
TEG®, Microclot and Platelet Mapping for Guiding Early Management of Severe COVID-19 Coagulopathy

 , and
, and
Abstract
:1. Introduction
1.1. Disseminated Intravascular Coagulopathy (DIC) and COVID-19: An Uncommon Phenomenon?
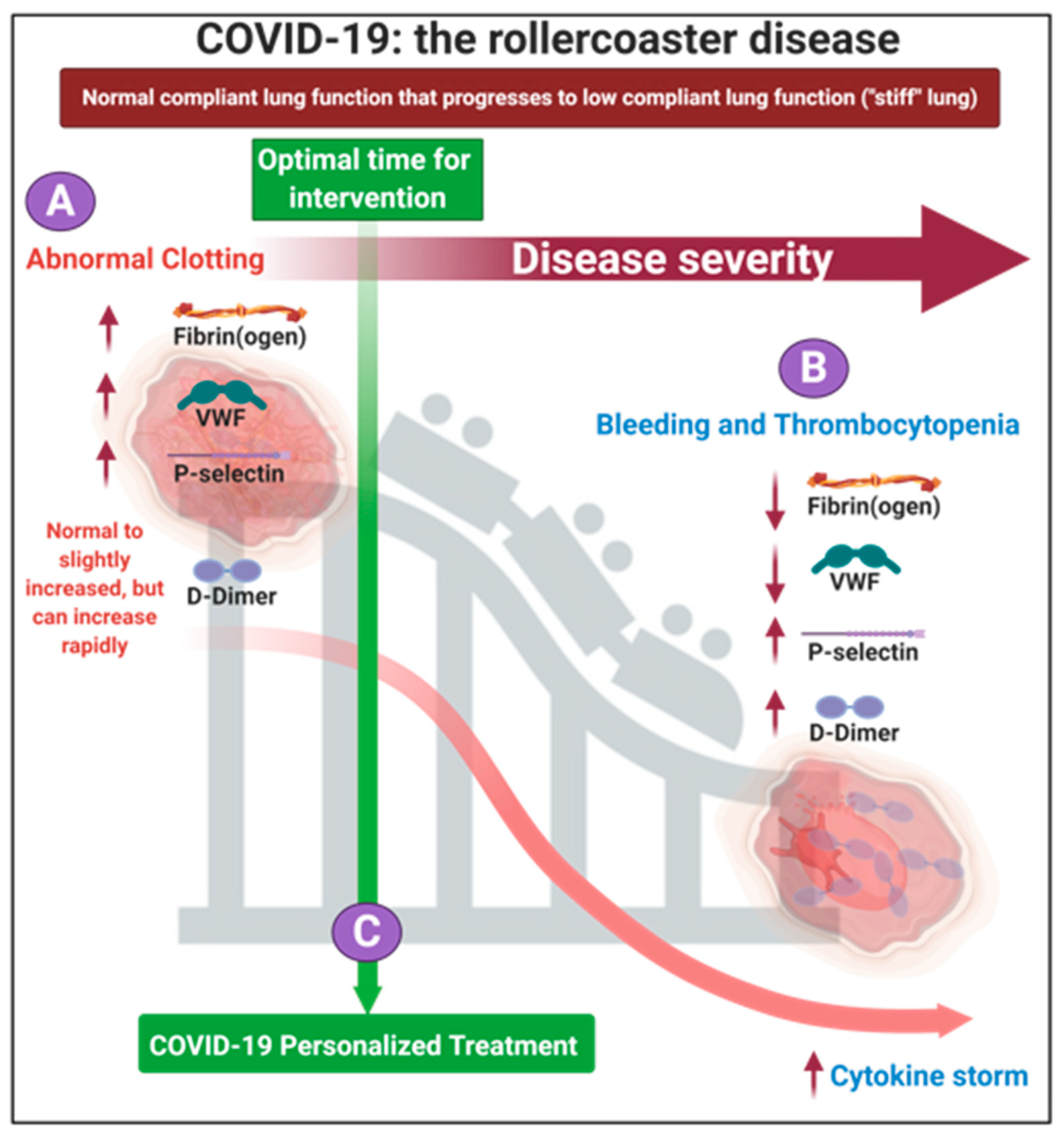
1.2. The Progression of the Disease, If Untreated Is a Two-Phase ‘Rollercoaster’ of Events, Characterized by Thrombotic Pathology Followed by Bleeding or Thrombocytopenia Pathologies
1.3. A Place for Existing Point-Of Care Techniques to Guide COVID-19 Treatment
2. The Early Treatment of Patients with Anticoagulation Medication
Anticoagulation Trails
- Therapeutic anticoagulation was in-hospital oral rivaroxaban (20 mg or 15 mg daily) for stable patients, or initial subcutaneous enoxaparin (1 mg/kg twice per day);
- or intravenous unfractionated heparin (to achieve a 0.3–0.7 IU/mL anti-Xa concentration) for clinically unstable patients, followed by rivaroxaban to day 30;
- Prophylactic anticoagulation was standard in-hospital enoxaparin or unfractionated heparin.
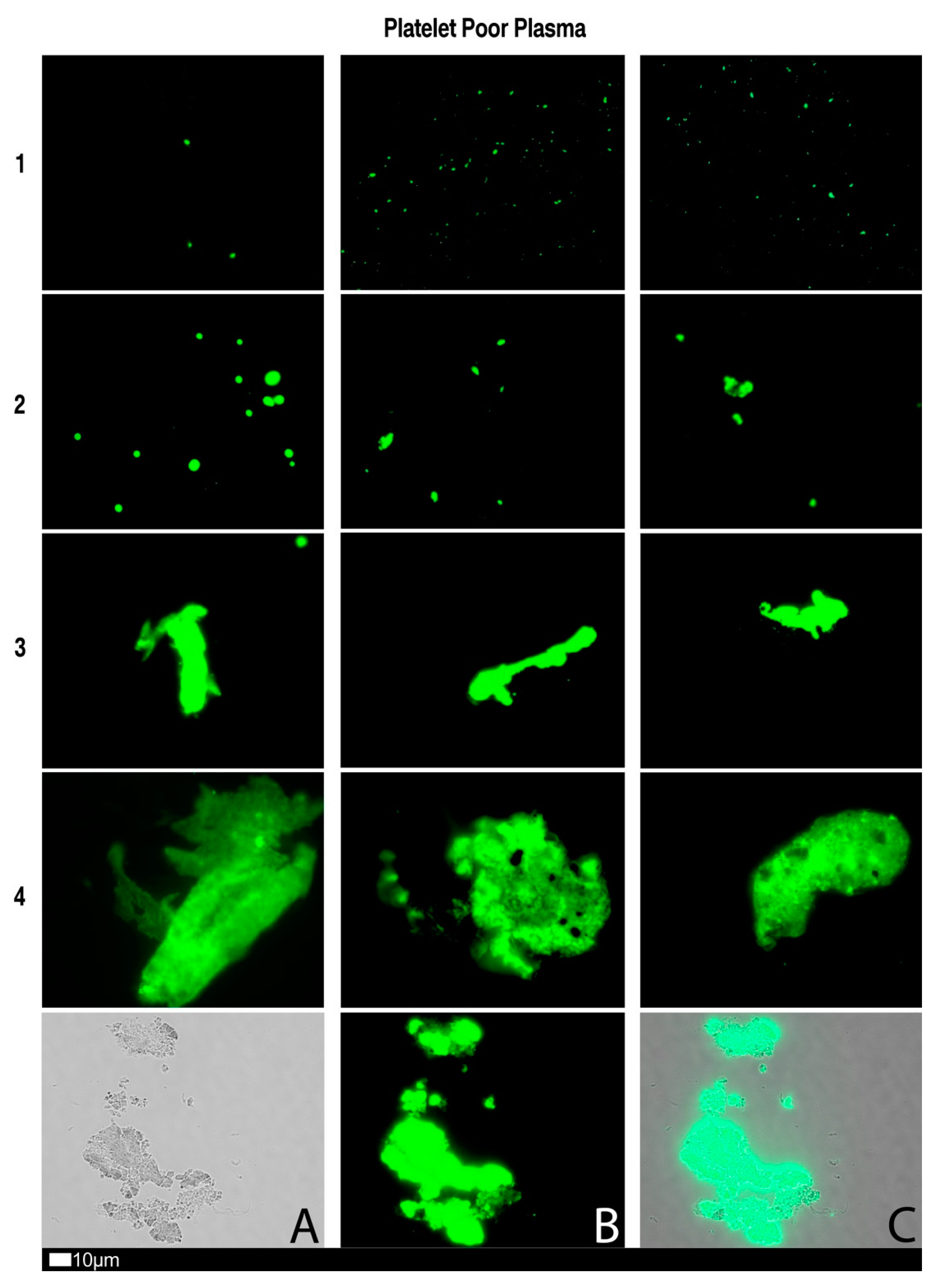
3. Fluorescence Microscopy Can Be Used to Provide a Marker of Microclot Formation and Platelet Hyperactivation
3.1. Methods Used to Analyse Microclots in Platelet Poor Plasma
3.2. Methods Used to Prepare Platelets
3.3. A Grading System for Plasma Microclot Formation and Platelet Activation, Spreading and Clumping
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Wynants, L.; Van Calster, B.; Collins, G.S.; Riley, R.D.; Heinze, G.; Schuit, E.; Bonten, M.M.J.; Damen, J.A.A.; Debray, T.P.A.; De Vos, M.; et al. Prediction models for diagnosis and prognosis of COVID-19 infection: Systematic review and critical appraisal. BMJ 2020, 369, m1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Docherty, A.B.; Harrison, E.M.; Green, C.A.; Hardwick, H.E.; Pius, R.; Norman, L.; Holden, K.A.; Read, J.M.; Dondelinger, F.; Carson, G.; et al. Features of 20 133 UK patients in hospital with COVID-19 using the ISARIC WHO Clinical Characterisation Protocol: Prospective observational cohort study. BMJ 2020, 369, m1985. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef]
- Siddiqi, H.K.; Libby, P.; Ridker, P.M. COVID-19—A vascular disease. Trends Cardiovasc. Med. 2021, 31, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Meizlish, M.; Pine, A.; Goshua, G.; Chang, C.H.; Zhang, H.; Bishai, J.; Bahel, P.; Patel, A.; Gbyli, R.; Kwan, J.; et al. Circulating Markers of Angiogenesis and Endotheliopathy in COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Benigni, A.; Casiraghi, F.; Ng, L.F.P.; Renia, L.; Remuzzi, G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat. Rev. Nephrol. 2021, 17, 46–64. [Google Scholar] [CrossRef]
- Smadja, D.M.; Mentzer, S.J.; Fontenay, M.; Laffan, M.A.; Ackermann, M.; Helms, J.; Jonigk, D.; Chocron, R.; Pier, G.B.; Gendron, N.; et al. COVID-19 is a systemic vascular hemopathy: Insight for mechanistic and clinical aspects. Angiogenesis 2021, 24, 755–788. [Google Scholar] [CrossRef] [PubMed]
- Wool, G.D.; Miller, J.L. The Impact of COVID-19 Disease on Platelets and Coagulation. Pathobiology 2021, 88, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C. Disseminated intravascular coagulation: Is it fact or fancy? Blood Coagul. Fibrinolysis 2018, 29, 330–337. [Google Scholar] [CrossRef]
- Gąsecka, A.; Borovac, J.A.; Guerreiro, R.A.; Giustozzi, M.; Parker, W.; Caldeira, D.; Chiva-Blanch, G. Thrombotic Complications in Patients with COVID-19: Pathophysiological Mechanisms, Diagnosis, and Treatment. Cardiovasc. Drugs Ther. 2021, 35, 215–229. [Google Scholar] [CrossRef]
- Paternoster, G.; Bertini, P.; Innelli, P.; Trambaiolo, P.; Landoni, G.; Franchi, F.; Scolletta, S.; Guarracino, F. Right Ventricular Dysfunction in Patients With COVID-19: A Systematic Review and Meta-analysis. J. Cardiothorac. Vasc. Anesth. 2021, 35, 3319–3324. [Google Scholar] [CrossRef]
- Lan, Y.; Liu, W.; Zhou, Y. Right Ventricular Damage in COVID-19: Association between Myocardial Injury and COVID-19. Front. Cardiovasc. Med. 2021, 8, 606318. [Google Scholar] [CrossRef] [PubMed]
