
Sodium–Glucose Cotransporter Inhibitors: Cellular Mechanisms Involved in the Lipid Metabolism and the Treatment of Chronic Kidney Disease Associated with Metabolic Syndrome

 , , ,
, , , 
Abstract

1. Introduction
2. Methodology
3. Risk Factors Involved in Target Organ Damage
Obesity, Dyslipidemia, and Insulin Resistance as Pathological Mechanisms
4. Role of Lipids in Renal Damage
4.1. Cellular Mechanisms Involved in Kidney Lipotoxicity
4.2. The Role of Mitochondria in Lipid-Induced Renal Damage in Metabolic Syndrome
5. Treatment of Chronic Kidney Disease Induced by Lipotoxicity
5.1. Traditional Medicine Utilizing Sodium–Glucose Cotransporter Modulation as an Alternative Therapy for RFs
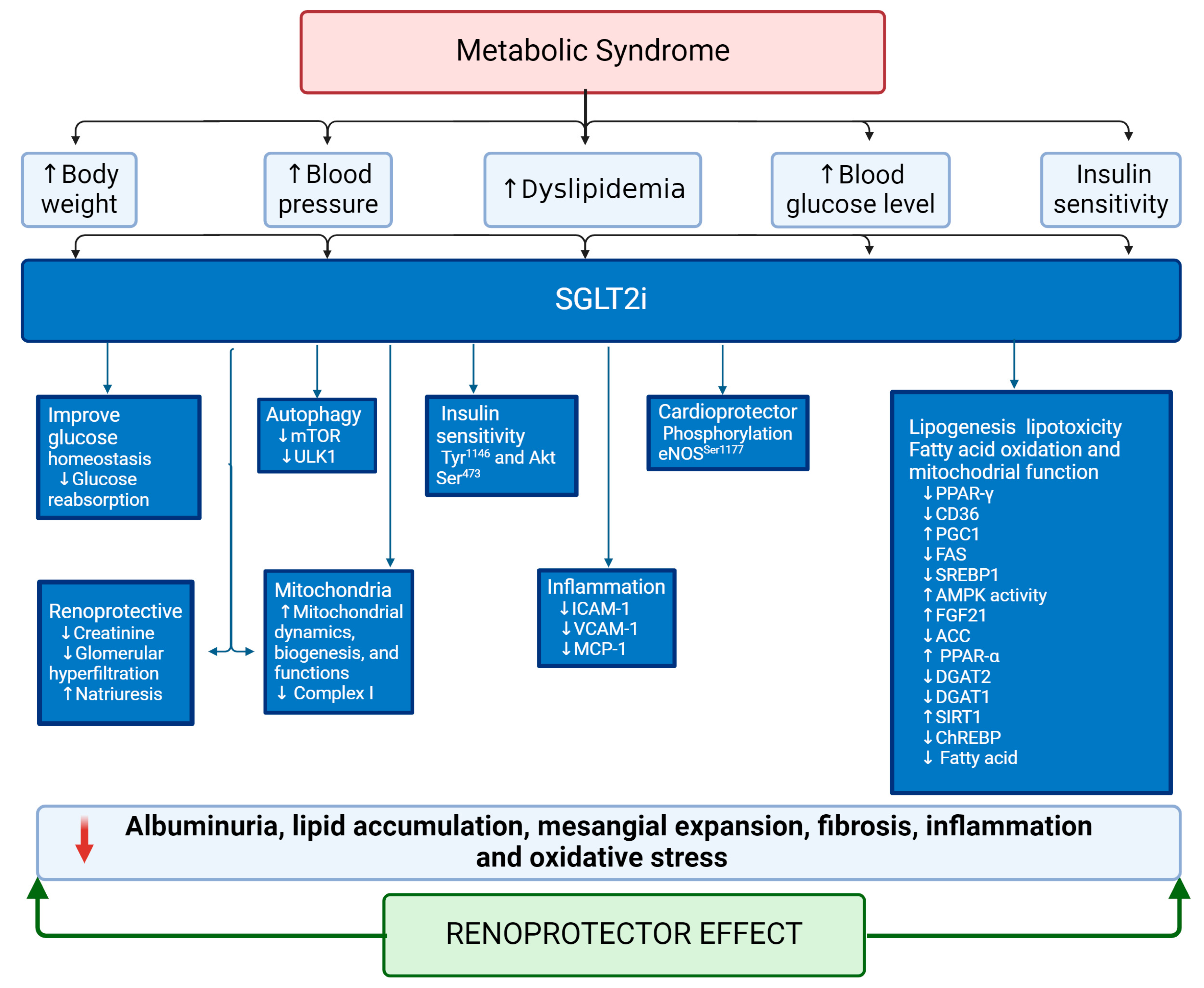
5.2. Sodium–Glucose Cotransporter 2 Inhibitors (SGLT2i) as an Emergent Treatment for MetS
5.3. Sodium–glucose Cotransporter 2 Inhibitors (SGLT2i) as an Emerging Treatment for Renal Lipotoxicity
6. Discussion
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rizvi, A.A.; Stoian, A.P.; Rizzo, M. Metabolic Syndrome: From Molecular Mechanisms to Novel Therapies. Int. J. Mol. Sci. 2021, 22, 38. [Google Scholar] [CrossRef]
- Bovolini, A.; Garcia, J.; Andrade, M.A.; Duarte, J.A. Metabolic Syndrome Pathophysiology and Predisposing Factors. Int. J. Sports Med. 2021, 42, 199–214. [Google Scholar] [CrossRef]
- Arellano Buendia, A.S.; Juárez Rojas, J.G.; García-Arroyo, F.; Aparicio Trejo, O.E.; Sánchez-Muñoz, F.; Argüello-García, R.; Sánchez-Lozada, L.G.; Bojalil, R.; Osorio-Alonso, H. Antioxidant and anti-inflammatory effects of allicin in the kidney of an experimental model of metabolic syndrome. PeerJ 2023, 11, e16132. [Google Scholar] [CrossRef]
- Xu, H.; Li, X.; Adams, H.; Kubena, K.; Guo, S. Etiology of Metabolic Syndrome and Dietary Intervention. Int. J. Mol. Sci. 2018, 20, 128. [Google Scholar] [CrossRef] [PubMed]
- Noubiap, J.J.; Nansseu, J.R.; Lontchi-Yimagou, E.; Nkeck, J.R.; Nyaga, U.F.; Ngouo, A.T.; Tounouga, D.N.; Tianyi, F.L.; Foka, A.J.; Ndoadoumgue, A.L.; et al. Geographic distribution of metabolic syndrome and its components in the general adult population: A meta-analysis of global data from 28 million individuals. Diabetes Res. Clin. Pract. 2022, 188, 109924. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.; Bhuket, T.; Torres, S.; Liu, B.; Wong, R.J. Prevalence of the metabolic syndrome in the United States, 2003–2012. Jama 2015, 313, 1973–1974. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Martínez, R.; Aguilar-Salinas, C.A.; Romero-Martínez, M.; Castro-Porras, L.; Gómez-Velasco, D.; Mehta, R. Trends in the prevalence of metabolic syndrome and its components in Mexican adults, 2006-2018. Salud Publica Mex 2021, 63, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chen, D.; Peng, Y.; Lu, Z.; Kwan, M.P.; Tse, L.A. Association Between Metabolic Syndrome and Mortality: Prospective Cohort Study. JMIR Public. Health Surveill. 2023, 9, e44073. [Google Scholar] [CrossRef]
- Campos-Nonato, I.; Hernández-Barrera, L.; Pedroza-Tobías, A.; Medina, C.; Barquera, S. Hypertension in Mexican adults: Prevalence, diagnosis and type of treatment. Ensanut MC 2016. Salud Publica Mex. 2018, 60, 233–243. [Google Scholar] [CrossRef]
- Wahl, P.; Ducasa, G.M.; Fornoni, A. Systemic and renal lipids in kidney disease development and progression. Am. J. Physiol. Renal Physiol. 2016, 310, F433–F445. [Google Scholar] [CrossRef]
- Zhang, X.; Lerman, L.O. The metabolic syndrome and chronic kidney disease. Transl. Res. 2017, 183, 14–25. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, A.; Gratteri, S.; Gualtieri, P.; Cammarano, A.; Bertucci, P.; Di Renzo, L. Why primary obesity is a disease? J. Transl. Med. 2019, 17, 169. [Google Scholar] [CrossRef] [PubMed]
- Neves, R.O.; Rocha, A.D.S.; de Vargas, B.O.; Kretzer, D.C.; de Matos, S.; Goldani, M.Z.; von Diemen, L.; Magalhães, J.A.A.; Bernardi, J.R. Obesity Cut-Off Points Using Prepregnancy Body Mass Index according to Cardiometabolic Conditions in Pregnancy. J. Pregnancy 2023, 2023, 6669700. [Google Scholar] [CrossRef] [PubMed]
- Barquera, S.; Hernández-Barrera, L.; Trejo-Valdivia, B.; Shamah, T.; Campos-Nonato, I.; Rivera-Dommarco, J. Obesity in Mexico, prevalence andtrends in adults. Ensanut 2018-19. Salud Publica Mex. 2020, 62, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Koenen, M.; Hill, M.A.; Cohen, P.; Sowers, J.R. Obesity, Adipose Tissue and Vascular Dysfunction. Circ. Res. 2021, 128, 951–968. [Google Scholar] [CrossRef] [PubMed]
- Unamuno, X.; Gómez-Ambrosi, J.; Rodríguez, A.; Becerril, S.; Frühbeck, G.; Catalán, V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur. J. Clin. Investig. 2018, 48, e12997. [Google Scholar] [CrossRef] [PubMed]
