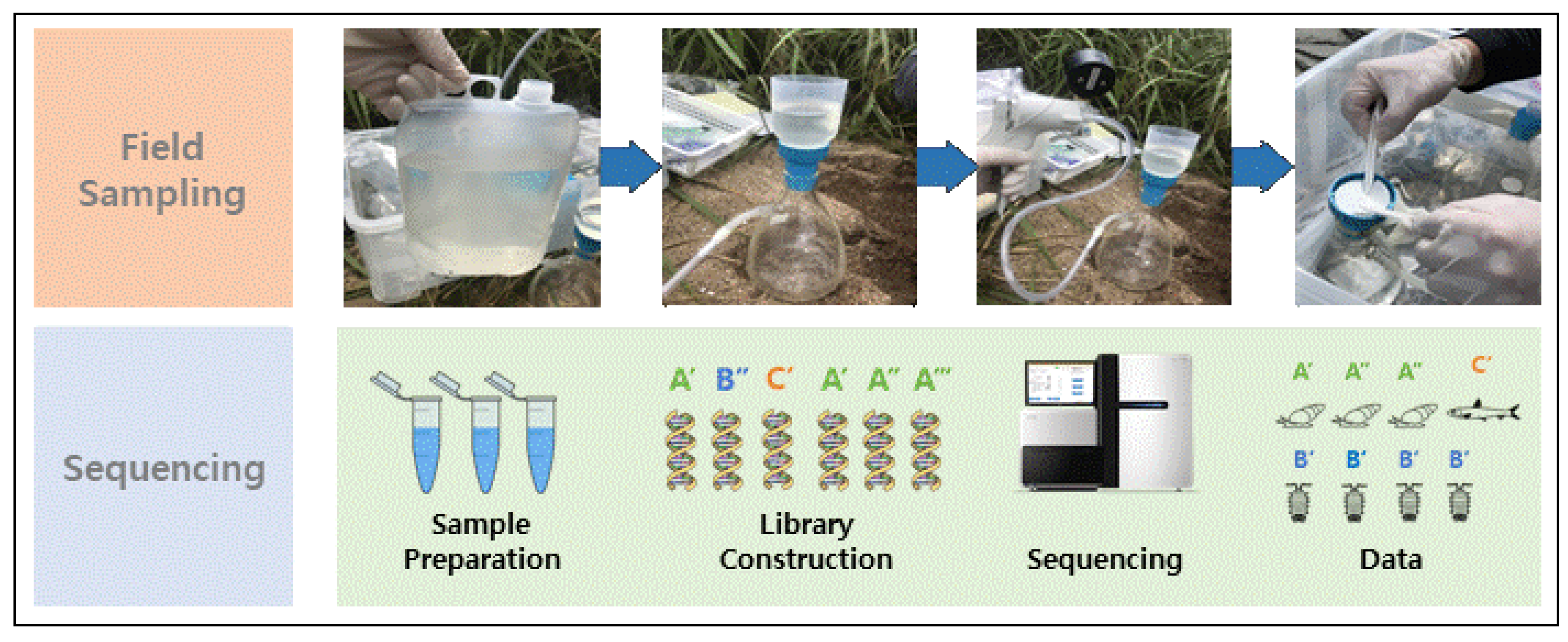
2.2. Sampling and Data Collection
2.3. DNA Extraction and Metagenomic Sequencing
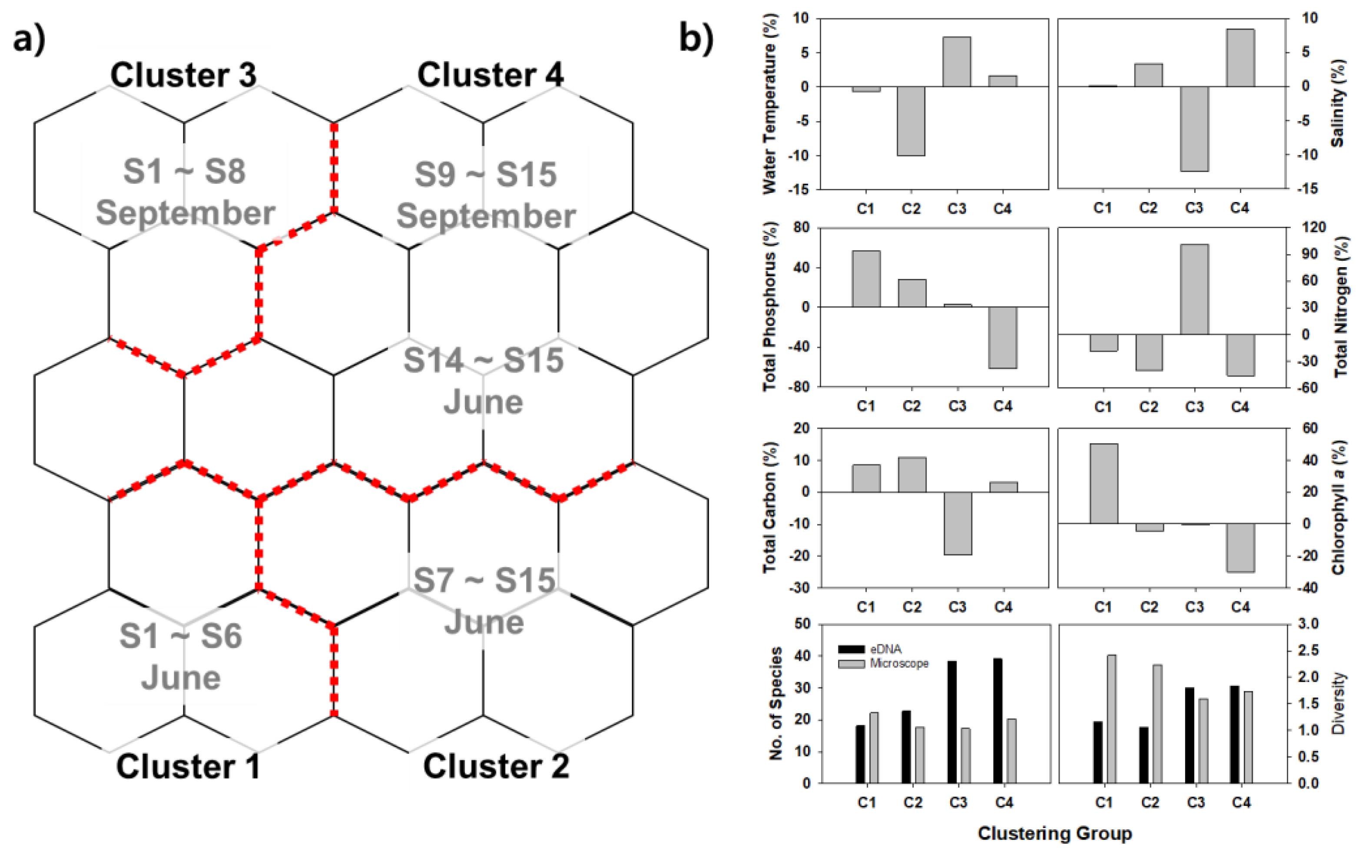
2.4. Analytical Methods
3. Results
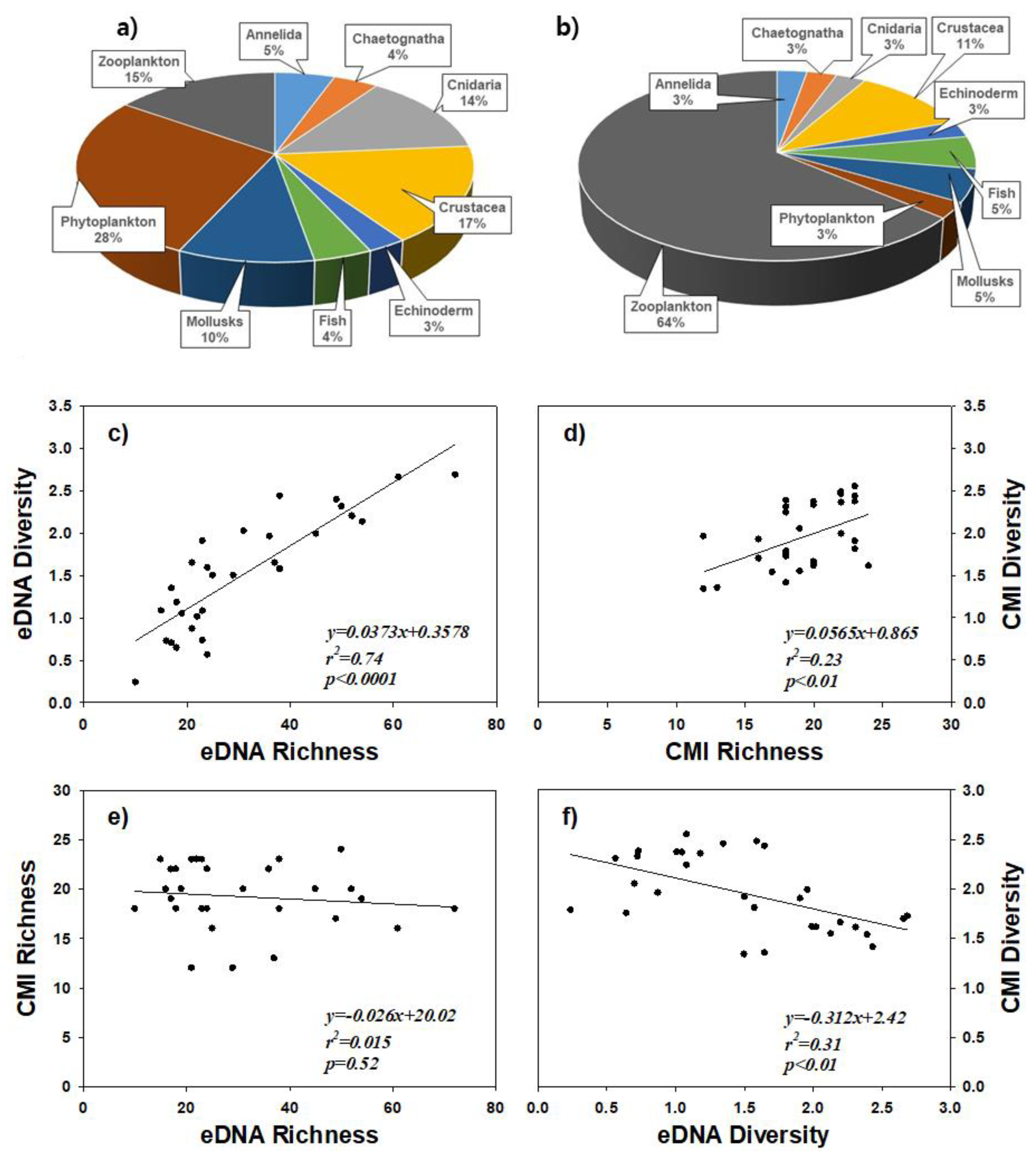
3.1. Comparative Estimation of Coastal Biota between eDNA Metabarcoding and CMI
3.2. Relationships of Biotic Information between eDNA and CMI Samples
3.3. Assessment of Biogeochemical Characteristics in Gwangyang Bay
4. Discussion
4.1. Congruence of Taxonomic Information between eDNA Metabarcoding and CMI
4.2. Potential Values of an eDNA Approach for Biological Monitoring and Assessment
Author Contributions
Funding
Conflicts of Interest
Appendix A. Rarefaction Curves of the 18S rDNA V9 Samples in May (A) and September (B)

Appendix B. Visualization of Explanatory Variables Derived from Self-Organizing Maps


References
- Taberlet, P.; Coissac, E.; Hajibabaei, M.; Rieseberg, L.H. Environmental DNA. Mol. Ecol. 2012, 21, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, P.F.; Willerslev, E. Environmental DNA—An emerging tool in conservation for monitoring past and present biodiversity. Biol. Conserv. 2015, 183, 4–18. [Google Scholar] [CrossRef]
- Ogram, A.; Sayler, G.S.; Barkay, T. The extraction and purification of microbial DNA from sediments. J. Microbiol. Methods 1987, 7, 57–66. [Google Scholar] [CrossRef]
- Pace, N.R.; Stahl, D.A.; Lane, D.J.; Olsen, G.J. The analysis of natural microbial populations by ribosomal RNA sequences. In Advances in Microbial Ecology; Marshall, K.C., Ed.; Springer US: Boston, MA, USA, 1986; pp. 1–55. [Google Scholar]