- Soulat-Dufour, L.; Fauvel, C.; Weizman, O.; Barbe, T.; Pezel, T.; Mika, D.; Cellier, J.; Geneste, L.; Panagides, V.; Marsou, W.; et al. Prognostic value of right ventricular dilatation in patients with COVID-19: A multicentre study. Eur. Heart J. Cardiovasc. Imaging 2021. [Google Scholar] [CrossRef] [PubMed]
- Mancini, I.; Baronciani, L.; Artoni, A.; Colpani, P.; Biganzoli, M.; Cozzi, G.; Novembrino, C.; Boscolo Anzoletti, M.; De Zan, V.; Pagliari, M.T.; et al. The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J. Thromb. Haemost. 2021, 19, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil Extracellular Traps (NETs) Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Deigendesch, H.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; Bassetti, S.; et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology 2020, 77, 198–209. [Google Scholar] [CrossRef]
- Meizoso, J.P.; Moore, H.B.; Moore, E.E. Fibrinolysis Shutdown in COVID-19: Clinical Manifestations, Molecular Mechanisms, and Therapeutic Implications. J. Am. Coll. Surg. 2021, 232, 995–1003. [Google Scholar] [CrossRef]
- Pretorius, E.; Venter, C.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B. Prevalence of readily detected amyloid blood clots in ‘unclotted’ Type 2 Diabetes Mellitus and COVID-19 plasma: A preliminary report. Cardiovasc. Diabetol. 2020, 19, 193. [Google Scholar] [CrossRef] [PubMed]
- Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. Erythrocyte, Platelet, Serum Ferritin, and P-Selectin Pathophysiology Implicated in Severe Hypercoagulation and Vascular Complications in COVID-19. Int. J. Mol. Sci. 2020, 21, 8234. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Vlok, M.; Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Steenkamp, J.; Kell, D.B. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc. Diabetol. 2021, 20, 172. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.M.; Khan, R.; Kwaan, H.C.; Neal, M.D. Fibrinolysis Shutdown in COVID-19-Associated Coagulopathy: A Crosstalk among Immunity, Coagulation, and Specialists in Medicine and Surgery. J. Am. Coll. Surg. 2021, 232, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Giannis, D.; Ziogas, I.A.; Gianni, P. Coagulation disorders in coronavirus infected patients: COVID-19, SARS-CoV-1, MERS-CoV and lessons from the past. J. Clin. Virol. 2020, 127, 104362. [Google Scholar] [CrossRef] [PubMed]
- Kollias, A.; Kyriakoulis, K.G.; Dimakakos, E.; Poulakou, G.; Stergiou, G.S.; Syrigos, K. Thromboembolic risk and anticoagulant therapy in COVID-19 patients: Emerging evidence and call for action. Br. J. Haematol. 2020, 189, 846–847. [Google Scholar] [CrossRef]
- Middeldorp, S.; Coppens, M.; van Haaps, T.F.; Foppen, M.; Vlaar, A.P.; Müller, M.C.A.; Bouman, C.C.S.; Beenen, L.F.M.; Kootte, R.S.; Heijmans, J.; et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1995–2002. [Google Scholar] [CrossRef]
- Miesbach, W.; Makris, M. COVID-19: Coagulopathy, Risk of Thrombosis, and the Rationale for Anticoagulation. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620938149. [Google Scholar] [CrossRef]
- Levi, M.; Thachil, J.; Iba, T.; Levy, J.H. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol 2020, 7, e438–e440. [Google Scholar] [CrossRef]
- Liu, P.P.; Blet, A.; Smyth, D.; Li, H. The Science Underlying COVID-19: Implications for the Cardiovascular System. Circulation 2020, 142, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Kabrhel, C.; Mark Courtney, D.; Camargo, C.A., Jr.; Plewa, M.C.; Nordenholz, K.E.; Moore, C.L.; Richman, P.B.; Smithline, H.A.; Beam, D.M.; Kline, J.A. Factors associated with positive D-dimer results in patients evaluated for pulmonary embolism. Acad. Emerg. Med. 2010, 17, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Pretorius, E. The simultaneous occurrence of both hypercoagulability and hypofibrinolysis in blood and serum during systemic inflammation, and the roles of iron and fibrin(ogen). Integr. Biol. 2015, 7, 24–52. [Google Scholar] [CrossRef] [Green Version]
