
Protective Efficacy of the Epitope-Conjugated Antigen N-Tc52/TSkb20 in Mitigating Trypanosoma cruzi Infection through CD8+ T-Cells and IFNγ Responses

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. N-Tc52/TSkb20 Chimeric Protein Design and Genetic Manipulation
2.2. N-Tc52/TSkb20 Chimeric Protein Expression and Purification
2.3. Characterization of N-Tc52/TSkb20 Chimeric Protein by Liquid Chromatography–Mass Spectrometry (HPLC–MS)
2.4. Animal Model and Immunization Protocol
2.5. T. cruzi Challenge and Parasitemia Assessment
2.6. Assessment of Tissue Parasite Burden
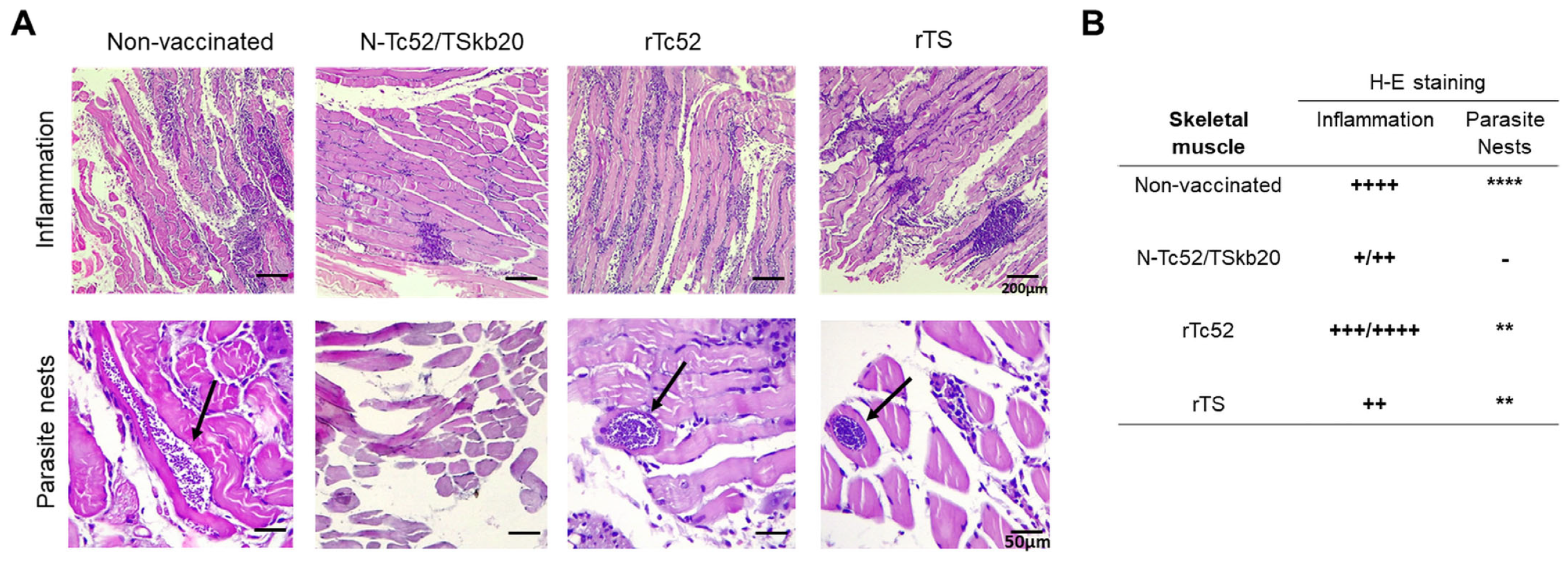
2.7. Histological Analysis
2.8. Detection of Specific IgG Subclasses
2.9. Western Blot Characterization of N-Tc52/TSkb20 via Serum Antibody Detection
2.10. Splenocyte Cell Culture
2.11. Measurement of IL-10 Cytokine Response
2.12. ELISPOT Assay for IFNγ Determination
2.13. Activation-Induced Marker (AIM) Assay
2.14. In-Silico Prediction of CD8+ T-Cell and B-Cell Potential Epitopes
2.15. Statistical Analysis