- Sakers, A.; De Siqueira, M.K.; Seale, P.; Villanueva, C.J. Adipose-tissue plasticity in health and disease. Cell 2022, 185, 419–446. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, D.; Montecucco, F.; Dallegri, F.; Carbone, F. Impact of different ectopic fat depots on cardiovascular and metabolic diseases. J. Cell Physiol. 2019, 234, 21630–21641. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.; Cline, D.L.; Glavas, M.M.; Covey, S.D.; Kieffer, T.J. Tissue-Specific Effects of Leptin on Glucose and Lipid Metabolism. Endocr. Rev. 2021, 42, 1–28. [Google Scholar] [CrossRef]
- Stenkula, K.G.; Erlanson-Albertsson, C. Adipose cell size: Importance in health and disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R284–R295. [Google Scholar] [CrossRef]
- Vekic, J.; Stefanovic, A.; Zeljkovic, A. Obesity and Dyslipidemia: A Review of Current Evidence. Curr. Obes. Rep. 2023, 12, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-García, A.; Arranz-Martínez, E.; López-Uriarte, B.; Rivera-Teijido, M.; Palacios-Martínez, D.; Dávila-Blázquez, G.M.; Rosillo-González, A.; González-Posada Delgado, J.A.; Mariño-Suárez, J.E.; Revilla-Pascual, E.; et al. Prevalence of hypertriglyceridemia in adults and related cardiometabolic factors. SIMETAP-HTG study. Clin. Investig. Arterioscler. 2020, 32, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Samadder, M.; Shourove, J.H.; Taher, A.; Islam, F. Prevalence and factors associated with metabolic syndrome in university students and academic staff in Bangladesh. Sci. Rep. 2023, 13, 19912. [Google Scholar] [CrossRef] [PubMed]
- Márquez, A.B.; Nazir, S.; van der Vorst, E.P.C. High-Density Lipoprotein Modifications: A Pathological Consequence or Cause of Disease Progression? Biomedicines 2020, 8, 549. [Google Scholar] [CrossRef] [PubMed]
- Denimal, D. Antioxidant and Anti-Inflammatory Functions of High-Density Lipoprotein in Type 1 and Type 2 Diabetes. Antioxidants 2024, 13, 57. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C.; Yuhanna, I.S.; Quon, M.J.; Shaul, P.W. High density lipoprotein-induced endothelial nitric-oxide synthase activation is mediated by Akt and MAP kinases. J. Biol. Chem. 2003, 278, 9142–9149. [Google Scholar] [CrossRef] [PubMed]
- Khatana, C.; Saini, N.K.; Chakrabarti, S.; Saini, V.; Sharma, A.; Saini, R.V.; Saini, A.K. Mechanistic Insights into the Oxidized Low-Density Lipoprotein-Induced Atherosclerosis. Oxid. Med. Cell Longev. 2020, 2020, 5245308. [Google Scholar] [CrossRef] [PubMed]
- Noels, H.; Lehrke, M.; Vanholder, R.; Jankowski, J. Lipoproteins and fatty acids in chronic kidney disease: Molecular and metabolic alterations. Nat. Rev. Nephrol. 2021, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Dincer, N.; Dagel, T.; Afsar, B.; Covic, A.; Ortiz, A.; Kanbay, M. The effect of chronic kidney disease on lipid metabolism. Int. Urol. Nephrol. 2019, 51, 265–277. [Google Scholar] [CrossRef]
- Cignarelli, A.; Genchi, V.A.; Perrini, S.; Natalicchio, A.; Laviola, L.; Giorgino, F. Insulin and Insulin Receptors in Adipose Tissue Development. Int. J. Mol. Sci. 2019, 20, 759. [Google Scholar] [CrossRef]
- Thongnak, L.; Pongchaidecha, A.; Lungkaphin, A. Renal Lipid Metabolism and Lipotoxicity in Diabetes. Am. J. Med. Sci. 2020, 359, 84–99. [Google Scholar] [CrossRef] [PubMed]
- Fahed, G.; Aoun, L.; Bou Zerdan, M.; Allam, S.; Bou Zerdan, M.; Bouferraa, Y.; Assi, H.I. Metabolic Syndrome: Updates on Pathophysiology and Management in 2021. Int. J. Mol. Sci. 2022, 23, 786. [Google Scholar] [CrossRef]
- Pantoja-Torres, B.; Toro-Huamanchumo, C.J.; Urrunaga-Pastor, D.; Guarnizo-Poma, M.; Lazaro-Alcantara, H.; Paico-Palacios, S.; Del Carmen Ranilla-Seguin, V.; Benites-Zapata, V.A. High triglycerides to HDL-cholesterol ratio is associated with insulin resistance in normal-weight healthy adults. Diabetes Metab. Syndr. 2019, 13, 382–388. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, S.Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef]
- Reaven, G.M.; Greenfield, M.S. Diabetic hypertriglyceridemia: Evidence for three clinical syndromes. Diabetes 1981, 30, 66–75. [Google Scholar] [CrossRef]
- Afshinnia, F.; Nair, V.; Lin, J.; Rajendiran, T.M.; Soni, T.; Byun, J.; Sharma, K.; Fort, P.E.; Gardner, T.W.; Looker, H.C.; et al. Increased lipogenesis and impaired β-oxidation predict type 2 diabetic kidney disease progression in American Indians. JCI Insight 2019, 4, e130317. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.A.; do Carmo, J.M.; Li, X.; Wang, Z.; Mouton, A.J.; Hall, J.E. Role of Hyperinsulinemia and Insulin Resistance in Hypertension: Metabolic Syndrome Revisited. Can. J. Cardiol. 2020, 36, 671–682. [Google Scholar] [CrossRef] [PubMed]
- García-Carrasco, A.; Izquierdo-Lahuerta, A.; Medina-Gómez, G. The Kidney-Heart Connection in Obesity. Nephron 2021, 145, 604–608. [Google Scholar] [CrossRef]
- Whaley-Connell, A.; Sowers, J.R. Insulin Resistance in Kidney Disease: Is There a Distinct Role Separate from That of Diabetes or Obesity? Cardiorenal Med. 2017, 8, 41–49. [Google Scholar] [CrossRef]