- Kell, D.B.; Pretorius, E. No effects without causes: The Iron Dysregulation and Dormant Microbes hypothesis for chronic, inflammatory diseases. Biol. Rev. Camb. Philos. Soc. 2018, 93, 1518–1557. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Pretorius, E. To What Extent Are the Terminal Stages of Sepsis, Septic Shock, Systemic Inflammatory Response Syndrome, and Multiple Organ Dysfunction Syndrome Actually Driven by a Prion/Amyloid Form of Fibrin? Semin. Thromb. Hemost. 2018, 44, 224–238. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, S.; Sharma, K.; Singh, P.K. Von Willebrand factor: A key glycoprotein involved in thrombo-inflammatory complications of COVID-19. Chem. Biol. Interact. 2021, 348, 109657. [Google Scholar] [CrossRef]
- Liao, D.; Zhou, F.; Luo, L.; Xu, M.; Wang, H.; Xia, J.; Gao, Y.; Cai, L.; Wang, Z.; Yin, P.; et al. Haematological characteristics and risk factors in the classification and prognosis evaluation of COVID-19: A retrospective cohort study. Lancet Haematol. 2020, 7, e671–e678. [Google Scholar] [CrossRef]
- Spiezia, L.; Boscolo, A.; Poletto, F.; Cerruti, L.; Tiberio, I.; Campello, E.; Navalesi, P.; Simioni, P. COVID-19-Related Severe Hypercoagulability in Patients Admitted to Intensive Care Unit for Acute Respiratory Failure. Thromb. Haemost. 2020, 120, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Costello, R.A.; Nehring, S.M. Disseminated Intravascular Coagulation (DIC). In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2020. [Google Scholar]
- Aigner, L.; Pietrantonio, F.; Bessa de Sousa, D.M.; Michael, J.; Schuster, D.; Reitsamer, H.A.; Zerbe, H.; Studnicka, M. The Leukotriene Receptor Antagonist Montelukast as a Potential COVID-19 Therapeutic. Front. Mol. Biosci. 2020, 7, 610132. [Google Scholar] [CrossRef]
- Grobler, C.; Bredenkamp, J.; Grobbelaar, M.; Maphumulo, S.; Laubscher, J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. COVID-19: The Rollercoaster of Fibrin(ogen), D-dimer, von Willebrand Factor, P-selectin and Their Interactions with Endothelial Cells, Platelets and Erythrocytes. Int. J. Mol. Sci. 2020, 21, 5168. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Karp Leaf, R.S.; Dzik, W.H.; Carlson, J.C.; Fogerty, A.E.; Waheed, A.; Goodarzi, K.; Bendapudi, P.; Bornikova, L.; Gupta, S.; et al. COVID and Coagulation: Bleeding and Thrombotic Manifestations of SARS-CoV-2 Infection. Blood 2020, 136, 489–500. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef]
- Boccia, M.; Aronne, L.; Celia, B.; Mazzeo, G.; Ceparano, M.; D’Agnano, V.; Parrella, R.; Valente, T.; Bianco, A.; Perrotta, F. COVID-19 and coagulative axis: Review of emerging aspects in a novel disease. Monaldi Arch. Chest Dis. 2020, 90. [Google Scholar] [CrossRef]
- Li, C.; Hu, B.; Zhang, Z.; Qin, W.; Zhu, Z.; Zhai, Z.; Davidson, B.L.; Wang, C. D-dimer triage for COVID-19. Acad. Emerg. Med. 2020, 27, 612–613. [Google Scholar] [CrossRef] [PubMed]
- Wright, F.L.; Vogler, T.O.; Moore, E.E.; Moore, H.B.; Wohlauer, M.V.; Urban, S.; Nydam, T.L.; Moore, P.K.; McIntyre, R.C., Jr. Fibrinolysis Shutdown Correlation with Thromboembolic Events in Severe COVID-19 Infection. J. Am. Coll. Surg. 2020, 231, 193–203.e1. [Google Scholar] [CrossRef]
- Zou, Y.; Guo, H.; Zhang, Y.; Zhang, Z.; Liu, Y.; Wang, J.; Lu, H.; Qian, Z. Analysis of coagulation parameters in patients with COVID-19 in Shanghai, China. Biosci. Trends 2020, 14, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef]
- Sinha, P.; Matthay, M.A.; Calfee, C.S. Is a “Cytokine Storm” Relevant to COVID-19? JAMA Intern Med. 2020, 180, 1152–1154. [Google Scholar] [CrossRef] [PubMed]
- Dwiputra Hernugrahanto, K.; Novembri Utomo, D.; Hariman, H.; Budhiparama, N.C.; Medika Hertanto, D.; Santoso, D.; Hogendoorn, P.C.W. Thromboembolic involvement and its possible pathogenesis in COVID-19 mortality: Lesson from post-mortem reports. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1670–1679. [Google Scholar]
- Neri, T.; Nieri, D.; Celi, A. P-selectin blockade in COVID-19-related ARDS. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L1237–L1238. [Google Scholar] [CrossRef]
- Panigada, M.; Bottino, N.; Tagliabue, P.; Grasselli, G.; Novembrino, C.; Chantarangkul, V.; Pesenti, A.; Peyvandi, F.; Tripodi, A. Hypercoagulability of COVID-19 patients in Intensive Care Unit. A Report of Thromboelastography Findings and other Parameters of Hemostasis. J. Thromb. Haemost. 2020, 18, 1738–1842. [Google Scholar] [CrossRef]