- Olsen, G.J.; Lane, D.J.; Giovannoni, S.J.; Pace, N.R.; Stahl, D.A. Microbial Ecology and Evolution: A Ribosomal RNA Approach. Annu. Rev. Microbiol. 1986, 40, 337–365. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D. The human genome project: Past, present, and future. Science 1990, 248, 44–49. [Google Scholar] [CrossRef]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental Genome Shotgun Sequencing of the Sargasso Sea. Science 2004, 304, 66. [Google Scholar] [CrossRef] [PubMed]
- Valentini, A.; Taberlet, P.; Miaud, C.; Civade, R.; Herder, J.; Thomsen, P.F.; Bellemain, E.; Besnard, A.; Coissac, E.; Boyer, F.; et al. Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Mol. Ecol. 2016, 25, 929–942. [Google Scholar] [CrossRef] [Green Version]
- Cristescu, M.E. From barcoding single individuals to metabarcoding biological communities: Towards an integrative approach to the study of global biodiversity. Trends Ecol. Evol. 2014, 29, 566–571. [Google Scholar] [CrossRef]
- Shokralla, S.; Spall, J.L.; Gibson, J.F.; Hajibabaei, M. Next-generation sequencing technologies for environmental DNA research. Mol. Ecol. 2012, 21, 1794–1805. [Google Scholar] [CrossRef]
- Taberlet, P.; Coissac, E.; Pompanon, F.; Brochmann, C.; Willerslev, E. Towards next-generation biodiversity assessment using DNA metabarcoding. Mol. Ecol. 2012, 21, 2045–2050. [Google Scholar] [CrossRef]
- Bohmann, K.; Evans, A.; Gilbert, M.T.P.; Carvalho, G.R.; Creer, S.; Knapp, M.; Yu, D.W.; de Bruyn, M. Environmental DNA for wildlife biology and biodiversity monitoring. Trends Ecol. Evol. 2014, 29, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Sigsgaard, E.E.; Carl, H.; Møller, P.R.; Thomsen, P.F. Monitoring the near-extinct European weather loach in Denmark based on environmental DNA from water samples. Biol. Conserv. 