- Opazo-Ríos, L.; Mas, S.; Marín-Royo, G.; Mezzano, S.; Gómez-Guerrero, C.; Moreno, J.A.; Egido, J. Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 2632. [Google Scholar] [CrossRef]
- Hall, J.E.; Mouton, A.J.; da Silva, A.A.; Omoto, A.C.M.; Wang, Z.; Li, X.; do Carmo, J.M. Obesity, kidney dysfunction, and inflammation: Interactions in hypertension. Cardiovasc. Res. 2021, 117, 1859–1876. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.Z.; Szeto, C.C. Mitochondrial dysfunction in diabetic kidney disease. Clin. Chim. Acta 2019, 496, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Miricescu, D.; Balan, D.G.; Tulin, A.; Stiru, O.; Vacaroiu, I.A.; Mihai, D.A.; Popa, C.C.; Enyedi, M.; Nedelea, A.S.; Nica, A.E.; et al. Impact of adipose tissue in chronic kidney disease development (Review). Exp. Ther. Med. 2021, 21, 539. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.E.; Yoo, K.H. Obesity and chronic kidney disease: Prevalence, mechanism, and management. Clin. Exp. Pediatr. 2021, 64, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Kon, V.; Yang, H.C.; Smith, L.E.; Vickers, K.C.; Linton, M.F. High-Density Lipoproteins in Kidney Disease. Int. J. Mol. Sci. 2021, 22, 8201. [Google Scholar] [CrossRef] [PubMed]
- Strazzella, A.; Ossoli, A.; Calabresi, L. High-Density Lipoproteins and the Kidney. Cells 2021, 10, 764. [Google Scholar] [CrossRef] [PubMed]
- Stadler, J.T.; Marsche, G. Obesity-Related Changes in High-Density Lipoprotein Metabolism and Function. Int. J. Mol. Sci. 2020, 21, 8985. [Google Scholar] [CrossRef]
- Miljkovic, M.; Stefanovic, A.; Simic-Ogrizovic, S.; Vekic, J.; Bogavac-Stanojevic, N.; Cerne, D.; Kocbek, P.; Marc, J.; Jelic-Ivanovic, Z.; Spasojevic-Kalimanovska, V.; et al. Association of Dyslipidemia, Oxidative Stress, and Inflammation With Redox Status in VLDL, LDL, and HDL Lipoproteins in Patients With Renal Disease. Angiology 2018, 69, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Lin, Y.; Liang, B.; Zhao, X.; Qiu, M.; Huang, H.; Li, C.; Wang, W.; Kong, Y. Statins attenuate cholesterol-induced ROS via inhibiting NOX2/NOX4 and mitochondrial pathway in collecting ducts of the kidney. BMC Nephrol. 2022, 23, 184. [Google Scholar] [CrossRef]
- Rogacka, D. Insulin resistance in glomerular podocytes: Potential mechanisms of induction. Arch. Biochem. Biophys. 2021, 710, 109005. [Google Scholar] [CrossRef]
- Merscher, S.; Pedigo, C.E.; Mendez, A.J. Metabolism, energetics, and lipid biology in the podocyte—Cellular cholesterol-mediated glomerular injury. Front. Endocrinol. 2014, 5, 169. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, S.; Guo, J. Lipotoxic Proximal Tubular Injury: A Primary Event in Diabetic Kidney Disease. Front. Med. 2021, 8, 751529. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in chronic kidney disease: Novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Wilbon, S.S.; Fornoni, A. Podocyte Lipotoxicity in CKD. Kidney360 2021, 2, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Cui, S.; Hou, Y.; Yi, F. The Updates of Podocyte Lipid Metabolism in Proteinuric Kidney Disease. Kidney Dis. 2021, 7, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Ebnet, K. Junctional Adhesion Molecules (JAMs): Cell Adhesion Receptors With Pleiotropic Functions in Cell Physiology and Development. Physiol. Rev. 2017, 97, 1529–1554. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sun, Y.; Wang, M.; Hou, Y.; Huang, W.; Zhou, D.; Wang, Z.; Yang, S.; Tang, W.; Zhen, J.; et al. Elevation of JAML Promotes Diabetic Kidney Disease by Modulating Podocyte Lipid Metabolism. Cell Metab. 2020, 32, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, K. A unified model for regulating lipoprotein lipase activity. Trends Endocrinol. Metab. 2024, 35, 490–504. [Google Scholar] [CrossRef] [PubMed]
- Sylvers-Davie, K.L.; Davies, B.S.J. Regulation of lipoprotein metabolism by ANGPTL3, ANGPTL4, and ANGPTL8. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E493–E508. [Google Scholar] [CrossRef]
- Clement, L.C.; Macé, C.; Avila-Casado, C.; Joles, J.A.; Kersten, S.; Chugh, S.S. Circulating angiopoietin-like 4 links proteinuria with hypertriglyceridemia in nephrotic syndrome. Nat. Med. 2014, 20, 37–46. [Google Scholar] [CrossRef]