3. Results
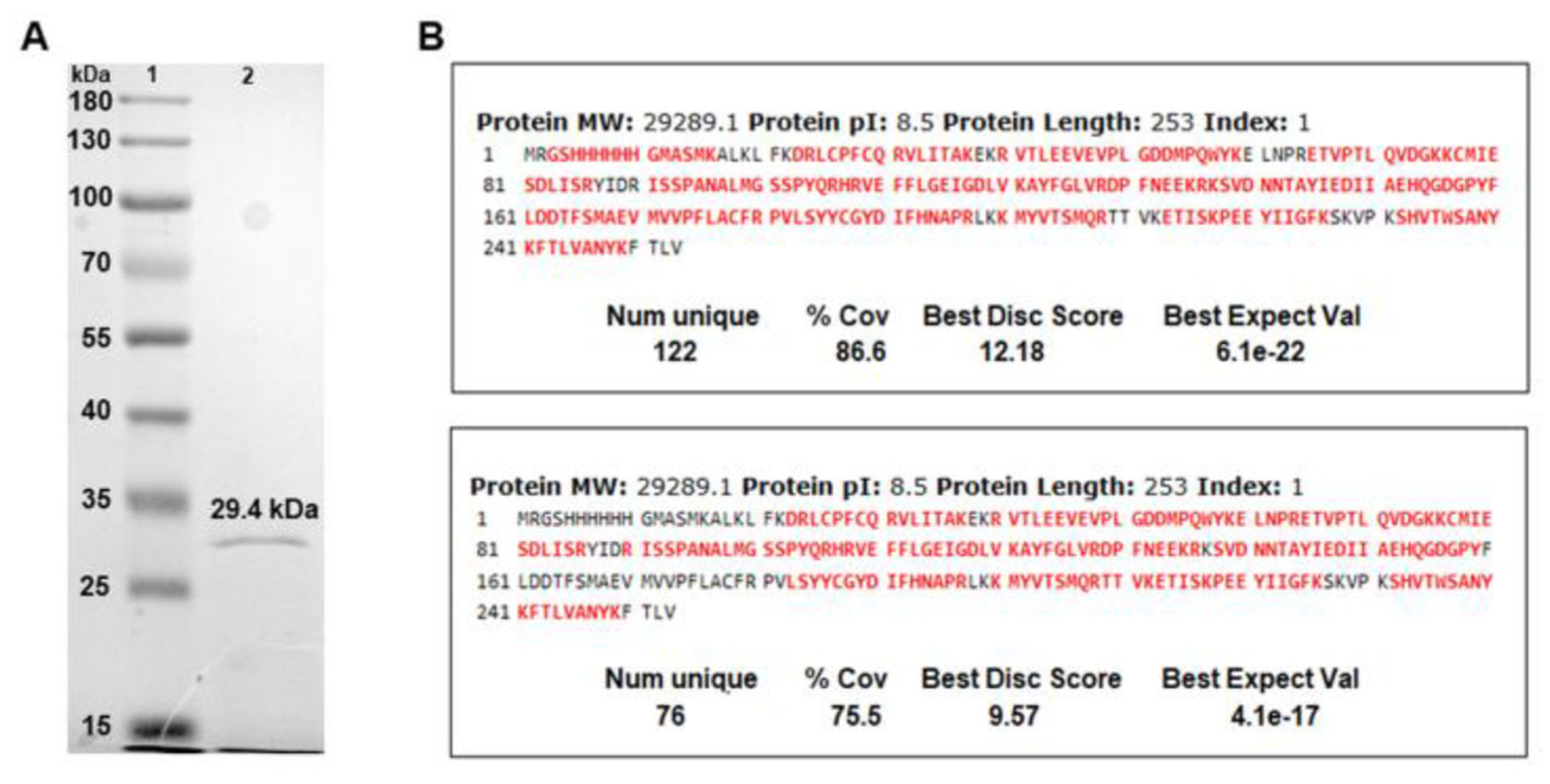
3.1. Development and Characterization of the N-Tc52/TSkb20 Chimeric Protein Construct
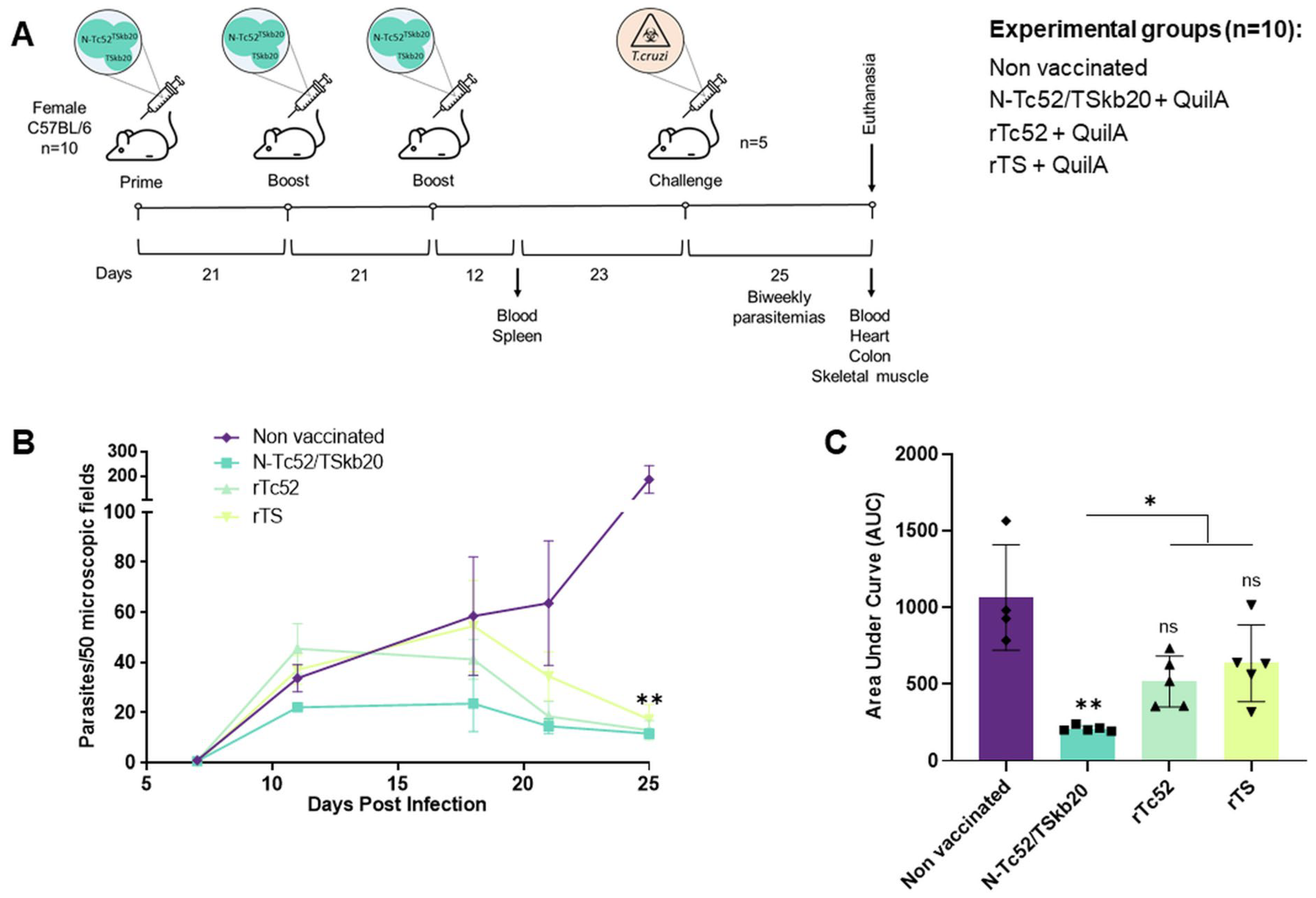
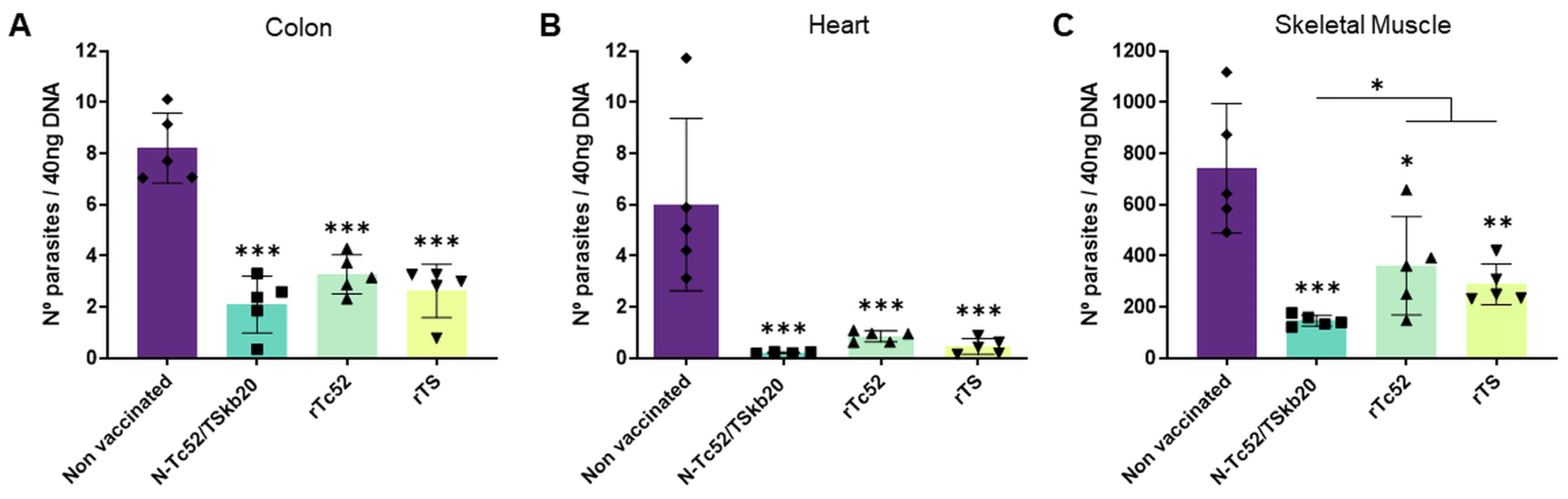
3.2. N-Tc52/TSkb20 Immunization Effectively Controls Parasitemia and Reduces Tissue Parasite Burden in a Mouse Model of T. cruzi Infection
3.3. N-Tc52/TSkb20 Immunization Induces a Moderate but Protective Anti- N-Tc52/TSkb20 Humoral Immune Response in a Mouse Model of T. cruzi Infection
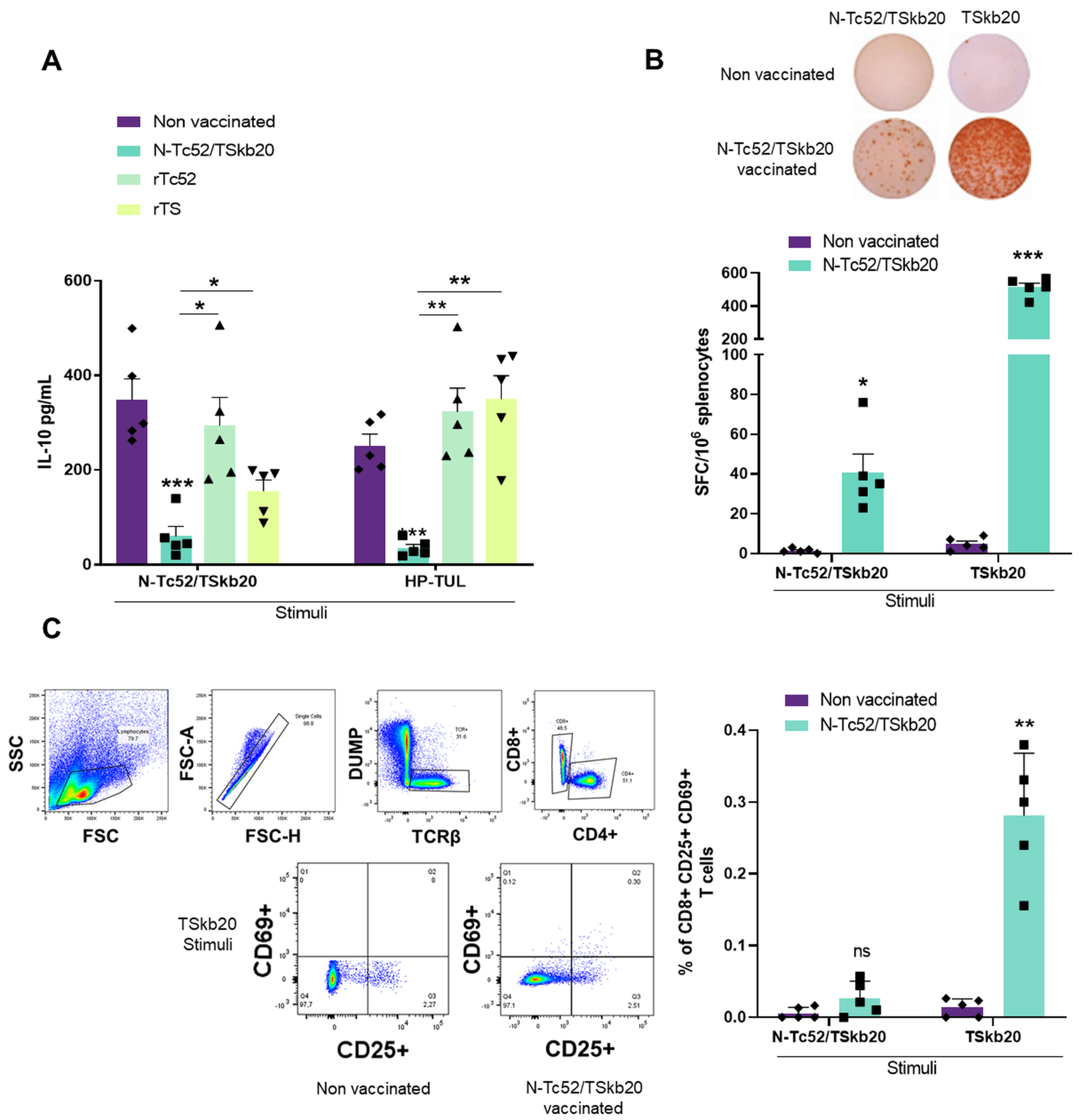
3.4. Elucidating the Combined CD8+, IFNγ and Regulatory Cytokine Facets of the Immune Response Induced by the N-Tc52/TSkb20 Vaccine
3.5. In-Silico Prediction of Novel CD8+ T-Cell and B-Cell Epitopes within the N-Tc52/TSkb20 Chimeric Vaccine Correlated with Its Enhanced Protective Efficacy
4. Discussion
4.1. Generation and characterization of the N-Tc52/TSkb20 chimera
4.2. Evaluation of N-Tc52/TSkb20 Protective Efficacy in a Mouse Model of Acute T. cruzi Infection
4.3. Immunogenicity and Protective Mechanisms of N-Tc52/TSkb20 Vaccination
4.4. Significance and Future Study Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Symptoms, Transmission, and Current Treatments for Chagas Disease | DNDi. Available online: https://dndi.org/diseases/chagas/facts/ (accessed on 21 November 2023).
- World Chagas Disease Day 2023 to Focus on Integrating Universal Care and Surveillance at the Primary Care Level. Available online: https://www.who.int/news/item/14-04-2023-world-chagas-disease-day-2023-to-focus-on-integrating-universal-care-and-surveillance-at-the-primary-care-level (accessed on 21 November 2023).
- The Epidemiology, Clinical Manifestations, and Management of Chagas Heart Disease—Malik—2015—Clinical Cardiology—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/clc.22421 (accessed on 21 November 2023).
- Gastrointestinal Manifestations of Chagas’ Disease—ScienceDirect. Available online: https://www.sciencedirect.com/science/article/pii/S0002927098001518 (accessed on 21 November 2023).