2015, 183, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Pawlowski, J.; Kelly-Quinn, M.; Altermatt, F.; Apothéloz-Perret-Gentil, L.; Beja, P.; Boggero, A.; Borja, A.; Bouchez, A.; Cordier, T.; Domaizon, I.; et al. The future of biotic indices in the ecogenomic era: Integrating (e)DNA metabarcoding in biological assessment of aquatic ecosystems. Sci. Total Environ. 2018, 637–638, 1295–1310. [Google Scholar] [CrossRef] [PubMed]
- Darling, J.A.; Mahon, A.R. From molecules to management: Adopting DNA-based methods for monitoring biological invasions in aquatic environments. Environ. Res. 2011, 111, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Baird, D.J.; Hajibabaei, M. Biomonitoring 2.0: A new paradigm in ecosystem assessment made possible by next-generation DNA sequencing. Mol. Ecol. 2012, 21, 2039–2044. [Google Scholar] [CrossRef]
- Goldberg, C.S.; Strickler, K.M.; Pilliod, D.S. Moving environmental DNA methods from concept to practice for monitoring aquatic macroorganisms. Biol. Conserv. 2015, 183, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Ruppert, K.M.; Kline, R.J.; Rahman, M.S. Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: A systematic review in methods, monitoring, and applications of global eDNA. Glob. Ecol. Conserv. 2019, 17, e00547. [Google Scholar] [CrossRef]
- Thomsen, P.F.; Kielgast, J.; Iversen, L.L.; Wiuf, C.; Rasmussen, M.; Gilbert, M.T.P.; Orlando, L.; Willerslev, E. Monitoring endangered freshwater biodiversity using environmental DNA. Mol. Ecol. 2012, 21, 2565–2573. [Google Scholar] [CrossRef]
- Quéré, C.L.; Harrison, S.P.; Colin Prentice, I.; Buitenhuis, E.T.; Aumont, O.; Bopp, L.; Claustre, H.; Cotrim Da Cunha, L.; Geider, R.; Giraud, X.; et al. Ecosystem dynamics based on plankton functional types for global ocean biogeochemistry models. Glob. Chang. Biol. 2005, 11, 2016–2040. [Google Scholar]