- Aryal, B.; Singh, A.K.; Zhang, X.; Varela, L.; Rotllan, N.; Goedeke, L.; Chaube, B.; Camporez, J.P.; Vatner, D.F.; Horvath, T.L.; et al. Absence of ANGPTL4 in adipose tissue improves glucose tolerance and attenuates atherogenesis. JCI Insight 2018, 3, e97918. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Lu, D.; Wang, J.; Yue, S.; Tan, M.; Liu, M.; Gao, X. ANGPTL3 is involved in kidney injury in high-fat diet-fed mice by suppressing ACTN4 expression. Lipids Health Dis. 2022, 21, 90. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gong, W.; Liu, J.; Chen, X.; Suo, Y.; Yang, H.; Gao, X. Angiopoietin-like protein 4 promotes hyperlipidemia-induced renal injury by down-regulating the expression of ACTN4. Biochem. Biophys. Res. Commun. 2022, 595, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Huang, L.; Li, Y.; Liu, Q.; Lv, Y. Dysregulation of Angiopoietin-like-4 Associated with Hyperlipidemia-induced Renal Injury by AMPK/ACC Pathway. Curr. Pharm. Des. 2023, 29, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, J.; Li, F.; Gao, F.; Zhu, L.; Hao, J. FBXW7 mediates high glucose-induced SREBP-1 expression in renal tubular cells of diabetic nephropathy under PI3K/Akt pathway regulation. Mol. Med. Rep. 2021, 23, 233. [Google Scholar] [CrossRef]
- Jun, H.; Song, Z.; Chen, W.; Zanhua, R.; Yonghong, S.; Shuxia, L.; Huijun, D. In vivo and in vitro effects of SREBP-1 on diabetic renal tubular lipid accumulation and RNAi-mediated gene silencing study. Histochem. Cell Biol. 2009, 131, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, Z.; Xia, J.; Xin, L.; Chen, Y.; Yang, S.; Li, K. A Long-Term High-Fat/High-Sucrose Diet Promotes Kidney Lipid Deposition and Causes Apoptosis and Glomerular Hypertrophy in Bama Minipigs. PLoS ONE 2015, 10, e0142884. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, T.; Dutta, R.K.; Joladarashi, D.; Doi, T.; Reddy, J.K.; Kanwar, Y.S. Transcriptional and Translational Modulation of myo-Inositol Oxygenase (Miox) by Fatty Acids: Implications in Renal Tubular Injury Induced in Obesity and Diabetes. J. Biol. Chem. 2016, 291, 1348–1367. [Google Scholar] [CrossRef]
- Lu, J.; Li, X.Q.; Chen, P.P.; Zhang, J.X.; Li, L.; Wang, G.H.; Liu, X.Q.; Jiang, C.M.; Ma, K.L. Acetyl-CoA synthetase 2 promotes diabetic renal tubular injury in mice by rewiring fatty acid metabolism through SIRT1/ChREBP pathway. Acta Pharmacol. Sin. 2024, 45, 366–377. [Google Scholar] [CrossRef]
- Katz, L.S.; Xu, S.; Ge, K.; Scott, D.K.; Gershengorn, M.C. T3 and Glucose Coordinately Stimulate ChREBP-Mediated Ucp1 Expression in Brown Adipocytes From Male Mice. Endocrinology 2018, 159, 557–569. [Google Scholar] [CrossRef]
- Jin, K.; Norris, K.; Vaziri, N.D. Dysregulation of hepatic fatty acid metabolism in chronic kidney disease. Nephrol. Dial. Transplant. 2013, 28, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Mu, L.; Yang, Z.; Du, C.; Wu, M.; Song, S.; Yuan, C.; Shi, Y. Carbohydrate response element-binding protein regulates lipid metabolism via mTOR complex1 in diabetic nephropathy. J. Cell Physiol. 2021, 236, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.A.; Wu, V.C.; Wang, C.Y. Autophagy in Chronic Kidney Diseases. Cells 2019, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, J.; Miao, X.; Cui, W.; Miao, L.; Cai, L. A minireview: Role of AMP-activated protein kinase (AMPK) signaling in obesity-related renal injury. Life Sci. 2021, 265, 118828. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, F.; Vlassembrouck, M.; Botton, O.; Zwakhals, T.; Decarnoncle, M.; Tassin, A.; Caron, N.; Declèves, A.-E. Delayed Exercise Training Improves Obesity-Induced Chronic Kidney Disease by Activating AMPK Pathway in High-Fat Diet-Fed Mice. Int. J. Mol. Sci. 2021, 22, 350. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Kwon, M.H.; Kim, H.M.; Kim, N.; Kim, Y.M.; Kim, H.S.; Lee, E.Y.; Chung, C.H. Dibenzoylmethane ameliorates lipid-induced inflammation and oxidative injury in diabetic nephropathy. J. Endocrinol. 2019, 240, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, Z.; Zhou, H.; Niu, B.; Liu, J.; Li, Y.; Mi, Y.; Li, P. ACT001 Alleviates chronic kidney injury induced by a high-fat diet in mice through the GPR43/AMPK pathway. Lipids Health Dis. 2023, 22, 198. [Google Scholar] [CrossRef] [PubMed]
- Corrales, P.; Vidal-Puig, A.; Medina-Gómez, G. PPARs and Metabolic Disorders Associated with Challenged Adipose Tissue Plasticity. Int. J. Mol. Sci. 2018, 19, 2124. [Google Scholar] [CrossRef] [PubMed]
- Libby, A.E.; Jones, B.; Lopez-Santiago, I.; Rowland, E.; Levi, M. Nuclear receptors in the kidney during health and disease. Mol. Aspects Med. 2021, 78, 100935. [Google Scholar] [CrossRef]
- Corrales, P.; Izquierdo-Lahuerta, A.; Medina-Gómez, G. Maintenance of Kidney Metabolic Homeostasis by PPAR Gamma. Int. J. Mol. Sci. 2018, 19, 2063. [Google Scholar] [CrossRef]
- Gao, J.; Gu, Z. The Role of Peroxisome Proliferator-Activated Receptors in Kidney Diseases. Front. Pharmacol. 2022, 13, 832732. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Tang, B.; Zhang, C. Signaling pathways of chronic kidney diseases, implications for therapeutics. Signal Transduct. Target. Ther. 2022, 7, 182. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef] [PubMed]
- Console, L.; Scalise, M.; Giangregorio, N.; Tonazzi, A.; Barile, M.; Indiveri, C. The Link Between the Mitochondrial Fatty Acid Oxidation Derangement and Kidney Injury. Front. Physiol. 2020, 11, 794. [Google Scholar] [CrossRef]