- Hidron, A.; Vogenthaler, N.; Santos-Preciado, J.I.; Rodriguez-Morales, A.J.; Franco-Paredes, C.; Rassi, A. Cardiac Involvement with Parasitic Infections. Clin. Microbiol. Rev. 2010, 23, 324–349. [Google Scholar] [CrossRef]
- Dumonteil, E.; Herrera, C. The Case for the Development of a Chagas Disease Vaccine: Why? How? When? Trop. Med. Infect. Dis. 2021, 6, 16. [Google Scholar] [CrossRef]
- Martín-Escolano, J.; Marín, C.; Rosales, M.J.; Tsaousis, A.D.; Medina-Carmona, E.; Martín-Escolano, R. An Updated View of the Trypanosoma cruzi Life Cycle: Intervention Points for an Effective Treatment. ACS Infect. Dis. 2022, 8, 1107–1115. [Google Scholar] [CrossRef]
- Burleigh, B.A.; Woolsey, A.M. Cell Signalling and Trypanosoma cruzi Invasion. Cell. Microbiol. 2002, 4, 701–711. [Google Scholar] [CrossRef]
- Cardoso, M.S.; Reis-Cunha, J.L.; Bartholomeu, D.C. Evasion of the Immune Response by Trypanosoma cruzi during Acute Infection. Front. Immunol. 2016, 6, 174016. [Google Scholar] [CrossRef]
- Pérez Brandán, C.; Mesías, A.C.; Parodi, C.; Cimino, R.O.; Pérez Brandán, C.; Diosque, P.; Basombrío, M.Á. Effects of IFN-γ Coding Plasmid Supplementation in the Immune Response and Protection Elicited by Trypanosoma cruzi Attenuated Parasites. BMC Infect. Dis. 2017, 17, 732. [Google Scholar] [CrossRef]
- Brandan, C.P.; Basombrío, M.Á. Genetically Attenuated Trypanosoma cruzi Parasites as a Potential Vaccination Tool. Bioengineered 2012, 3, 242–246. [Google Scholar] [CrossRef]
- Brandan, C.P.; Padilla, A.M.; Xu, D.; Tarleton, R.L.; Basombrio, M.A. Knockout of the Dhfr-Ts Gene in Trypanosoma cruzi Generates Attenuated Parasites Able to Confer Protection against a Virulent Challenge. PLoS Neglected Trop. Dis. 2011, 5, e1418. [Google Scholar] [CrossRef]
- Schijns, V.; Majhen, D.; van der Ley, P.; Thakur, A.; Summerfield, A.; Berisio, R.; Nativi, C.; Fernández-Tejada, A.; Alvarez-Dominguez, C.; Gizurarson, S.; et al. Rational Vaccine Design in Times of Emerging Diseases: The Critical Choices of Immunological Correlates of Protection, Vaccine Antigen and Immunomodulation. Pharmaceutics 2021, 13, 501. [Google Scholar] [CrossRef]
- Ouaissi, A.; Guilvard, E.; Delneste, Y.; Caron, G.; Magistrelli, G.; Herbault, N.; Thieblemont, N.; Jeannin, P. The Trypanosoma cruzi Tc52-Released Protein Induces Human Dendritic Cell Maturation, Signals Via Toll-Like Receptor 2, and Confers Protection Against Lethal Infection1. J. Immunol. 2002, 168, 6366–6374. [Google Scholar] [CrossRef]
- Matos, M.N.; Cazorla, S.I.; Schulze, K.; Ebensen, T.; Guzmán, C.A.; Malchiodi, E.L. Immunization with Tc52 or Its Amino Terminal Domain Adjuvanted with C-Di-AMP Induces Th17+ Th1 Specific Immune Responses and Confers Protection against Trypanosoma cruzi. PLoS Neglected Trop. Dis. 2017, 11, e0005300. [Google Scholar] [CrossRef]
- Bontempi, I.A.; Vicco, M.H.; Cabrera, G.; Villar, S.R.; González, F.B.; Roggero, E.A.; Ameloot, P.; Callewaert, N.; Pérez, A.R.; Marcipar, I.S. Efficacy of a Trans-Sialidase-ISCOMATRIX Subunit Vaccine Candidate to Protect against Experimental Chagas Disease. Vaccine 2015, 33, 1274–1283. [Google Scholar] [CrossRef]
- Fontanella, G.H.; De Vusser, K.; Laroy, W.; Daurelio, L.; Nocito, A.L.; Revelli, S.; Contreras, R. Immunization with an Engineered Mutant Trans-Sialidase Highly Protects Mice from Experimental Trypanosoma cruzi Infection: A Vaccine Candidate. Vaccine 2008, 26, 2322–2334. [Google Scholar] [CrossRef]
- Borges, M.; Da Silva, A.C.; Sereno, D.; Ouaissi, A. Peptide-based Analysis of the Amino Acid Sequence Important to the Immunoregulatory Function of Trypanosoma Cruzi Tc52 Virulence Factor. Immunology 2003, 109, 147–155. [Google Scholar] [CrossRef]
- Moutiez, M.; Quéméneur, E.; Sergheraert, C.; Lucas, V.; Tartar, A.; Davioud-Charvet, E. Glutathione-Dependent Activities of Trypanosoma cruzi P52 Makes It a New Member of the Thiol: Disulphide Oxidoreductase Family. Biochem. J. 1997, 322, 43–48. [Google Scholar] [CrossRef]
- Oury, B.; Tarrieu, F.; Monte-Alegre, A.; Ouaissi, A. Trypanosoma cruzi: Sequence Polymorphism of the Gene Encoding the Tc52 Immunoregulatory-Released Factor in Relation to the Phylogenetic Diversity of the Species. Exp. Parasitol. 2005, 111, 198–206. [Google Scholar] [CrossRef]
- Matos, M.N.; Cazorla, S.I.; Bivona, A.E.; Morales, C.; Guzmán, C.A.; Malchiodi, E.L. Tc52 Amino-Terminal-Domain DNA Carried by Attenuated Salmonella Enterica Serovar Typhimurium Induces Protection against a Trypanosoma cruzi Lethal Challenge. Infect. Immun. 2014, 82, 4265–4275. [Google Scholar] [CrossRef]
- Matos, M.N.; Alberti, A.S.; Morales, C.; Cazorla, S.I.; Malchiodi, E.L. A Prime-Boost Immunization with Tc52 N-Terminal Domain DNA and the Recombinant Protein Expressed in Pichia Pastoris Protects against Trypanosoma cruzi Infection. Vaccine 2016, 34, 3243–3251. [Google Scholar] [CrossRef]
- Pereira-Chioccola, V.L.; Acosta-Serrano, A.; de Almeida, I.C.; Ferguson, M.A.; Souto-Padron, T.; Rodrigues, M.M.; Travassos, L.R.; Schenkman, S. Mucin-like Molecules Form a Negatively Charged Coat That Protects Trypanosoma cruzi trypomastigotes from Killing by Human Anti-α-Galactosyl Antibodies. J. Cell Sci. 2000, 113, 1299–1307. [Google Scholar] [CrossRef]