- Romare, P.; Bergman, E.; Hansson, L.-A. The impact of larval and juvenile fish on zooplankton and algal dynamics. Limnol. Oceanogr. 1999, 44, 1655–1666. [Google Scholar] [CrossRef]
- Descy, J.-P.; Leitao, M.; Everbecq, E.; Smitz, J.S.; Deliège, J.-F. Phytoplankton of the River Loire, France: A biodiversity and modelling study. J. Plankton Res. 2012, 34, 120–135. [Google Scholar] [CrossRef]
- Wetzel, R.G.; Likens, G.E. Limnological Analysis; Springer-Verlag: New York, NY, USA, 1991; p. 429. [Google Scholar]
- Zimmermann, J.; Glöckner, G.; Jahn, R.; Enke, N.; Gemeinholzer, B. Metabarcoding vs. morphological identification to assess diatom diversity in environmental studies. Mol. Ecol. Resour. 2015, 15, 526–542. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.C.; Maddison, B.C.; Middleditch, D.J.; Patmore, J.R.M.; Gough, K.C. The detection of aquatic animal species using environmental DNA—A review of eDNA as a survey tool in ecology. J. Appl. Ecol. 2014, 51, 1450–1459. [Google Scholar] [CrossRef]
- Kang, C.-K.; Kim, J.B.; Lee, K.-S.; Kim, J.B.; Lee, P.-Y.; Hong, J.-S. Trophic importance of benthic microalgae to macrozoobenthos in coastal bay systems in Korea: Dual stable C and N isotope analyses. Mar. Ecol. Prog. Ser. 2003, 259, 79–92. [Google Scholar] [CrossRef]
- Lee, M.; Park, B.S.; Baek, S.H. Tidal Influences on Biotic and Abiotic Factors in the Seomjin River Estuary and Gwangyang Bay, Korea. Estuaries Coasts 2018, 41, 1977–1993. [Google Scholar] [CrossRef]
- Korean Statistical Information Service (KOSIS). Available online: http://kosis.kr (accessed on 10 July 2018).