- Lee, M.; Katerelos, M.; Gleich, K.; Galic, S.; Kemp, B.E.; Mount, P.F.; Power, D.A. Phosphorylation of Acetyl-CoA Carboxylase by AMPK Reduces Renal Fibrosis and Is Essential for the Anti-Fibrotic Effect of Metformin. J. Am. Soc. Nephrol. 2018, 29, 2326–2336. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Parikh, S.M. Targeting energy pathways in kidney disease: The roles of sirtuins, AMPK, and PGC1α. Kidney Int. 2021, 99, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Tan, E.; Shi, H.; Ren, X.; Wan, X.; Wu, W.; Chen, Y.; Niu, H.; Zhu, G.; Li, J.; et al. Mitochondrial oxidative damage reprograms lipid metabolism of renal tubular epithelial cells in the diabetic kidney. Cell Mol. Life Sci. 2024, 81, 23. [Google Scholar] [CrossRef]
- Ceja-Galicia, Z.A.; García-Arroyo, F.E.; Aparicio-Trejo, O.E.; El-Hafidi, M.; Gonzaga-Sánchez, G.; León-Contreras, J.C.; Hernández-Pando, R.; Guevara-Cruz, M.; Tovar, A.R.; Rojas-Morales, P.; et al. Therapeutic Effect of Curcumin on 5/6Nx Hypertriglyceridemia: Association with the Improvement of Renal Mitochondrial β-Oxidation and Lipid Metabolism in Kidney and Liver. Antioxidants 2022, 11, 2195. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Susztak, K. The Role of Peroxisome Proliferator-Activated Receptor γ Coactivator 1α (PGC-1α) in Kidney Disease. Semin. Nephrol. 2018, 38, 121–126. [Google Scholar] [CrossRef]
- Jang, S.; Jang, B.H.; Ko, Y.; Sasaki, Y.; Park, J.S.; Hwang, E.H.; Song, Y.K.; Shin, Y.C.; Ko, S.G. Herbal Medicines for Treating Metabolic Syndrome: A Systematic Review of Randomized Controlled Trials. Evid. Based Complement. Alternat Med. 2016, 2016, 5936402. [Google Scholar] [CrossRef]
- Ahmed, M.; Kumari, N.; Mirgani, Z.; Saeed, A.; Ramadan, A.; Ahmed, M.H.; Almobarak, A.O. Metabolic syndrome; Definition, Pathogenesis, Elements, and the Effects of medicinal plants on it’s elements. J. Diabetes Metab. Disord. 2022, 21, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Noce, A.; Di Lauro, M.; Di Daniele, F.; Pietroboni Zaitseva, A.; Marrone, G.; Borboni, P.; Di Daniele, N. Natural Bioactive Compounds Useful in Clinical Management of Metabolic Syndrome. Nutrients 2021, 13, 630. [Google Scholar] [CrossRef] [PubMed]
- Chaachouay, N.; Zidane, L. Plant-Derived Natural Products: A Source for Drug Discovery and Development. Drugs Drug Candidates 2024, 3, 184–207. [Google Scholar] [CrossRef]
- Qu, Y.; Chan, J.Y.; Wong, C.W.; Cheng, L.; Xu, C.; Leung, A.W.; Lau, C.B. Antidiabetic Effect of Schisandrae Chinensis Fructus Involves Inhibition of the Sodium Glucose Cotransporter. Drug Dev. Res. 2015, 76, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, C.; Wang, S.; Gao, S.; Liu, J.; Dong, Z.; Zhang, B.; Liu, M.; Sun, X.; Guo, P. Extracts and lignans of Schisandra chinensis fruit alter lipid and glucose metabolism in vivo and in vitro. J. Funct. Foods 2015, 19, 296–307. [Google Scholar] [CrossRef]
- Sato, S.; Takeo, J.; Aoyama, C.; Kawahara, H. Na+-glucose cotransporter (SGLT) inhibitory flavonoids from the roots of Sophora flavescens. Bioorg Med. Chem. 2007, 15, 3445–3449. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, J.; Xu, C.; Huang, M.; Zhou, Q.; Lv, J.; Ma, X.; Ke, C.; Ye, Y.; Shu, G.; et al. Antidiabetic effects of flavonoids from Sophora flavescens EtOAc extract in type 2 diabetic KK-ay mice. J. Ethnopharmacol. 2015, 171, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Y.; Gao, Y.; Zhao, K.; Li, Z.; Luo, Y.; Chen, L. Exploring the anti-diabetic effects and the underlying mechanisms of ethyl acetate extract from Sophora flavescens by integrating network pharmacology and pharmacological evaluation. Tradit. Med. Res. 2022, 7, 3. [Google Scholar] [CrossRef]
- Moradi-Marjaneh, R.; Paseban, M.; Sahebkar, A. Natural products with SGLT2 inhibitory activity: Possibilities of application for the treatment of diabetes. Phytother. Res. 2019, 33, 2518–2530. [Google Scholar] [CrossRef]
- Shimokawa, Y.; Akao, Y.; Hirasawa, Y.; Awang, K.; Hadi, A.H.; Sato, S.; Aoyama, C.; Takeo, J.; Shiro, M.; Morita, H. Gneyulins A and B, stilbene trimers, and noidesols A and B, dihydroflavonol-C-glucosides, from the bark of Gnetum gnemonoides. J. Nat. Prod. 2010, 73, 763–767. [Google Scholar] [CrossRef]
- Morita, H.; Deguchi, J.; Motegi, Y.; Sato, S.; Aoyama, C.; Takeo, J.; Shiro, M.; Hirasawa, Y. Cyclic diarylheptanoids as Na+-glucose cotransporter (SGLT) inhibitors from Acer nikoense. Bioorganic Med. Chem. Lett. 2010, 20, 1070–1074. [Google Scholar] [CrossRef]
- Arai, H.; Hirasawa, Y.; Rahman, A.; Kusumawati, I.; Zaini, N.C.; Sato, S.; Aoyama, C.; Takeo, J.; Morita, H. Alstiphyllanines E-H, picraline and ajmaline-type alkaloids from Alstonia macrophylla inhibiting sodium glucose cotransporter. Bioorg. Med. Chem. 2010, 18, 2152–2158. [Google Scholar] [CrossRef]
- Khanam, S.; Mishra, D.A.; Shahid, A.; Pujari, N.M. Therapeutic indication of Phloridzin: A new Gleam for metabolic disorders. Phytomed. Plus 2022, 2, 100200. [Google Scholar] [CrossRef]