- Garcia-Olivé, I.; Sintes, H.; Radua, J.; Abad Capa, J.; Rosell, A. D-dimer in patients infected with COVID-19 and suspected pulmonary embolism. Respir. Med. 2020, 169, 106023. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, K.; Wei, H.; Chen, W.; Wang, W.; Jia, L.; Liu, Q.; Zhang, J.; Shan, T.; Peng, Z.; et al. Dynamic relationship between D-dimer and COVID-19 severity. Br. J. Haematol. 2020, 190, e24–e27. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, E.J.; Thachil, J. Reporting of D-dimer data in COVID-19: Some confusion and potential for misinformation. Clin. Chem. Lab. Med. 2020, 58, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Favaloro, E.J. D-dimer is Associated with Severity of Coronavirus Disease 2019: A Pooled Analysis. Thromb. Haemost. 2020, 120, 876–878. [Google Scholar] [CrossRef] [Green Version]
- Pretorius, E.; Page, M.J.; Engelbrecht, L.; Ellis, G.C.; Kell, D.B. Substantial fibrin amyloidogenesis in type 2 diabetes assessed using amyloid-selective fluorescent stains. Cardiovasc. Diabetol. 2017, 16, 141. [Google Scholar] [CrossRef] [Green Version]
- Lopes, R.D.; de Barros, E.S.P.G.M.; Furtado, R.H.M.; Macedo, A.V.S.; Bronhara, B.; Damiani, L.P.; Barbosa, L.M.; de Aveiro Morata, J.; Ramacciotti, E.; de Aquino Martins, P.; et al. Therapeutic versus prophylactic anticoagulation for patients admitted to hospital with COVID-19 and elevated D-dimer concentration (ACTION): An open-label, multicentre, randomised, controlled trial. Lancet 2021, 397, 2253–2263. [Google Scholar] [CrossRef]
- Escher, R.; Breakey, N.; Lämmle, B. Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 2020, 190, 62. [Google Scholar] [CrossRef]
- Zachariah, U.; Nair, S.C.; Goel, A.; Balasubramanian, K.A.; Mackie, I.; Elias, E.; Eapen, C.E. Targeting raised von Willebrand factor levels and macrophage activation in severe COVID-19: Consider low volume plasma exchange and low dose steroid. Thromb. Res. 2020, 192, 2. [Google Scholar] [CrossRef]
- Chapman, M.P.; Moore, E.E.; Ramos, C.R.; Ghasabyan, A.; Harr, J.N.; Chin, T.L.; Stringham, J.R.; Sauaia, A.; Silliman, C.C.; Banerjee, A. Fibrinolysis greater than 3% is the critical value for initiation of antifibrinolytic therapy. J. Trauma Acute Care Surg. 2013, 75, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Görlinger, K.; Levy, J.H. COVID-19–associated Coagulopathy: Less Fibrinolysis Can Be More Harmful! Anesthesiology 2021, 134, 366–369. [Google Scholar] [CrossRef]
- Chandel, A.; Patolia, S.; Looby, M.; Bade, N.; Khangoora, V.; King, C.S. Association of D-dimer and Fibrinogen with Hypercoagulability in COVID-19 Requiring Extracorporeal Membrane Oxygenation. J. Intensive Care Med. 2021, 36, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Hranjec, T.; Estreicher, M.; Rogers, B.; Kohler, L.; Solomon, R.; Hennessy, S.; Cibulas, M.; Hurst, D.; Hegazy, M.; Lee, J.; et al. Integral Use of Thromboelastography With Platelet Mapping to Guide Appropriate Treatment, Avoid Complications, and Improve Survival of Patients With Coronavirus Disease 2019-Related Coagulopathy. Crit. Care Explor. 2020, 2, e0287. [Google Scholar] [CrossRef] [PubMed]
- Smolarz, A.; McCarthy, P.; Shmookler, A.; Badhwar, V.; Hayanga, A.J.; Sakhuja, A. Utilization of Thromboelastogram and Inflammatory Markers in the Management of Hypercoagulable State in Patients with COVID-19 Requiring ECMO Support. Case Rep. Crit. Care 2021, 2021, 8824531. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, E.J.; Bonar, R. An update on quality control for the PFA-100/PFA-200. Platelets 2018, 29, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, V.G.; Kirklin, H.K.; Hoogendoorn, H.; Ellis, T.C.; Holman, W.L. Thromboelastographic method to quantify the contribution of factor XIII to coagulation kinetics. Blood Coagul. Fibrinolysis Int. J. Haemost. Thromb. 2007, 18, 145–150. [Google Scholar] [CrossRef]
- Nielsen, V.G. Beyond cell based models of coagulation: Analyses of coagulation with clot “lifespan” resistance-time relationships. Thromb. Res. 2008, 122, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, V.G. Effects of purified human fibrinogen modified with carbon monoxide and iron on coagulation in rabbits injected with Crotalus atrox venom. J. Thromb. Thrombolysis 2017, 44, 481–488. [Google Scholar] [CrossRef]