- Rubin-de-Celis, S.S.C.; Uemura, H.; Yoshida, N.; Schenkman, S. Expression of Trypomastigote Trans-Sialidase in Metacyclic Forms of Trypanosoma cruzi Increases Parasite Escape from Its Parasitophorous Vacuole. Cell Microbiol. 2006, 8, 1888–1898. [Google Scholar] [CrossRef]
- Martin, D.L.; Weatherly, D.B.; Laucella, S.A.; Cabinian, M.A.; Crim, M.T.; Sullivan, S.; Heiges, M.; Craven, S.H.; Rosenberg, C.S.; Collins, M.H. CD8+ T-Cell Responses to Trypanosoma cruzi Are Highly Focused on Strain-Variant Trans-Sialidase Epitopes. PLoS Pathog. 2006, 2, e77. [Google Scholar] [CrossRef]
- Padilla, A.M.; Yao, P.Y.; Landry, T.J.; Cooley, G.M.; Mahaney, S.M.; Ribeiro, I.; VandeBerg, J.L.; Tarleton, R.L. High Variation in Immune Responses and Parasite Phenotypes in Naturally Acquired Trypanosoma cruzi Infection in a Captive Non-Human Primate Breeding Colony in Texas, USA. PLoS Neglected Trop. Dis. 2021, 15, e0009141. [Google Scholar] [CrossRef]
- El-Sayed, N.M.; Myler, P.J.; Bartholomeu, D.C.; Nilsson, D.; Aggarwal, G.; Tran, A.-N.; Ghedin, E.; Worthey, E.A.; Delcher, A.L.; Blandin, G.; et al. The Genome Sequence of Trypanosoma cruzi, Etiologic Agent of Chagas Disease. Science 2005, 309, 409–415. [Google Scholar] [CrossRef]
- Braun, R.; Babiuk, L.A.; Littel-van den Hurk, S.V.D. Compatibility of Plasmids Expressing Different Antigens in a Single DNA Vaccine Formulation. J. Gen. Virol. 1998, 79, 2965–2970. [Google Scholar] [CrossRef]
- Sanchez Alberti, A.; Bivona, A.E.; Cerny, N.; Schulze, K.; Weißmann, S.; Ebensen, T.; Morales, C.; Padilla, A.M.; Cazorla, S.I.; Tarleton, R.L. Engineered Trivalent Immunogen Adjuvanted with a STING Agonist Confers Protection against Trypanosoma cruzi Infection. NPJ Vaccines 2017, 2, 9. [Google Scholar] [CrossRef]
- Castro, J.T.; Brito, R.; Hojo-Souza, N.S.; Azevedo, B.; Salazar, N.; Ferreira, C.P.; Junqueira, C.; Fernandes, A.P.; Vasconcellos, R.; Cardoso, J.M.; et al. ASP-2/Trans-Sialidase Chimeric Protein Induces Robust Protective Immunity in Experimental Models of Chagas’ Disease. npj Vaccines 2023, 8, 81. [Google Scholar] [CrossRef]
- Lage, D.P.; Vale, D.L.; Linhares, F.P.; Freitas, C.S.; Machado, A.S.; Cardoso, J.M.O.; de Oliveira, D.; Galvani, N.C.; de Oliveira, M.P.; Oliveira-da-Silva, J.A.; et al. A Recombinant Chimeric Protein-Based Vaccine Containing T-Cell Epitopes from Amastigote Proteins and Combined with Distinct Adjuvants, Induces Immunogenicity and Protection against Leishmania Infantum Infection. Vaccines 2022, 10, 1146. [Google Scholar] [CrossRef]
- Clímaco, M.d.C.; de Figueiredo, L.A.; Lucas, R.C.; Pinheiro, G.R.G.; Dias Magalhães, L.M.; de Oliveira, A.L.G.; Almeida, R.M.; Barbosa, F.S.; Castanheira Bartholomeu, D.; Bueno, L.L.; et al. Development of Chimeric Protein as a Multivalent Vaccine for Human Kinetoplastid Infections: Chagas Disease and Leishmaniasis. Vaccine 2023, 41, 5400–5411. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; CSHL Press: New York, NY, USA, 2001; ISBN 978-0-87969-576-7. [Google Scholar]
- Acuña, L.; Picariello, G.; Sesma, F.; Morero, R.D.; Bellomio, A. A New Hybrid Bacteriocin, Ent35–MccV, Displays Antimicrobial Activity against Pathogenic Gram-Positive and Gram-Negative Bacteria. FEBS Open Bio 2012, 2, 12–19. [Google Scholar] [CrossRef]
- Acuña, L.; Corbalán, N.; Quintela-Baluja, M.; Barros-Velázquez, J.; Bellomio, A. Expression of the Hybrid Bacteriocin Ent35-MccV in Lactococcus lactis and Its Use for Controlling Listeria monocytogenes and Escherichia coli in Milk. Int. Dairy J. 2020, 104, 104650. [Google Scholar] [CrossRef]
- Magalhães, P.O.; Lopes, A.M.; Rangel-Yagui, C.; Penna, T.C.V. Methods of Endotoxin Removal from Biological Preparations: A Review; Faculty of Pharmacy & Pharmaceutical Sciences—Katz Centre for Pharmacy & Health Research—Canadá: Edmonton, Camada, 2007. [Google Scholar]
- George, R.; Gsottberger, F.; Ammon, A.; Wendland, K.; Mellenthin, L.; Mackensen, A.; Müller, F. Triton X-114 and Amine-Based Wash Strategy Reduces Lipopolysaccharides to FDA Limit and Achieves Purer, More Potent Recombinant Immunotoxin. Bioconjugate Chem. 2021, 32, 713–720. [Google Scholar] [CrossRef]
- Anspach, F.B.; Hilbeck, O. Removal of Endotoxins by Affinity Sorbents. J. Chromatogr. A 1995, 711, 81–92. [Google Scholar] [CrossRef]
- do Prado, J.C., Jr.; Leal, M.P.; Anselmo-Franci, J.A.; de Andrade, H.F., Jr.; Kloetzel, J.K. Influence of Female Gonadal Hormones on the Parasitemia of Female Calomys Callosus Infected with the “Y” Strain of Trypanosoma cruzi. Parasitol. Res. 1997, 84, 100–105. [Google Scholar]
- do Prado, J.C., Jr.; de Apparecida Levy, A.M.; de Paula Leal, M.; Bernard, E.; Kardos Kloetzel, J. Influence of Male Gonadal Hormones on the Parasitemia and Humoral Response of Male Calomys Callosus Infected with the Y Strain of Trypanosoma cruzi. Parasitol. Res. 1999, 85, 826–829. [Google Scholar] [CrossRef]
- Biscari, L.; Kaufman, C.D.; Farré, C.; Huhn, V.; Pacini, M.F.; Balbi, C.B.; Gómez, K.A.; Pérez, A.R.; Alloatti, A. Immunization with Lipopolysaccharide-Activated Dendritic Cells Generates a Specific CD8+ T Cell Response That Confers Partial Protection against Infection with Trypanosoma cruzi. Front. Cell. Infect. Microbiol. 2022, 12, 897133. [Google Scholar] [CrossRef]