- Maisto, M.; Piccolo, V.; Novellino, E.; Schiano, E.; Iannuzzo, F.; Ciampaglia, R.; Summa, V.; Tenore, G.C. Optimization of Phlorizin Extraction from Annurca Apple Tree Leaves Using Response Surface Methodology. Antioxidants 2022, 11, 1933. [Google Scholar] [CrossRef]
- Osorio, H.; Coronel, I.; Arellano, A.; Pacheco, U.; Bautista, R.; Franco, M.; Escalante, B. Sodium-glucose cotransporter inhibition prevents oxidative stress in the kidney of diabetic rats. Oxid. Med. Cell Longev. 2012, 2012, 542042. [Google Scholar] [CrossRef]
- Osorio, H.; Bautista, R.; Rios, A.; Franco, M.; Arellano, A.; Vargas-Robles, H.; Romo, E.; Escalante, B. Effect of phlorizin on SGLT2 expression in the kidney of diabetic rats. J. Nephrol. 2010, 23, 541–546. [Google Scholar]
- Mei, X.; Zhang, X.; Wang, Z.; Gao, Z.; Liu, G.; Hu, H.; Zou, L.; Li, X. Insulin Sensitivity-Enhancing Activity of Phlorizin Is Associated with Lipopolysaccharide Decrease and Gut Microbiota Changes in Obese and Type 2 Diabetes (db/db) Mice. J. Agric. Food Chem. 2016, 64, 7502–7511. [Google Scholar] [CrossRef] [PubMed]
- Castañeda, A.M.; Dutra-Rufato, A.; Juarez, M.J.; Grosembacher, L.; Gonzalez-Torres, H.; Musso, C.G. Sodium-glucose cotransporter 2 inhibitors (SGLT2i): Renal implications. Int. Urol. Nephrol. 2021, 53, 291–299. [Google Scholar] [CrossRef]
- Pedraza-Chaverri, J.; Sánchez-Lozada, L.G.; Osorio-Alonso, H.; Tapia, E.; Scholze, A. New Pathogenic Concepts and Therapeutic Approaches to Oxidative Stress in Chronic Kidney Disease. Oxid. Med. Cell Longev. 2016, 2016, 6043601. [Google Scholar] [CrossRef]
- Andreea, M.M.; Surabhi, S.; Razvan-Ionut, P.; Lucia, C.; Camelia, N.; Emil, T.; Tiberiu, N.I. Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitors: Harms or Unexpected Benefits? Medicina 2023, 59, 742. [Google Scholar] [CrossRef]
- Zhang, J.; Huan, Y.; Leibensperger, M.; Seo, B.; Song, Y. Comparative Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Serum Electrolyte Levels in Patients with Type 2 Diabetes: A Pairwise and Network Meta-Analysis of Randomized Controlled Trials. Kidney360 2022, 3, 477–487. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhou, S.; Liu, L. Efficacy and safety evaluation of SGLT2i on blood pressure control in patients with type 2 diabetes and hypertension: A new meta-analysis. Diabetol. Metab. Syndr. 2023, 15, 118. [Google Scholar] [CrossRef]
- Ni, L.; Yuan, C.; Chen, G.; Zhang, C.; Wu, X. SGLT2i: Beyond the glucose-lowering effect. Cardiovasc. Diabetol. 2020, 19, 98. [Google Scholar] [CrossRef]
- Chino, Y.; Samukawa, Y.; Sakai, S.; Nakai, Y.; Yamaguchi, J.; Nakanishi, T.; Tamai, I. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm. Drug Dispos. 2014, 35, 391–404. [Google Scholar] [CrossRef]
- Sánchez-Briales, P.; Marques Vidas, M.; López-Sánchez, P.; López-Illázquez, M.V.; Martín-Testillano, L.; Vedat-Ali, A.; Portolés, J. The Uricosuric Effect of SGLT2 Inhibitors Is Maintained in the Long Term in Patients with Chronic Kidney Disease and Type 2 Diabetes Mellitus. J. Clin. Med. 2024, 13, 1360. [Google Scholar] [CrossRef]
- Somagutta, M.K.R.; Luvsannyam, E.; Jain, M.; Cuddapah, G.V.; Pelluru, S.; Mustafa, N.; Nasereldin, D.S.; Pendyala, S.K.; Jarapala, N.; Padamati, B. Sodium glucose co-transport 2 inhibitors for gout treatment. Discoveries 2022, 10, e152. [Google Scholar] [CrossRef]
- Szekeres, Z.; Sandor, B.; Bognar, Z.; Ramadan, F.H.J.; Palfi, A.; Bodis, B.; Toth, K.; Szabados, E. Clinical Study of Metabolic Parameters, Leptin and the SGLT2 Inhibitor Empagliflozin among Patients with Obesity and Type 2 Diabetes. Int. J. Mol. Sci. 2023, 24, 4405. [Google Scholar] [CrossRef]
- Kohlmorgen, C.; Gerfer, S.; Feldmann, K.; Twarock, S.; Hartwig, S.; Lehr, S.; Klier, M.; Krüger, I.; Helten, C.; Keul, P.; et al. Dapagliflozin reduces thrombin generation and platelet activation: Implications for cardiovascular risk reduction in type 2 diabetes mellitus. Diabetologia 2021, 64, 1834–1849. [Google Scholar] [CrossRef]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 Inhibition by Empagliflozin Promotes Fat Utilization and Browning and Attenuates Inflammation and Insulin Resistance by Polarizing M2 Macrophages in Diet-induced Obese Mice. EBioMedicine 2017, 20, 137–149. [Google Scholar] [CrossRef]
- Ji, W.; Zhao, M.; Wang, M.; Yan, W.; Liu, Y.; Ren, S.; Lu, J.; Wang, B.; Chen, L. Effects of canagliflozin on weight loss in high-fat diet-induced obese mice. PLoS ONE 2017, 12, e0179960. [Google Scholar] [CrossRef]