- Nielsen, V.G.; Audu, P.; Cankovic, L.; Lyerly, R.T., 3rd; Steenwyk, B.L.; Armstead, V.; Powell, G. Qualitative thrombelastographic detection of tissue factor in human plasma. Anesth. Analg. 2007, 104, 59–64. [Google Scholar] [CrossRef]
- Pretorius, E.; Swanepoel, A.C.; DeVilliers, S.; Bester, J. Blood clot parameters: Thromboelastography and scanning electron microscopy in research and clinical practice. Thromb. Res. 2017, 154, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Bester, J.; Matshailwe, C.; Pretorius, E. Simultaneous presence of hypercoagulation and increased clot lysis time due to IL-1β, IL-6 and IL-8. Cytokine 2018, 110, 237–242. [Google Scholar] [CrossRef]
- Bester, J.; Pretorius, E. Effects of IL-1β, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci. Rep. 2016, 6, 32188. [Google Scholar] [CrossRef]
- Bester, J.; Soma, P.; Kell, D.B.; Pretorius, E. Viscoelastic and ultrastructural characteristics of whole blood and plasma in Alzheimer-type dementia, and the possible role of bacterial lipopolysaccharides (LPS). Oncotarget 2015, 6, 35284–35303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pretorius, L.; Thomson, G.J.A.; Adams, R.C.M.; Nell, T.A.; Laubscher, W.A.; Pretorius, E. Platelet activity and hypercoagulation in type 2 diabetes. Cardiovasc. Diabetol. 2018, 17, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randeria, S.N.; Thomson, G.J.A.; Nell, T.A.; Roberts, T.; Pretorius, E. Inflammatory cytokines in type 2 diabetes mellitus as facilitators of hypercoagulation and abnormal clot formation. Cardiovasc. Diabetol. 2019, 18, 72. [Google Scholar] [CrossRef] [Green Version]
- Sayyadi, M.; Khosravi, M.; Ghaznavi-Rad, E. Contribution value of coagulation abnormalities in COVID-19 prognosis: A bright perspective on the laboratory pattern of patients with coronavirus disease 2019. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 518–522. [Google Scholar]
- Amgalan, A.; Allen, T.; Othman, M.; Ahmadzia, H.K. Systematic review of viscoelastic testing (TEG/ROTEM) in obstetrics and recommendations from the women’s SSC of the ISTH. J. Thromb. Haemost. 2020, 18, 1813–1838. [Google Scholar] [CrossRef] [PubMed]
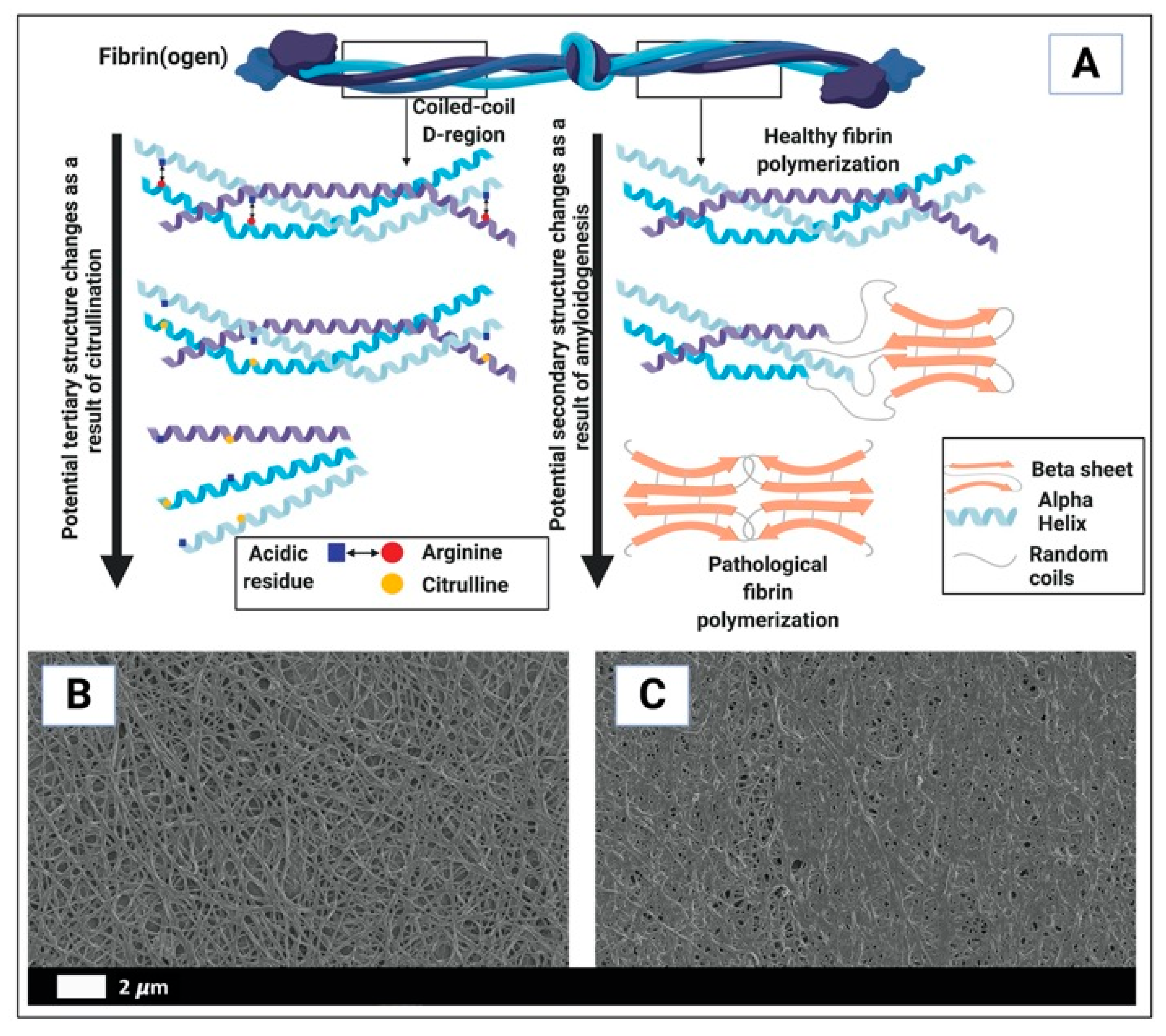
- Kell, D.B.; Pretorius, E. Proteins behaving badly. Substoichiometric molecular control and amplification of the initiation and nature of amyloid fibril formation: Lessons from and for blood clotting. Prog. Biophys. Mol. Biol. 2017, 123, 16–41. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; Thomson, G.J.A.; Nunes, J.M.; Engelbrecht, A.M.; Nell, T.A.; de Villiers, W.J.S.; de Beer, M.C.; Engelbrecht, L.; Kell, D.B.; Pretorius, E. Serum amyloid A binds to fibrin(ogen), promoting fibrin amyloid formation. Sci. Rep. 2019, 9, 3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pretorius, E.; Mbotwe, S.; Bester, J.; Robinson, C.J.; Kell, D.B. Acute induction of anomalous and amyloidogenic blood clotting by molecular amplification of highly substoichiometric levels of bacterial lipopolysaccharide. J. R. Soc. Interface 2016, 13, 20160539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, J.M.; Fillis, T.; Page, M.J.; Venter, C.; Lancry, O.; Kell, D.B.; Windberger, U.; Pretorius, E. Gingipain R1 and lipopolysaccharide from Porphyromonas gingivalis have major effects on blood clot morphology and mechanics. Front. Immunol. 2020, 11, 1551. [Google Scholar] [CrossRef]
- Alhazzani, W.; Møller, M.H.; Arabi, Y.M.; Loeb, M.; Gong, M.N.; Fan, E.; Oczkowski, S.; Levy, M.M.; Derde, L.; Dzierba, A.; et al. Surviving Sepsis Campaign: Guidelines on the Management of Critically Ill Adults with Coronavirus Disease 2019 (COVID-19). Crit. Care Med. 2020, 48, e440–e469. [Google Scholar] [CrossRef] [PubMed]
- American Thoracic Society-led International Task Force; Wilson, K.C.; Chotirmall, S.H.; Bai, C.; Rello, J. COVID-19: Interim Guidance on Management Pending Empirical Evidence. 2020. Available online: https://www.thoracic.org/covid/covid-19-guidance.pdf (accessed on 15 October 2021).