- Pérez, A.R.; Morrot, A.; Carvalho, V.F.; De Meis, J.; Savino, W. Role of Hormonal Circuitry upon T Cell Development in Chagas Disease: Possible Implications on T Cell Dysfunctions. Front. Endocrinol. 2018, 9, 334. [Google Scholar] [CrossRef]
- Schuster, J.P.; Schaub, G.A. Experimental Chagas Disease: The Influence of Sex and Psychoneuroimmunological Factors. Parasitol. Res. 2001, 87, 994–1000. [Google Scholar] [CrossRef]
- Vázquez, M.E.; Mesías, A.C.; Acuña, L.; Spangler, J.; Zabala, B.; Parodi, C.; Thakur, M.; Oh, E.; Walper, S.A.; Brandán, C.P. Exploring the Performance of Escherichia coli Outer Membrane Vesicles as a Tool for Vaccine Development against Chagas Disease. Memórias Do Inst. Oswaldo Cruz 2023, 118, e220263. [Google Scholar] [CrossRef]
- Dumonteil, E.; Herrera, C.; Tu, W.; Goff, K.; Fahlberg, M.; Haupt, E.; Kaur, A.; Marx, P.A.; Ortega-Lopez, J.; Hotez, P.J.; et al. Safety and Immunogenicity of a Recombinant Vaccine against Trypanosoma cruzi in Rhesus Macaques. Vaccine 2020, 38, 4584–4591. [Google Scholar] [CrossRef]
- Poveda, C.; Leão, A.C.; Mancino, C.; Taraballi, F.; Chen, Y.-L.; Adhikari, R.; Villar, M.J.; Kundu, R.; Nguyen, D.M.; Versteeg, L. Heterologous mRNA-Protein Vaccination with Tc24 Induces a Robust Cellular Immune Response against Trypanosoma cruzi, Characterized by an Increased Level of Polyfunctional CD8+ T-Cells. Curr. Res. Immunol. 2023, 4, 100066. [Google Scholar] [CrossRef]
- Singh, M.; T O’Hagan, D. Recent Advances in Veterinary Vaccine Adjuvants. Int. J. Parasitol. 2003, 33, 469–478. [Google Scholar] [CrossRef]
- Petrovsky, N.; Aguilar, J.C. Vaccine Adjuvants: Current State and Future Trends. Immunol. Cell Biol. 2004, 82, 488–496. [Google Scholar] [CrossRef]
- Greenfield, E.A. Standard Immunization of Mice, Rats, and Hamsters. Cold Spring Harb. Protoc. 2020, 2020, pdb-prot100297. [Google Scholar] [CrossRef]
- Duffy, T.; Bisio, M.; Altcheh, J.; Burgos, J.M.; Diez, M.; Levin, M.J.; Favaloro, R.R.; Freilij, H.; Schijman, A.G. Accurate Real-Time PCR Strategy for Monitoring Bloodstream Parasitic Loads in Chagas Disease Patients. PLoS Neglected Trop. Dis. 2009, 3, e419. [Google Scholar] [CrossRef]
- Davies, C.; Poma, R.H.; Marino Cardozo, R.; Mora, M.C.; Ramos, F.; Rajal, V.B.; Basombrío, M.Á. Detección de Trypanosoma cruzi En Tejido y Sangre Murina Por PCR Convencional y En Tiempo Real. Acta Bioquímica Clínica Latinoam. 2014, 48, 421–428. [Google Scholar]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving Sequence-Based B-Cell Epitope Prediction Using Conformational Epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef]
- Reed, S.G.; Brownell, C.E.; Russo, D.M.; Silva, J.S.; Grabstein, K.H.; Morrissey, P.J. IL-10 Mediates Susceptibility to Trypanosoma cruzi Infection. J. Immunol. 1994, 153, 3135–3140. [Google Scholar] [CrossRef]
- Soto, C.D.A.; Solana, M.E.; Poncini, C.V.; Pino-Martinez, A.M.; Tekiel, V.; González-Cappa, S.M. Dendritic Cells Devoid of IL-10 Induce Protective Immunity against the Protozoan Parasite Trypanosoma cruzi. Vaccine 2010, 28, 7407–7413. [Google Scholar] [CrossRef]
- Martin, D.; Tarleton, R. Generation, Specificity, and Function of CD8+ T Cells in Trypanosoma cruzi Infection. Immunol. Rev. 2004, 201, 304–317. [Google Scholar] [CrossRef]
- Hoft, D.F.; Eickhoff, C.S.; Giddings, O.K.; Vasconcelos, J.R.; Rodrigues, M.M. Trans-Sialidase Recombinant Protein Mixed with CpG Motif-Containing Oligodeoxynucleotide Induces Protective Mucosal and Systemic Trypanosoma cruzi Immunity Involving CD8+ CTL and B Cell-Mediated Cross-Priming. J. Immunol. 2007, 179, 6889–6900. [Google Scholar] [CrossRef]
- Alves, M.J.M.; Kawahara, R.; Viner, R.; Colli, W.; Mattos, E.C.; Thaysen-Andersen, M.; Larsen, M.R.; Palmisano, G. Comprehensive Glycoprofiling of the Epimastigote and Trypomastigote Stages of Trypanosoma cruzi. J. Proteom. 2017, 151, 182–192. [Google Scholar] [CrossRef]
- Almedia, I.C.; Ferguson, M.A.J.; Schenkman, S.; Travassos, L.R. Lytic Anti-Alpha-Galatosyl Antibodies from Patients with Chronic Chagas’ Disease Recognize Novel O-Linked Oligosaccharides on Mucin-like Glycosyl-Phosphatidylinositol-Anchored Glycoproteins of Trypanosoma cruzi. Biochem. J. 1994, 304, 793–802. [Google Scholar] [CrossRef]
- Buscaglia, C.A.; Campo, V.A.; Di Noia, J.M.; Torrecilhas, A.C.; De Marchi, C.R.; Ferguson, M.A.; Frasch, A.C.; Almeida, I.C. The Surface Coat of the Mammal-Dwelling Infective Trypomastigote Stage of Trypanosoma cruzi Is Formed by Highly Diverse Immunogenic Mucins. J. Biol. Chem. 2004, 279, 15860–15869. [Google Scholar] [CrossRef]
- Portillo, S.; Zepeda, B.G.; Iniguez, E.; Olivas, J.J.; Karimi, N.H.; Moreira, O.C.; Marques, A.F.; Michael, K.; Maldonado, R.A.; Almeida, I.C. A Prophylactic α-Gal-Based Glycovaccine Effectively Protects against Murine Acute Chagas Disease. npj Vaccines 2019, 4, 1–15. [Google Scholar] [CrossRef]
- De Wachter, C.; Van Landuyt, L.; Callewaert, N. Engineering of Yeast Glycoprotein Expression. In Advances in Glycobiotechnology; Rapp, E., Reichl, U., Eds.; Advances in Biochemical Engineering/Biotechnology; Springer International Publishing: Cham, Switzerland, 2021; pp. 93–135. ISBN 978-3-030-69590-3. [Google Scholar]