- Osataphan, S.; Macchi, C.; Singhal, G.; Chimene-Weiss, J.; Sales, V.; Kozuka, C.; Dreyfuss, J.M.; Pan, H.; Tangcharoenpaisan, Y.; Morningstar, J.; et al. SGLT2 inhibition reprograms systemic metabolism via FGF21-dependent and -independent mechanisms. JCI Insight 2019, 4, e123130. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Lu, J.H.; Lu, R.; Lundenberg, L.; Desjardins, E.M.; Green, A.E.; Lally, J.S.V.; Schertzer, J.D.; Steinberg, G.R. The SGLT2 inhibitor canagliflozin suppresses lipid synthesis and interleukin-1 beta in ApoE deficient mice. Biochem. J. 2020, 477, 2347–2361. [Google Scholar] [CrossRef] [PubMed]
- Salim, H.M.; Fukuda, D.; Yagi, S.; Soeki, T.; Shimabukuro, M.; Sata, M. Glycemic Control with Ipragliflozin, a Novel Selective SGLT2 Inhibitor, Ameliorated Endothelial Dysfunction in Streptozotocin-Induced Diabetic Mouse. Front. Cardiovasc. Med. 2016, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Levi, J.; Luo, Y.; Myakala, K.; Herman-Edelstein, M.; Qiu, L.; Wang, D.; Peng, Y.; Grenz, A.; Lucia, S.; et al. SGLT2 Protein Expression Is Increased in Human Diabetic Nephropathy: SGLT2 Protein Inhibition Decreases Renal Lipid Accumulation, Inflammation, and the Development of Nephropathy in Diabetic Mice. J. Biol. Chem. 2017, 292, 5335–5348. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Chou, C.A.; Chen, W.Y.; Yang, J.L.; Lee, W.C.; Chen, J.B.; Lee, C.T.; Li, L.C. Empagliflozin Ameliorates Free Fatty Acid Induced-Lipotoxicity in Renal Proximal Tubular Cells via the PPARγ/CD36 Pathway in Obese Mice. Int. J. Mol. Sci. 2021, 22, 12408. [Google Scholar] [CrossRef]
- Thongnak, L.; Chatsudthipong, V.; Kongkaew, A.; Lungkaphin, A. Effects of dapagliflozin and statins attenuate renal injury and liver steatosis in high-fat/high-fructose diet-induced insulin resistant rats. Toxicol. Appl. Pharmacol. 2020, 396, 114997. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Ke, Q.; Fang, Y.; Wen, P.; Chen, H.; Yuan, Q.; Luo, J.; Zhang, Y.; Sun, Q.; Lv, Y.; et al. Sodium-glucose cotransporter 2 inhibition suppresses HIF-1α-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis. 2020, 11, 390. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, S.H.; Kang, J.M.; Heo, J.H.; Kim, D.J.; Park, S.H.; Sung, M.; Kim, J.; Oh, J.; Yang, D.H.; et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am. J. Physiol. Renal Physiol. 2019, 317, F767–F780. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Ford, R.J.; Smith, B.K.; Gowans, G.J.; Mancini, S.J.; Pitt, R.D.; Day, E.A.; Salt, I.P.; Steinberg, G.R.; Hardie, D.G. The Na+/Glucose Cotransporter Inhibitor Canagliflozin Activates AMPK by Inhibiting Mitochondrial Function and Increasing Cellular AMP Levels. Diabetes 2016, 65, 2784–2794. [Google Scholar] [CrossRef]
- Feder, D.; de Fatima Veiga Gouveia, M.R.; Govato, T.C.P.; Nassis, C.D.Z. SGLT2 Inhibitors and the Mechanisms Involved in Weight Loss. Curr. Pharmacol. Rep. 2020, 6, 346–353. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Maleki, M.; Butler, A.E.; Jamialahmadi, T.; Sahebkar, A. Sodium-glucose cotransporter 2 inhibitors and mitochondrial functions: State of the art. Excli J. 2023, 22, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, A.; Lazareth, H.; Poindessous, V.; Nemazanyy, I.; Sampaio, J.L.; Malpetti, D.; Bignon, Y.; Naesens, M.; Rabant, M.; Anglicheau, D.; et al. Impaired fatty acid metabolism perpetuates lipotoxicity along the transition to chronic kidney injury. JCI Insight 2022, 7, e161783. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shang, L.; Deng, S.; Li, P.; Chen, K.; Gao, T.; Zhang, X.; Chen, Z.; Zeng, J. Peroxisomal oxidation of erucic acid suppresses mitochondrial fatty acid oxidation by stimulating malonyl-CoA formation in the rat liver. J. Biol. Chem. 2020, 295, 10168–10179. [Google Scholar] [CrossRef] [PubMed]
- Ndumele, C.E.; Neeland, I.J.; Tuttle, K.R.; Chow, S.L.; Mathew, R.O.; Khan, S.S.; Coresh, J.; Baker-Smith, C.M.; Carnethon, M.R.; Després, J.P.; et al. A Synopsis of the Evidence for the Science and Clinical Management of Cardiovascular-Kidney-Metabolic (CKM) Syndrome: A Scientific Statement From the American Heart Association. Circulation 2023, 148, 1636–1664. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sanchez, F.D.; Medina-Urrutia, A.X.; Jorge-Galarza, E.; Martínez-Alvarado, M.D.R.; Reyes-Barrera, J.; Osorio-Alonso, H.; Arellano-Buendía, A.S.; Del Carmen González-Salazar, M.; Posadas-Sánchez, R.; Vargas-Alarcón, G.; et al. Effect of metabolic control on recurrent major adverse cardiovascular events and cardiovascular mortality in patients with premature coronary artery disease: Results of the Genetics of Atherosclerotic Disease study. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 2227–2237. [Google Scholar] [CrossRef] [PubMed]
- Doroszko, A.; Janus, A.; Szahidewicz-Krupska, E.; Mazur, G.; Derkacz, A. Resistant Hypertension. Adv. Clin. Exp. Med. 2016, 25, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Osorio, H.; Bautista, R.; Rios, A.; Franco, M.; Santamaría, J.; Escalante, B. Effect of treatment with losartan on salt sensitivity and SGLT2 expression in hypertensive diabetic rats. Diabetes Res. Clin. Pract. 2009, 86, e46–e49. [Google Scholar] [CrossRef] [PubMed]