- Ayerbe, L.; Risco, C.; Ayis, S. The association between treatment with heparin and survival in patients with COVID-19. J. Thromb. Thrombolysis 2020, 50, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Menezes-Rodrigues, F.S.; Padrão Tavares, J.G.; Pires de Oliveira, M.; Guzella de Carvalho, R.; Ruggero Errante, P.; Omar Taha, M.; Fagundes, D.J.; Caricati-Neto, A. Anticoagulant and antiarrhythmic effects of heparin in the treatment of COVID-19 patients. J. Thromb. Haemost. 2020, 18, 2073–2075. [Google Scholar] [CrossRef]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Lawler, P.R.; Goligher, E.C.; Berger, J.S.; Neal, M.D.; McVerry, B.J.; Nicolau, J.C.; Gong, M.N.; Carrier, M.; Rosenson, R.S.; Reynolds, H.R.; et al. Therapeutic Anticoagulation with Heparin in Noncritically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 385, 790–802. [Google Scholar]
- Ten Cate, H. Surviving COVID-19 with Heparin? N. Engl. J. Med. 2021, 385, 845–846. [Google Scholar] [CrossRef]
- Viecca, M.; Radovanovic, D.; Forleo, G.B.; Santus, P. Enhanced platelet inhibition treatment improves hypoxemia in patients with severe COVID-19 and hypercoagulability. A case control, proof of concept study. Pharmacol. Res. 2020, 158, 104950. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.R.; Misdary, C.; Yegya-Raman, N.; Kim, S.; Narayanan, N.; Siddiqui, S.; Salgame, P.; Radbel, J.; Groote, F.; Michel, C.; et al. Montelukast in hospitalized patients diagnosed with COVID-19. J. Asthma 2021, 1–7. [Google Scholar] [CrossRef]
- Barré, J.; Sabatier, J.M.; Annweiler, C. Montelukast Drug May Improve COVID-19 Prognosis: A Review of Evidence. Front. Pharmacol. 2020, 11, 1344. [Google Scholar] [CrossRef]
- Almerie, M.Q.; Kerrigan, D.D. The association between obesity and poor outcome after COVID-19 indicates a potential therapeutic role for montelukast. Med. Hypotheses 2020, 143, 109883. [Google Scholar] [CrossRef]
- Dey, M.; Singh, R.K. Possible Therapeutic Potential of Cysteinyl Leukotriene Receptor Antagonist Montelukast in Treatment of SARS-CoV-2-Induced COVID-19. Pharmacology 2021, 106, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Fidan, C.; Aydoğdu, A. As a potential treatment of COVID-19: Montelukast. Med. Hypotheses 2020, 142, 109828. [Google Scholar] [CrossRef] [PubMed]
- Sanghai, N.; Tranmer, G.K. Taming the cytokine storm: Repurposing montelukast for the attenuation and prophylaxis of severe COVID-19 symptoms. Drug Discov. Today 2020, 25, 2076–2079. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Almulaiky, Y.Q.; Cruz-Martins, N.; El-Saber Batiha, G. Role of leukotriene pathway and montelukast in pulmonary and extrapulmonary manifestations of COVID-19: The enigmatic entity. Eur. J. Pharmacol. 2021, 904, 174196. [Google Scholar] [CrossRef] [PubMed]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets Can Associate with SARS-CoV-2 RNA and Are Hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef]
- Lu, Q.; Malinauskas, R.A. Comparison of two platelet activation markers using flow cytometry after in vitro shear stress exposure of whole human blood. Artif. Organs 2011, 35, 137–144. [Google Scholar] [CrossRef]
- Oldenburg, C.E.; Doan, T. Rigorous Randomized Controlled Trial Implementation in the Era of COVID-19. Am. J. Trop. Med. Hyg. 2020, 102, 1154–1155. [Google Scholar] [CrossRef] [Green Version]
- Busch, C.; Dawes, J.; Pepper, D.S.; Wasteson, A. Binding of platelet factor 4 to cultured human umbilical vein endothelial cells. Thromb. Res. 1980, 19, 129–137. [Google Scholar] [CrossRef]