- You, Y.-S.; Cho, H.-S.; Choi, Y.-C. A study on the pollution of polycyclic aromatic hydrocarbons (PAHs) in the surface sediments around Gwangyang Bay. J. Korean Soc. Mar. Environ. Saf. 2007, 13, 9–20. [Google Scholar]
- Chihara, M.; Murano, M. An Illustrated Guide to Marine Plankton in Japan; Tokai University Press: Tokyo, Japan, 1997; p. 1574. [Google Scholar]
- Albaina, A.; Aguirre, M.; Abad, D.; Santos, M.; Estonba, A. 18S rRNA V9 metabarcoding for diet characterization: A critical evaluation with two sympatric zooplanktivorous fish species. Ecol. Evol. 2016, 6, 1809–1824. [Google Scholar] [CrossRef]
- Abad, D.; Albaina, A.; Aguirre, M.; Laza-Martínez, A.; Uriarte, I.; Iriarte, A.; Villate, F.; Estonba, A. Is metabarcoding suitable for estuarine plankton monitoring? A comparative study with microscopy. Mar. Biol. 2016, 163, 149. [Google Scholar] [CrossRef]
- Guo, L.; Sui, Z.; Liu, Y. Quantitative analysis of dinoflagellates and diatoms community via Miseq sequencing of actin gene and v9 region of 18S rDNA. Sci. Rep. 2016, 6, 34709. [Google Scholar] [CrossRef] [Green Version]
- Amaral-Zettler, L.A.; McCliment, E.A.; Ducklow, H.W.; Huse, S.M. A Method for Studying Protistan Diversity Using Massively Parallel Sequencing of V9 Hypervariable Regions of Small-Subunit Ribosomal RNA Genes. PLoS ONE 2009, 4, e6372. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335. [Google Scholar] [CrossRef]
- Kohonen, T. Self-Organizing Maps; Springer: New York, NY, USA, 1997; p. 426. [Google Scholar]
- Giraudel, J.L.; Lek, S. A comparison of self-organizing map algorithm and some conventional statistical methods for ecological community ordination. Ecol. Model. 2001, 146, 329–339. [Google Scholar] [CrossRef]
- Chon, T.-S. Self-Organizing Maps applied to ecological sciences. Ecol. Inform. 2011, 6, 50–61. [Google Scholar] [CrossRef]
- Kim, D.-K.; Jo, H.; Han, I.; Kwak, I.-S. Explicit Characterization of Spatial Heterogeneity Based on Water Quality, Sediment Contamination, and Ichthyofauna in a Riverine-to-Coastal Zone. Int. J. Environ. Res. Public Health 2019, 16, 409. [Google Scholar] [CrossRef]
- Kim, D.-K.; Javed, A.; Yang, C.; Arhonditsis, G.B. Development of a mechanistic eutrophication model for wetland management: Sensitivity analysis of the interplay among phytoplankton, macrophtyes, and sediment nutrient release. Ecol. Inform. 2018, 48, 198–214. [Google Scholar] [CrossRef]
- Chon, T.-S.; Park, Y.-S.; Cha, E.Y. Patterning of community changes in bentic macroinvertebrates collected from urbanized streams for the short term prediction by temporal artificial neuronal networks. In Artificial Neuronal Networks: Application to Ecology and Evolution; Lek, S., Guegan, J.F., Eds.; Springer: Berlin, Germany, 2000; pp. 99–114. [Google Scholar]
- Park, Y.-S.; Kwon, Y.-S.; Hwang, S.-J.; Park, S. Characterizing effects of landscape and morphometric factors on water quality of reservoirs using a self-organizing map. Environ. Model. Softw. 2014, 55, 214–221. [Google Scholar] [CrossRef]
- Kim, D.-K.; Jeong, K.-S.; Chang, K.-H.; La, G.-H.; Joo, G.-J.; Kim, H.-W. Patterning zooplankton communities in accordance with annual climatic conditions in a regulated river system (the Nakdong River, South Korea). Int. Rev. Hydrobiol. 2012, 97, 55–72. [Google Scholar] [CrossRef]