- Bautista, R.; Manning, R.; Martinez, F.; Avila-Casado Mdel, C.; Soto, V.; Medina, A.; Escalante, B. Angiotensin II-dependent increased expression of Na+-glucose cotransporter in hypertension. Am. J. Physiol. Renal Physiol. 2004, 286, F127–F133. [Google Scholar] [CrossRef] [PubMed]
- Veelen, A.; Andriessen, C.; Op den Kamp, Y.; Erazo-Tapia, E.; de Ligt, M.; Mevenkamp, J.; Jörgensen, J.A.; Moonen-Kornips, E.; Schaart, G.; Esterline, R.; et al. Effects of the sodium-glucose cotransporter 2 inhibitor dapagliflozin on substrate metabolism in prediabetic insulin resistant individuals: A randomized, double-blind crossover trial. Metabolism 2023, 140, 155396. [Google Scholar] [CrossRef]
- Jhuo, S.J.; Lin, Y.H.; Liu, I.H.; Lin, T.H.; Wu, B.N.; Lee, K.T.; Lai, W.T. Sodium Glucose Cotransporter 2 (SGLT2) Inhibitor Ameliorate Metabolic Disorder and Obesity Induced Cardiomyocyte Injury and Mitochondrial Remodeling. Int. J. Mol. Sci. 2023, 24, 6842. [Google Scholar] [CrossRef]
- Kim, H.S.; Yoon, T.; Jung, C.H.; Park, J.Y.; Lee, W.J. Clinical Efficacy of Sodium-Glucose Cotransporter 2 Inhibitor and Glucagon-Like Peptide-1 Receptor Agonist Combination Therapy in Type 2 Diabetes Mellitus: Real-World Study. Diabetes Metab. J. 2022, 46, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.d.S.; Souza, L.P.; Pereira, P.G.; Carvalho, J.J.d.; Moreno, A.M.; Castro-Faria-Neto, H.C.; Siqueira, R.d.A.; d’Avila, J.d.C.; Carlos, A.S. Combination of a Glucagon-Like Peptide 1 Analog and a Sodium-Glucose Cotransporter 2 Inhibitor Improves Lipid Metabolism Compared to the Monotherapies in Experimental Metabolic Syndrome. J. Endocrinol. Metab. 2022, 12, 168–177. [Google Scholar] [CrossRef]
- Shaheer, A.; Kumar, A.; Menon, P.; Jallo, M.; Basha, S. Effect of Add-On Therapy of Sodium-Glucose Cotransporter 2 Inhibitors and Dipeptidyl Peptidase 4 Inhibitors on Adipokines in Type 2 Diabetes Mellitus. J. Clin. Med. Res. 2021, 13, 355–362. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organization | NCEP ATPIII 2005 | IDF 2005 | WHO 1998 |
|---|---|---|---|
| Criteria | Any of three or more of the following five components | Central obesity plus any two other factors | IGT, IFG, T2DM, or reduced insulin sensitivity plus any two of the following |
| Obesity | WC ≥ 102 cm in men or ≥88 cm in women population-specific increased WC cutoffs | Men: WHR > 0.9 Women: WHR > 0.85 and/or BMI > 30 kg/m2 | |
| Blood Pressure | ≥130/85 mmHg or on antihypertensive therapy | ≥130/85 mmHg or on antihypertensive therapy | ≥140/90 mmHg |
| Lipid profile | TG ≥ 150 mg/dL or on therapy lowering TG | TG ≥ 150 mg/dL or chronic treatment for lipid abnormality and reduced HDL-c: < 40 mg/dL for males and < 50 mg/dL for females | TG ≥ 150 mg/dL |
| HDL-c < 40 mg/dL in men or HDL-c < 50 mg/dL in women on therapy increasing HDL-c | |||
| Glucose | ≥100 mg/dL (including T2DM) | ≥100 mg/dL (including T2DM) | IGT, IFG, or T2DM |
| Microalbuminuria | Urinary excretion rate > 20 mg/min or | ||
| Albumin/creatinine >30 mg/min | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortés-Camacho, F.; Zambrano-Vásquez, O.R.; Aréchaga-Ocampo, E.; Castañeda-Sánchez, J.I.; Gonzaga-Sánchez, J.G.; Sánchez-Gloria, J.L.; Sánchez-Lozada, L.G.; Osorio-Alonso, H. Sodium–Glucose Cotransporter Inhibitors: Cellular Mechanisms Involved in the Lipid Metabolism and the Treatment of Chronic Kidney Disease Associated with Metabolic Syndrome. Antioxidants 2024, 13, 768. https://doi.org/10.3390/antiox13070768
Cortés-Camacho F, Zambrano-Vásquez OR, Aréchaga-Ocampo E, Castañeda-Sánchez JI, Gonzaga-Sánchez JG, Sánchez-Gloria JL, Sánchez-Lozada LG, Osorio-Alonso H. Sodium–Glucose Cotransporter Inhibitors: Cellular Mechanisms Involved in the Lipid Metabolism and the Treatment of Chronic Kidney Disease Associated with Metabolic Syndrome. Antioxidants. 2024; 13(7):768. https://doi.org/10.3390/antiox13070768
Chicago/Turabian StyleCortés-Camacho, Fernando, Oscar René Zambrano-Vásquez, Elena Aréchaga-Ocampo, Jorge Ismael Castañeda-Sánchez, José Guillermo Gonzaga-Sánchez, José Luis Sánchez-Gloria, Laura Gabriela Sánchez-Lozada, and Horacio Osorio-Alonso. 2024. "Sodium–Glucose Cotransporter Inhibitors: Cellular Mechanisms Involved in the Lipid Metabolism and the Treatment of Chronic Kidney Disease Associated with Metabolic Syndrome" Antioxidants 13, no. 7: 768. https://doi.org/10.3390/antiox13070768
APA StyleCortés-Camacho, F., Zambrano-Vásquez, O. R., Aréchaga-Ocampo, E., Castañeda-Sánchez, J. I., Gonzaga-Sánchez, J. G., Sánchez-Gloria, J. L., Sánchez-Lozada, L. G., & Osorio-Alonso, H. (2024). Sodium–Glucose Cotransporter Inhibitors: Cellular Mechanisms Involved in the Lipid Metabolism and the Treatment of Chronic Kidney Disease Associated with Metabolic Syndrome. Antioxidants, 13(7), 768. https://doi.org/10.3390/antiox13070768