- Dawes, J.; Smith, R.C.; Pepper, D.S. The release, distribution, and clearance of human beta-thromboglobulin and platelet factor 4. Thromb. Res. 1978, 12, 851–861. [Google Scholar] [CrossRef]
- Kaplan, K.L.; Owen, J. Plasma levels of beta-thromboglobulin and platelet factor 4 as indices of platelet activation in vivo. Blood 1981, 57, 199–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windberger, U.; Dibiasi, C.; Lotz, E.M.; Scharbert, G.; Reinbacher-Koestinger, A.; Ivanov, I.; Ploszczanski, L.; Antonova, N.; Lichtenegger, H. The effect of hematocrit, fibrinogen concentration and temperature on the kinetics of clot formation of whole blood. Clin. Hemorheol. Microcirc. 2020, 75, 431–445. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Circulating Biomarkers | Selected References |
|---|---|
| P-selectin | [7,51] |
| Fibrin(ogen) and D-dimer | [38,42,45,47,52,53,54,55,56,57,58] |
| Von Willebrand Factor | [17,36,59,60] |
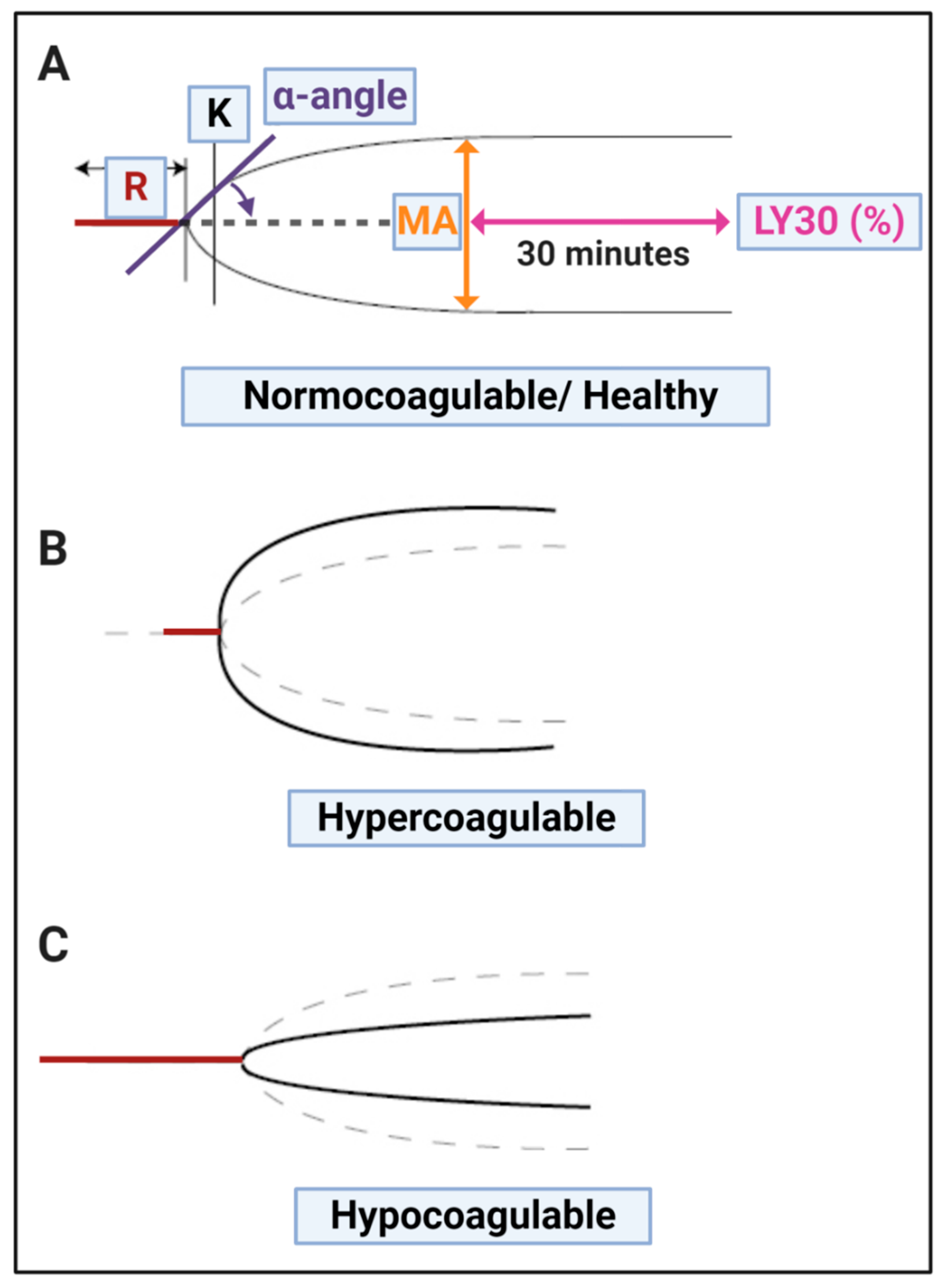
| (A) Thromboelastography® | ||
| TEG® Parameters | Explanation | |
| R value: reaction time measured in minutes | Time of latency from start of test to initial fibrin formation (amplitude of 2 mm); i.e., initiation time. | |
| K: kinetics measured in minutes | Time taken to achieve a certain level of clot strength (amplitude of 20 mm); i.e., amplification. | |
| A (Alpha): Angle (slope between the traces represented by R and K) Angle is measured in degrees | The angle measures the speed at which fibrin build up and cross linking takes place, hence assesses the rate of clot formation, i.e., thrombin burst. | |
| MA: Maximal Amplitude measured in mm | Maximum clot size: it reflects the ultimate strength of the fibrin clot, i.e., overall stability of the clot. The larger the MA the more hypercoagulable the clot. | |
| Maximum rate of thrombus generation (MRTG) measured in Dyn.cm−2.s−1 | The maximum velocity of clot growth observed or maximum rate of thrombus generation using G, where G is the elastic modulus strength of the thrombus in dynes per cm−2. | |
| Time to maximum rate of thrombusgeneration (TMRTG) measured in minutes | The time interval observed before the maximum speed of the clot growth. | |
| Total thrombus generation (TTG) measured in Dyn.cm−2 | The clot strength: the amount of total resistance (to movement of the cup and pin) generated during clot formation. This is the total area under the velocity curve during clot growth, representing the amount of clot strength generated during clot growth. | |
| Lysis at 30 min (LY30) measured in % | The LY30 parameter (measured in %) is recorded at 30 min, measured from the point where the maximum amplitude (MA) of the clot is reached. | |
| G value measured in Dyn.sec | G-value is a log-derivation of the MA and is meant to also represent the clot strength Elevated G-value is associated with a hypercoagulable state and therefore increases the risk for venous thromboembolic disease. | |
| (B) PFA-200 Platelet Test Interpretation: | ||
| Citrated whole blood is aspirated at high shear rates through disposable cartridges. These cartridges contain an aperture within a membrane coated agonist. The agonist cartridges are Col/EPI, Col/ADP and P2Y and they report data in closure time. The PFA-200 test induces platelet adhesion, activation and aggregation using the three cartridges. Closure times increase progressively as the platelet counts falls below 100 × 109/L. | ||
| Agonist cartridges [66] | Test principle | Closure time interpretations: measured in seconds |