- Rodríguez-Morales, O.; Monteón-Padilla, V.; Carrillo-Sánchez, S.C.; Rios-Castro, M.; Martínez-Cruz, M.; Carabarin-Lima, A.; Arce-Fonseca, M. Experimental Vaccines against Chagas Disease: A Journey through History. J. Immunol. Res. 2015, 2015, 489758. [Google Scholar] [CrossRef]
- Luhrs, K.A.; Fouts, D.L.; Manning, J.E. Immunization with Recombinant Paraflagellar Rod Protein Induces Protective Immunity against Trypanosoma cruzi Infection. Vaccine 2003, 21, 3058–3069. [Google Scholar] [CrossRef]
- Cazorla, S.I.; Frank, F.M.; Becker, P.D.; Arnaiz, M.; Mirkin, G.A.; Corral, R.S.; Guzmán, C.A.; Malchiodi, E.L. Redirection of the Immune Response to the Functional Catalytic Domain of the Cystein Proteinase Cruzipain Improves Protective Immunity against Trypanosoma cruzi Infection. J. Infect. Dis. 2010, 202, 136–144. [Google Scholar] [CrossRef]
- Pérez Brandán, C.; Mesias, A.C.; Acuña, L.; Teixeira, T.L.; da Silva, C.V. Evaluation of Pathogen P21 Protein as a Potential Modulator of the Protective Immunity Induced by Trypanosoma Cruzi Attenuated Parasites. Mem. Inst. Oswaldo Cruz 2019, 114, e180571. [Google Scholar] [CrossRef]
- Markine-Goriaynoff, D.; van der Logt, J.T.; Truyens, C.; Nguyen, T.D.; Heessen, F.W.; Bigaignon, G.; Carlier, Y.; Coutelier, J.-P. IFN-γ-Independent IgG2a Production in Mice Infected with Viruses and Parasites. Int. Immunol. 2000, 12, 223–230. [Google Scholar] [CrossRef]
- Buxbaum, L.U.; Thomas, B.N. IgG1, but Not IgG2a/c, Is Pathogenic in Leishmania Mexicana Infection (133.18). J. Immunol. 2009, 182, 133-18. [Google Scholar] [CrossRef]
- Aparicio-Burgos, J.E.; Ochoa-García, L.; Zepeda-Escobar, J.A.; Gupta, S.; Dhiman, M.; Martínez, J.S.; Oca-Jiménez, R.M.d.; Arreola, M.V.; Barbabosa-Pliego, A.; Vázquez-Chagoyán, J.C.; et al. Testing the Efficacy of a Multi-Component DNA-Prime/DNA-Boost Vaccine against Trypanosoma cruzi Infection in Dogs. PLoS Neglected Trop. Dis. 2011, 5, e1050. [Google Scholar] [CrossRef]
- Morell, M.; Thomas, M.C.; Caballero, T.; Alonso, C.; López, M.C. The Genetic Immunization with Paraflagellar Rod Protein-2 Fused to the HSP70 Confers Protection against Late Trypanosoma cruzi Infection. Vaccine 2006, 24, 7046–7055. [Google Scholar] [CrossRef]
- Arce-Fonseca, M.; Ballinas-Verdugo, M.A.; Zenteno, E.R.A.; Suárez-Flores, D.; Carrillo-Sánchez, S.C.; Alejandre-Aguilar, R.; Rosales-Encina, J.L.; Reyes, P.A.; Rodríguez-Morales, O. Specific Humoral and Cellular Immunity Induced by Trypanosoma cruzi DNA Immunization in a Canine Model. Vet. Res. 2013, 44, 15. [Google Scholar] [CrossRef]
- Rodríguez-Morales, O.; Pérez-Leyva, M.M.; Ballinas-Verdugo, M.A.; Carrillo-Sánchez, S.C.; Rosales-Encina, J.L.; Alejandre-Aguilar, R.; Reyes, P.A.; Arce-Fonseca, M. Plasmid DNA Immunization with Trypanosoma cruzi Genes Induces Cardiac and Clinical Protection against Chagas Disease in the Canine Model. Vet. Res. 2012, 43, 79. [Google Scholar] [CrossRef]
- Rodríguez-Morales, O.; Carrillo-Sánchez, S.C.; García-Mendoza, H.; Aranda-Fraustro, A.; Ballinas-Verdugo, M.A.; Alejandre-Aguilar, R.; Rosales-Encina, J.L.; Vallejo, M.; Arce-Fonseca, M. Effect of the Plasmid-DNA Vaccination on Macroscopic and Microscopic Damage Caused by the Experimental Chronic Trypanosoma cruzi Infection in the Canine Model. BioMed Res. Int. 2013, 2013, e826570. [Google Scholar] [CrossRef]
- Arce-Fonseca, M.; Rios-Castro, M.; Carrillo-Sánchez, S.D.C.; Martínez-Cruz, M.; Rodríguez-Morales, O. Prophylactic and Therapeutic DNA Vaccines against Chagas Disease. Parasites Vectors 2015, 8, 121. [Google Scholar] [CrossRef]
- Bivona, A.E.; Alberti, A.S.; Cerny, N.; Trinitario, S.N.; Malchiodi, E.L. Chagas Disease Vaccine Design: The Search for an Efficient Trypanosoma cruzi Immune-Mediated Control. Biochim. Et Biophys. Acta (BBA)—Mol. Basis Dis. 2020, 1866, 165658. [Google Scholar] [CrossRef]
- Gomes-Neto, J.F.; Sartorius, R.; Canto, F.B.; Almeida, T.S.; Dias, A.A.; Barbosa, C.-H.D.; Melo, G.A.; Oliveira, A.C.; Aguiar, P.-H.N.; Machado, C.R. Vaccination with Recombinant Filamentous Fd Phages against Parasite Infection Requires TLR9 Expression. Front. Immunol. 2018, 9, 1173. [Google Scholar] [CrossRef]
- Miyahira, Y.; Takashima, Y.; Kobayashi, S.; Matsumoto, Y.; Takeuchi, T.; Ohyanagi-Hara, M.; Yoshida, A.; Ohwada, A.; Akiba, H.; Yagita, H.; et al. Immune Responses against a Single CD8+-T-Cell Epitope Induced by Virus Vector Vaccination Can Successfully Control Trypanosoma cruzi Infection. Infect. Immun. 2005, 73, 7356–7365. [Google Scholar] [CrossRef]
- Takayama, E.; Ono, T.; Carnero, E.; Umemoto, S.; Yamaguchi, Y.; Kanayama, A.; Oguma, T.; Takashima, Y.; Tadakuma, T.; García-Sastre, A. Quantitative and Qualitative Features of Heterologous Virus-Vector-Induced Antigen-Specific CD8+ T Cells against Trypanosoma cruzi Infection. Int. J. Parasitol. 2010, 40, 1549–1561. [Google Scholar] [CrossRef]
- Rosenberg, C.S.; Martin, D.L.; Tarleton, R.L. CD8+ T Cells Specific for Immunodominant Trans-Sialidase Epitopes Contribute to Control of Trypanosoma cruzi Infection but Are Not Required for Resistance. J. Immunol. 2010, 185, 560–568. [Google Scholar] [CrossRef]