- Hadjisolomou, E.; Stefanidis, K.; Papatheodorou, G.; Papastergiadou, E. Assessment of the Eutrophication-Related Environmental Parameters in Two Mediterranean Lakes by Integrating Statistical Techniques and Self-Organizing Maps. Int. J. Environ. Res. Public Health 2018, 15, 547. [Google Scholar] [CrossRef]
- Várbíró, G.; Ács, É.; Borics, G.; Érces, K.; Fehér, G.; Grigorszky, I.; Japport, T.; Kocsis, G.; Krasznai, E.; Nagy, K.; et al. Use of self-organizing maps (SOM) for characterization of riverine phytoplankton associations in Hungary. Arch. Hydrobiol. 2007, 161, 388–394. [Google Scholar] [CrossRef]
- Tison, J.; Giraudel, J.L.; Coste, M.; Park, Y.S.; Delmas, F. Use of unsupervised neural networks for ecoregional zoning of hydrosystems through diatom communities: Case study of Adour-Garonne watershed (France). Arch. Hydrobiol. 2004, 159, 409–422. [Google Scholar] [CrossRef]
- Cuss, C.W.; Gueguen, C. Analysis of dissolved organic matter fluorescence using self-organizing maps: Mini-review and tutorial. Anal. Methods 2016, 8, 716–725. [Google Scholar] [CrossRef]
- Penczak, T.; Głowacki, Ł.; Kruk, A.; Galicka, W. Implementation of a self-organizing map for investigation of impoundment impact on fish assemblages in a large, lowland river: Long-term study. Ecol. Model. 2012, 227, 64–71. [Google Scholar] [CrossRef]
- Brosse, S.; Giraudel, J.L.; Lek, S. Utilisation of non-supervised neural networks and principal component analysis to study fish assemblages. Ecol. Model. 2001, 146, 159–166. [Google Scholar] [CrossRef]
- Ha, J.-Y.; Hanazato, T.; Chang, K.-H.; Jeong, K.-S.; Kim, D.-K. Assessment of the lake biomanipulation by introducing both piscivorous rainbow trout and herbivorous daphnids using self-organizing map analysis: A case study in Lake Shirakaba, Japan. Ecol. Inform. 2015, 29, 182–191. [Google Scholar] [CrossRef]
- Jeong, K.-S.; Kim, D.-K.; Pattnaik, A.; Bhatta, K.; Bhandari, B.; Joo, G.-J. Patterning limnological characteristics of the Chilika lagoon (India) using a self-organizing map. Limnology 2008, 9, 231–242. [Google Scholar] [CrossRef]
- Vesanto, J.; Alhoniemi, E. Clustering of the Self-Organizing Map. IEEE Trans. Neural Netw. 2000, 11, 586–600. [Google Scholar] [CrossRef]
- Shannon, C.E.; Weaver, W. The Mathematical Theory of Communication; The University of Illinois Press: Urbana, IL, USA, 1964; p. 125. [Google Scholar]
- Vasselon, V.; Rimet, F.; Tapolczai, K.; Bouchez, A. Assessing ecological status with diatoms DNA metabarcoding: Scaling-up on a WFD monitoring network (Mayotte island, France). Ecol. Indic. 2017, 82, 1–12. [Google Scholar] [CrossRef]
- Sun, C.; Zhao, Y.; Li, H.; Dong, Y.; MacIsaac, H.J.; Zhan, A. Unreliable quantitation of species abundance based on high-throughput sequencing data of zooplankton communities. Aquat. Biol. 2015, 24, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.P.; Port, J.A.; Yamahara, K.M.; Martone, R.G.; Lowell, N.; Thomsen, P.F.; Mach, M.E.; Bennett, M.; Prahler, E.; Caldwell, M.R.; et al. Harnessing DNA to improve environmental management. Science 2014, 344, 1455–1456. [Google Scholar] [CrossRef]
- Goldberg, C.S.; Turner, C.R.; Deiner, K.; Klymus, K.E.; Thomsen, P.F.; Murphy, M.A.; Spear, S.F.; McKee, A.; Oyler-McCance, S.J.; Cornman, R.S.; et al. Critical considerations for the application of environmental DNA methods to detect aquatic species. Methods Ecol. Evol. 2016, 7, 1299–1307. [Google Scholar] [CrossRef]