| Collagen and epinephrine (Col/EPI): This cartridge has a collagen (2 μg equine type I) and epinephrine (10 μg)-coated membrane (C/Epi). | Co-stimulation by shear stress, collagen, epinephrine. Gives an indication of effectiveness of aspirin and GP IIβ/IIIα inhibitor dosage. | Col/EPI closure time is 82–150 s with a value >150 s regarded as prolonged. |
| Collagen and ADP (Col/ADP): This cartridge has a collagen (2 μg equine type I) and adenosine-diphosphate (50 μg)-coated membrane (Col/ADP). | Co-stimulation by shear stress, collagen, ADP. Gives an indication of effectiveness of aspirin and clopidogrel and GP IIβ/IIIα inhibitor dosage. | Col/ADP closure time is 62–100 s with a value >100 s regarded as prolonged. |
| P2Y: This cartridge has a prostaglandin E1 (5 ng) and ADP (20 μg)-coated membrane. | Co-stimulation by shear stress, ADP, PGE1 and Ca2+. Gives an indication of effectiveness of clopidogrel and GP IIβ/IIIα inhibitor dosage | Shortened PFA P2Y closure times >106 s are viewed as prolonged. |
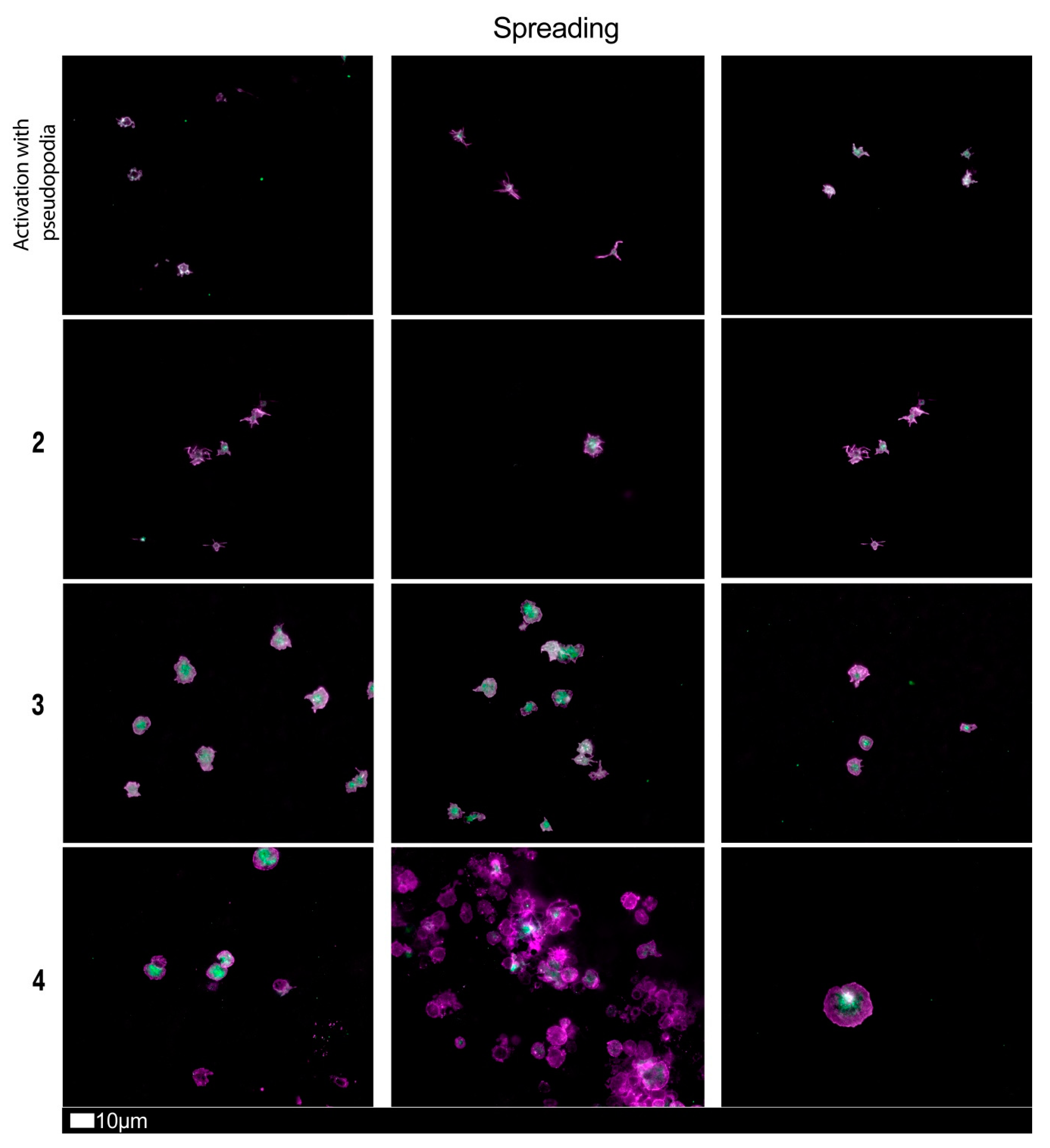
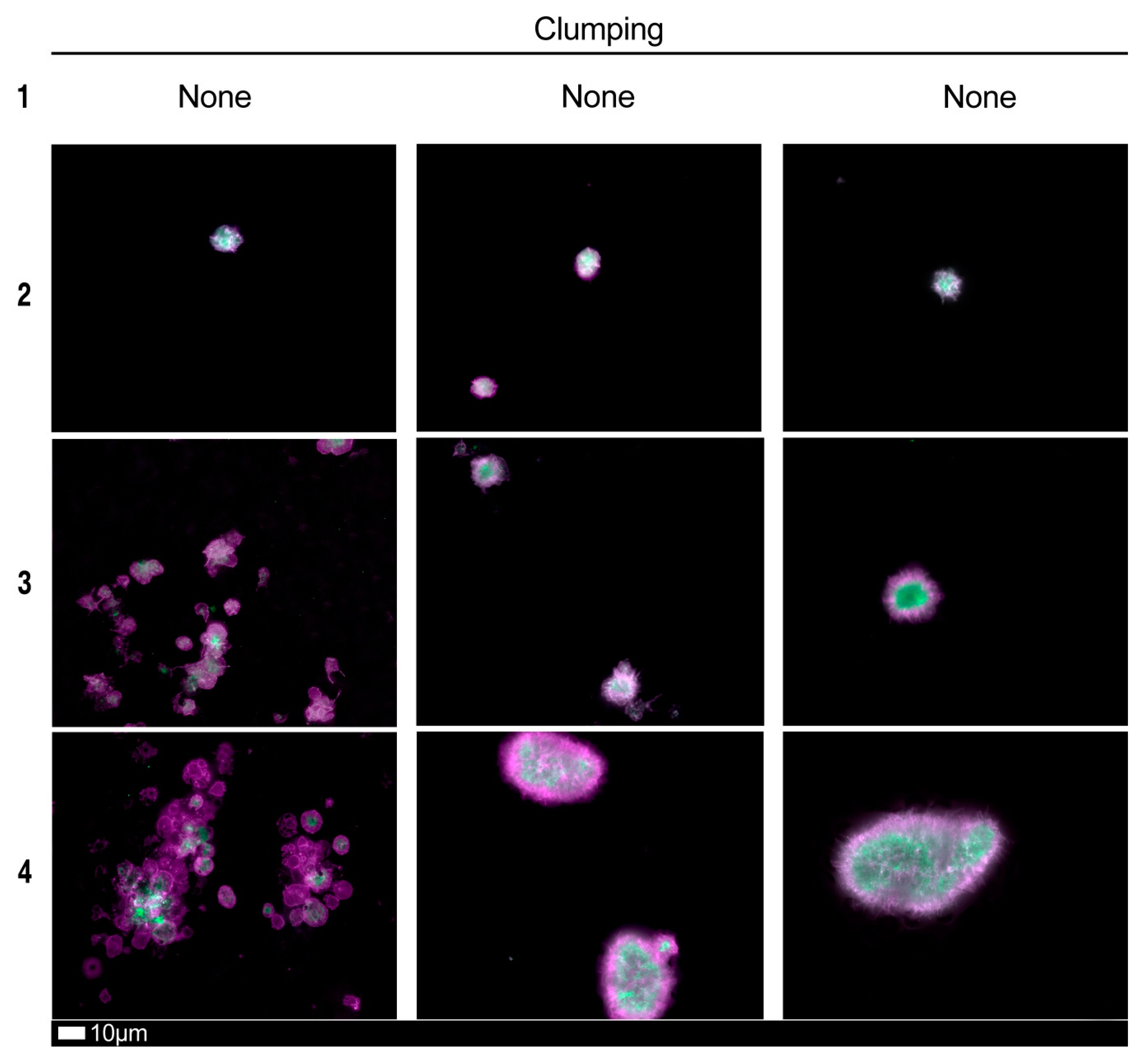
| Score | Spreading | Score | Clumping |
|---|---|---|---|
| 1 | Activation with pseudopodia | 1 | None |
| 2 | Mild | 2 | Mild |
| 3 | Moderate | 3 | Moderate |
| 4 | Severe | 4 | Severe |
| Score | Presence of Microclots in Platelet Poor Plasma |
|---|---|
| 1 | Very few areas of plasma protein misfolding (≤1 µm) visible with a few ≤10 µm microclots |
| 2 | Very few areas of plasma protein misfolding (≤1 µm) visible with scattered/mild ≤10 µm microclots |
| 3 | Moderate areas of plasma protein misfolding visible as microclots ≥15 µm |
| 4 | Severe areas of plasma protein misfolding visible as large microclots |
| Control/Healthy | Mild | Moderate | Severe |
|---|---|---|---|
| =3 | 4–7 | 8–10 | 11–12 |
| Platelets + PPP scores = | |||
| Prognostic Indicator | |||
|---|---|---|---|
| Points assigned | 0 | 1 | 2 |
| Age (years) | ≤44 | 45–64 | ≥65 |
| Effort intolerance above baseline | No | Yes | |
| Hypoxemia, O2 saturation | >95 | 92–95 | <92 |
| Chest X-ray/CT scan | Normal | ≤1 quad | ≥1 quad |
| Obesity (BMI) | <26 | 26–36 | >36 |
| Co-morbidities: 1 point for each comorbid condition | Type 2 Diabetes Mellitus, coronary artery disease (CAD), non-atrial fibrillation (AF) stroke, smoking, deep vein thrombosis (DVT), hyperparathyroidism (HPT), chronic kidney disease (CKD) | ||
| TEG: MA | <69 | 69–75 | >75 |
| TEG: G-score | <10 | 10–15 | >15 |
| TEG: Ly-30 | >0 | None (1 point) | |
| Score | |||
| Low Risk | Moderate Risk | High Risk | |
| 0–3 | 4–10 | ≥11 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laubscher, G.J.; Lourens, P.J.; Venter, C.; Kell, D.B.; Pretorius, E. TEG®, Microclot and Platelet Mapping for Guiding Early Management of Severe COVID-19 Coagulopathy. J. Clin. Med. 2021, 10, 5381. https://doi.org/10.3390/jcm10225381
Laubscher GJ, Lourens PJ, Venter C, Kell DB, Pretorius E. TEG®, Microclot and Platelet Mapping for Guiding Early Management of Severe COVID-19 Coagulopathy. Journal of Clinical Medicine. 2021; 10(22):5381. https://doi.org/10.3390/jcm10225381
Chicago/Turabian StyleLaubscher, Gert Jacobus, Petrus Johannes Lourens, Chantelle Venter, Douglas B Kell, and Etheresia Pretorius. 2021. "TEG®, Microclot and Platelet Mapping for Guiding Early Management of Severe COVID-19 Coagulopathy" Journal of Clinical Medicine 10, no. 22: 5381. https://doi.org/10.3390/jcm10225381
APA StyleLaubscher, G. J., Lourens, P. J., Venter, C., Kell, D. B., & Pretorius, E. (2021). TEG®, Microclot and Platelet Mapping for Guiding Early Management of Severe COVID-19 Coagulopathy. Journal of Clinical Medicine, 10(22), 5381. https://doi.org/10.3390/jcm10225381