- Dominguez, M.R.; Silveira, E.L.; de Vasconcelos, J.R.C.; de Alencar, B.C.; Machado, A.V.; Bruna-Romero, O.; Gazzinelli, R.T.; Rodrigues, M.M. Subdominant/Cryptic CD8 T Cell Epitopes Contribute to Resistance against Experimental Infection with a Human Protozoan Parasite. PLoS ONE 2011, 6, e22011. [Google Scholar] [CrossRef]
- Sanchez-Burgos, G.; Mezquita-Vega, R.G.; Escobedo-Ortegon, J.; Ramirez-Sierra, M.J.; Arjona-Torres, A.; Ouaissi, A.; Rodrigues, M.M.; Dumonteil, E. Comparative Evaluation of Therapeutic DNA Vaccines against Trypanosoma cruzi in Mice. FEMS Immunol. Med. Microbiol. 2007, 50, 333–341. [Google Scholar] [CrossRef]
- Costa, F.; Franchin, G.; Pereira-Chioccola, V.L.; Ribeirão, M.; Schenkman, S.; Rodrigues, M.M. Immunization with a Plasmid DNA Containing the Gene of Trans-Sialidase Reduces Trypanosoma cruzi Infection in Mice. Vaccine 1998, 16, 768–774. [Google Scholar] [CrossRef]
- Lúcia Pereira-Chioccola, V.; Costa, F.; Ribeirão, M.; Soares, I.S.; Arena, F.; Schenkman, S.; Rodrigues, M.M. Comparison of Antibody and Protective Immune Responses against Trypanosoma Cruzi Infection Elicited by Immunization with a Parasite Antigen Delivered as Naked DNA or Recombinant Protein. Parasite Immunol. 1999, 21, 103–110. [Google Scholar] [CrossRef]
- Palma, M. Epitopes and Mimotopes Identification Using Phage Display for Vaccine Development against Infectious Pathogens. Vaccines 2023, 11, 1176. [Google Scholar] [CrossRef]
- Prochetto, E.; Roldán, C.; Bontempi, I.A.; Bertona, D.; Peverengo, L.; Vicco, M.H.; Rodeles, L.M.; Pérez, A.R.; Marcipar, I.S.; Cabrera, G. Trans-Sialidase-Based Vaccine Candidate Protects against Trypanosoma cruzi Infection, Not Only Inducing an Effector Immune Response but Also Affecting Cells with Regulatory/Suppressor Phenotype. Oncotarget 2017, 8, 58003. [Google Scholar] [CrossRef]
- Pacini, M.F.; González, F.B.; Dinatale, B.; Balbi, C.B.; Villar, S.R.; Farré, C.; Lupi, G.; Espariz, M.; Blancato, V.S.; Magni, C. Nasal Immunization with a L. lactis-Derived Trans-Sialidase Antigen plus c-Di-AMP Protects against Acute Oral T. cruzi Infection. Vaccine 2022, 40, 2311–2323. [Google Scholar] [CrossRef]
- Queiroz, A.M.V.; Oliveira, J.W.d.F.; Moreno, C.J.; Guérin, D.M.A.; Silva, M.S. VLP-Based Vaccines as a Suitable Technology to Target Trypanosomatid Diseases. Vaccines 2021, 9, 220. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm1. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide Sequences (5′-3′) | |
|---|---|
| F-chimera | CTAGCTAGCATGAAGGCTTTGAAACTTTTTAAAG |
| R1-chimera | GTTCGCAACCAGGGTAAATTTATAATTAGCTGACCATGTGACGTGC |
| R2-chimera | CCGCTCGAGTTACACAAGAGTGAACTTGTAGTTCGCAACCAGGGTAAAT |
| F-TS | CTAGCTAGCAGAAGGTCAATGGGAAAG |
| R-TS | CCGCTCGAGTCACAGAGCCGCAAACCCCCAC |
| F-Tc52 | GGACTGCAGGAAGGCTTTGAAACTTTT |
| R-Tc52 | GGAAAGCTTTCAAGACGATGGACGCAAA |
| F-Sat | GCAGTCGGCKGATCGTTTTCG |
| R-Sat | TTCAGRGTTGTTTGGTGTCCAGTG |
| Amino Acid Sequences | Epitope Characteristics |
|---|---|
| Binding MHCI epitopes | Type of binding/allele |
| SANYKFTL | SB H2-Kb (New) |
| SANYKFTLV | WB H2-Db and SB H2-Kb (New) |
| ANYKFTLV | SB H2-Kb (TSkb20) |
| ANYKFTLVA | SB H2-Kb (New) |
| VANYKFTL | SB H2-Kb (New) |
| VANYKFTLV | SB H2-Kb (New) |
| B-cell epitopes | Origin |
| SHHHHHHGMASMK | Histidine tag |
| EVPLGDDMPQWYKELNPRE | Tc52 |
| RDPFNEEKRKSVDN | Tc52 |
| YCGYDIFHNAP | Tc52 |
| RTTVKETISKPE | Tc52 |
| PKSHVTWSANYK | New |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez, M.E.; Zabala, B.A.; Mesías, A.C.; Biscari, L.; Kaufman, C.D.; Alloatti, A.; Siano, F.; Picariello, G.; Corbalán, N.S.; Lenis, B.A.; et al. Protective Efficacy of the Epitope-Conjugated Antigen N-Tc52/TSkb20 in Mitigating Trypanosoma cruzi Infection through CD8+ T-Cells and IFNγ Responses. Vaccines 2024, 12, 621. https://doi.org/10.3390/vaccines12060621
Vázquez ME, Zabala BA, Mesías AC, Biscari L, Kaufman CD, Alloatti A, Siano F, Picariello G, Corbalán NS, Lenis BA, et al. Protective Efficacy of the Epitope-Conjugated Antigen N-Tc52/TSkb20 in Mitigating Trypanosoma cruzi Infection through CD8+ T-Cells and IFNγ Responses. Vaccines. 2024; 12(6):621. https://doi.org/10.3390/vaccines12060621
Chicago/Turabian StyleVázquez, María Elisa, Brenda A. Zabala, Andrea C. Mesías, Lucia Biscari, Cintia D. Kaufman, Andrés Alloatti, Francesco Siano, Gianluca Picariello, Natalia S. Corbalán, Bladimiro A. Lenis, and et al. 2024. "Protective Efficacy of the Epitope-Conjugated Antigen N-Tc52/TSkb20 in Mitigating Trypanosoma cruzi Infection through CD8+ T-Cells and IFNγ Responses" Vaccines 12, no. 6: 621. https://doi.org/10.3390/vaccines12060621
APA StyleVázquez, M. E., Zabala, B. A., Mesías, A. C., Biscari, L., Kaufman, C. D., Alloatti, A., Siano, F., Picariello, G., Corbalán, N. S., Lenis, B. A., Toscano, M. A., Parodi, C. M., Brandán, C. M. P., & Acuña, L. (2024). Protective Efficacy of the Epitope-Conjugated Antigen N-Tc52/TSkb20 in Mitigating Trypanosoma cruzi Infection through CD8+ T-Cells and IFNγ Responses. Vaccines, 12(6), 621. https://doi.org/10.3390/vaccines12060621