- Kembel, S.W.; Wu, M.; Eisen, J.A.; Green, J.L. Incorporating 16S Gene Copy Number Information Improves Estimates of Microbial Diversity and Abundance. PLoS Comp. Biol. 2012, 8, e1002743. [Google Scholar] [CrossRef]
- Kelly, R.P.; Closek, C.J.; O’Donnell, J.L.; Kralj, J.E.; Shelton, A.O.; Samhouri, J.F. Genetic and Manual Survey Methods Yield Different and Complementary Views of an Ecosystem. Front. Mar. Sci. 2017, 3, 283. [Google Scholar] [CrossRef]
- Stoeck, T.; Breiner, H.-W.; Filker, S.; Ostermaier, V.; Kammerlander, B.; Sonntag, B. A morphogenetic survey on ciliate plankton from a mountain lake pinpoints the necessity of lineage-specific barcode markers in microbial ecology. Environ. Microbiol. 2014, 16, 430–444. [Google Scholar] [CrossRef]
- Gonzalez, J.M.; Portillo, M.C.; Belda-Ferre, P.; Mira, A. Amplification by PCR Artificially Reduces the Proportion of the Rare Biosphere in Microbial Communities. PLoS ONE 2012, 7, e29973. [Google Scholar] [CrossRef]
- Elbrecht, V.; Leese, F. Can DNA-Based Ecosystem Assessments Quantify Species Abundance? Testing Primer Bias and Biomass—Sequence Relationships with an Innovative Metabarcoding Protocol. PLoS ONE 2015, 10, e0130324. [Google Scholar] [CrossRef]
- Piñol, J.; San Andrés, V.; Clare, E.L.; Mir, G.; Symondson, W.O.C. A pragmatic approach to the analysis of diets of generalist predators: The use of next-generation sequencing with no blocking probes. Mol. Ecol. Resour. 2014, 14, 18–26. [Google Scholar] [CrossRef]
- Chen, G.; Hare, M.P. Cryptic ecological diversification of a planktonic estuarine copepod. Acartia Tonsa. Mol. Ecol. 2008, 17, 1451–1468. [Google Scholar] [CrossRef]
- Lindeque, P.K.; Parry, H.E.; Harmer, R.A.; Somerfield, P.J.; Atkinson, A. Next Generation Sequencing Reveals the Hidden Diversity of Zooplankton Assemblages. PLoS ONE 2013, 8, e81327. [Google Scholar] [CrossRef]
- Hirai, J.; Kuriyama, M.; Ichikawa, T.; Hidaka, K.; Tsuda, A. A metagenetic approach for revealing community structure of marine planktonic copepods. Mol. Ecol. Resour. 2015, 15, 68–80. [Google Scholar] [CrossRef]
- Massana, R.; Gobet, A.; Audic, S.; Bass, D.; Bittner, L.; Boutte, C.; Chambouvet, A.; Christen, R.; Claverie, J.-M.; Decelle, J.; et al. Marine protist diversity in European coastal waters and sediments as revealed by high-throughput sequencing. Environ. Microbiol. 2015, 17, 4035–4049. [Google Scholar] [CrossRef] [Green Version]
- Coissac, E.; Riaz, T.; Puillandre, N. Bioinformatic challenges for DNA metabarcoding of plants and animals. Mol. Ecol. 2012, 21, 1834–1847. [Google Scholar] [CrossRef]
- Eichmiller, J.J.; Best, S.E.; Sorensen, P.W. Effects of Temperature and Trophic State on Degradation of Environmental DNA in Lake Water. Environ. Sci. Technol. 2016, 50, 1859–1867. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| June | September | |||||||
|---|---|---|---|---|---|---|---|---|
| eDNA | CMI | eDNA | CMI | |||||
| Site | Richness | Diversity | Richness | Diversity | Richness | Diversity | Richness | Diversity |
| GY1 | 28 (6) | 1.01 | 23 | 2.37 | 54 (9) | 1.99 | 20 | 1.62 |
| GY2 | 28 (7) | 1.65 | 23 | 2.43 | 59 (10) | 2.39 | 17 | 1.54 |
| GY3 | 20 (5) | 1.08 | 23 | 2.55 | 34 (5) | 1.50 | 12 | 1.34 |
| GY4 | 23 (6) | 1.35 | 22 | 2.46 | 72 (11) | 2.66 | 16 | 1.70 |
| GY5 | 25 (7) | 1.18 | 22 | 2.36 | 44 (7) | 1.65 | 13 | 1.35 |
| GY6 | 22 (6) | 0.72 | 20 | 2.33 | 62 (8) | 2.13 | 19 | 1.55 |
| GY7 | 29 (8) | 0.87 | 12 | 1.96 | 13 (3) | 0.24 | 18 | 1.78 |
| GY8 | 33 (10) | 1.08 | 18 | 2.24 | 29 (6) | 1.91 | 23 | 1.90 |
| GY9 | 35 (11) | 1.59 | 22 | 2.48 | 58 (8) | 2.31 | 24 | 1.61 |
| GY10 | 27 (8) | 1.05 | 20 | 2.37 | 48 (10) | 2.44 | 18 | 1.41 |
| GY11 | 36 (11) | 1.50 | 16 | 1.92 | 72 (11) | 2.69 | 18 | 1.72 |
| GY12 | 34 (10) | 0.56 | 18 | 2.31 | 35 (4) | 2.02 | 20 | 1.61 |
| GY13 | 31 (8) | 0.73 | 18 | 2.38 | 46 (8) | 1.57 | 23 | 1.81 |
| GY14 | 24 (6) | 0.64 | 18 | 1.75 | 45 (9) | 1.96 | 22 | 1.99 |
| GY15 | 24 (7) | 0.70 | 19 | 2.05 | 64 (12) | 2.20 | 20 | 1.66 |
| Mean | 27.9 (7.7) | 1.0 | 19.6 | 2.3 | 49.7 (8.1) | 2.0 | 18.9 | 1.6 |
| S.D. | 5.0 (1.9) | 0.4 | 3.1 | 0.2 | 17.8 (2.7) | 0.6 | 3.5 | 0.2 |
| Site | eDNA Metabarcoding | CMI |
|---|---|---|
| GY1 | Acropora, Candacia, Caprella, Oryzias | Acartia, Paracalanus |
| GY2 | Acropora, Candacia, Caprella, Corophium, Oryzias | Acartia, Corycaeus, Centropages, Corycaeus, Oithona, Paracalanus, Sagitta |
| GY3 | Acartia, Centropages | Acartia, Noctiluca, Oithona, Paracalanus, Sagitta |
| GY4 | Acartia, Acropora, Caprella, Corophium | Acartia, Corycaeus, Noctiluca, Oithona, Paracalanus, Sagitta |
| GY5 | Acartia, Acropora, Centropages | Acartia, Noctiluca, Paracalanus, Sagitta |
| GY6 | Acropora, Hematodinium | Acartia, Corycaeus, Noctiluca, Oithona, Paracalanus, Sagitta |
| GY7 | Acartia | Centropages |
| GY8 | Acropora, Caprella, | Centropages, Noctiluca |
| GY9 | Acartia, Acropora, Candacia, Centropages, Hematodinium | Centropages, Noctiluca |
| GY10 | Acropora, Thalassiosira | Centropages, Corycaeus, Sagitta |
| GY11 | Candacia, Caprella, Centropages | Centropages |
| GY12 | Centropages, Hematodinium, | Centropages, Paracalanus |
| GY13 | Candacia, Centropages | Centropages, Paracalanus |
| GY14 | Hematodinium | Oithona |
| GY15 | Hematodinium | Oithona |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.-K.; Park, K.; Jo, H.; Kwak, I.-S. Comparison of Water Sampling between Environmental DNA Metabarcoding and Conventional Microscopic Identification: A Case Study in Gwangyang Bay, South Korea. Appl. Sci. 2019, 9, 3272. https://doi.org/10.3390/app9163272
Kim D-K, Park K, Jo H, Kwak I-S. Comparison of Water Sampling between Environmental DNA Metabarcoding and Conventional Microscopic Identification: A Case Study in Gwangyang Bay, South Korea. Applied Sciences. 2019; 9(16):3272. https://doi.org/10.3390/app9163272
Chicago/Turabian StyleKim, Dong-Kyun, Kiyun Park, Hyunbin Jo, and Ihn-Sil Kwak. 2019. "Comparison of Water Sampling between Environmental DNA Metabarcoding and Conventional Microscopic Identification: A Case Study in Gwangyang Bay, South Korea" Applied Sciences 9, no. 16: 3272. https://doi.org/10.3390/app9163272
APA StyleKim, D.-K., Park, K., Jo, H., & Kwak, I.-S. (2019). Comparison of Water Sampling between Environmental DNA Metabarcoding and Conventional Microscopic Identification: A Case Study in Gwangyang Bay, South Korea. Applied Sciences, 9(16), 3272. https://doi.org/10.3390/app9163272