

The Design, Synthesis, and Evaluation of Diaminopimelic Acid Derivatives as Potential dapF Inhibitors Preventing Lysine Biosynthesis for Antibacterial Activity
 , , , , , ,
, , , , , ,  , , ,
, , ,  and
and
Abstract

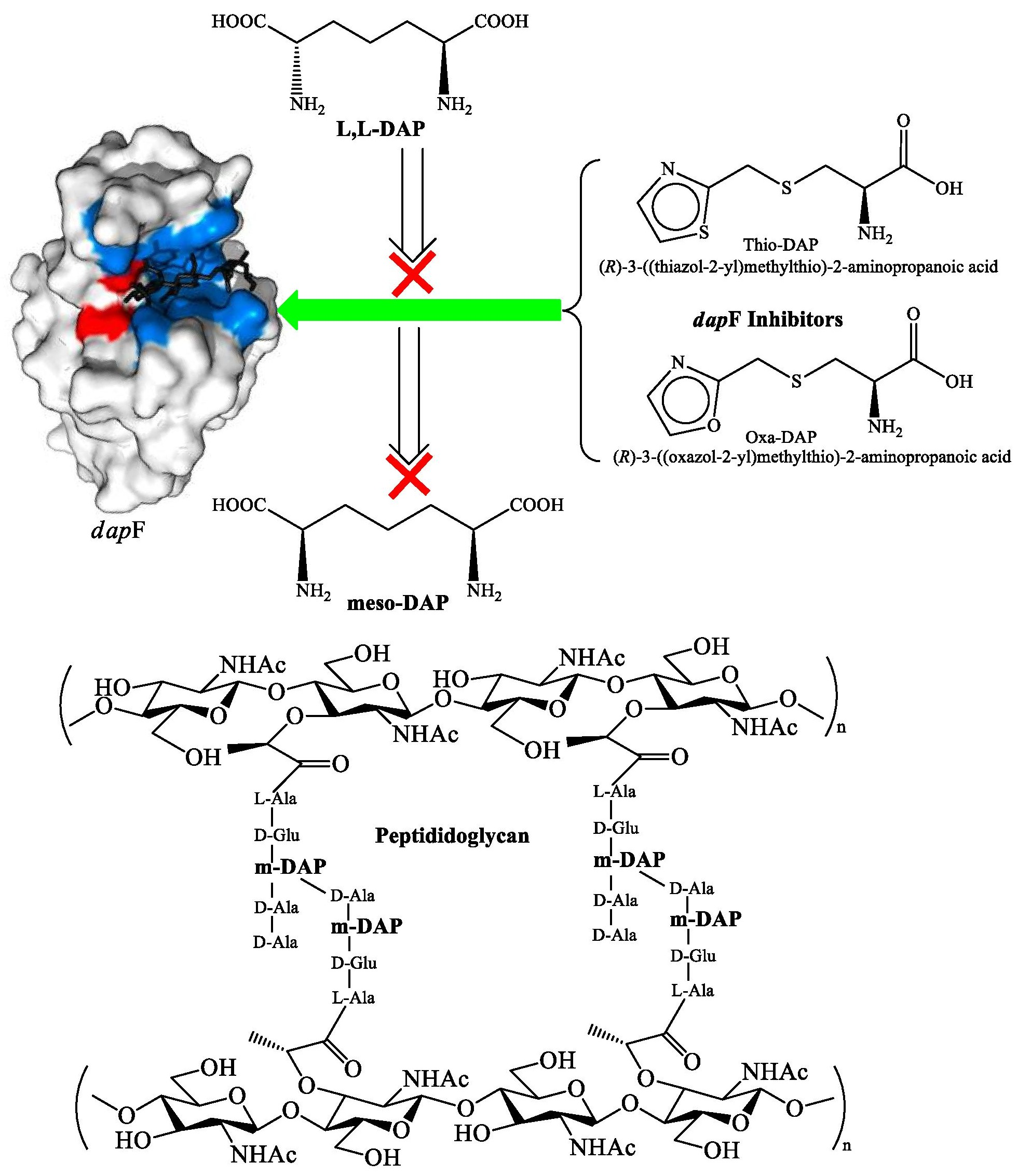
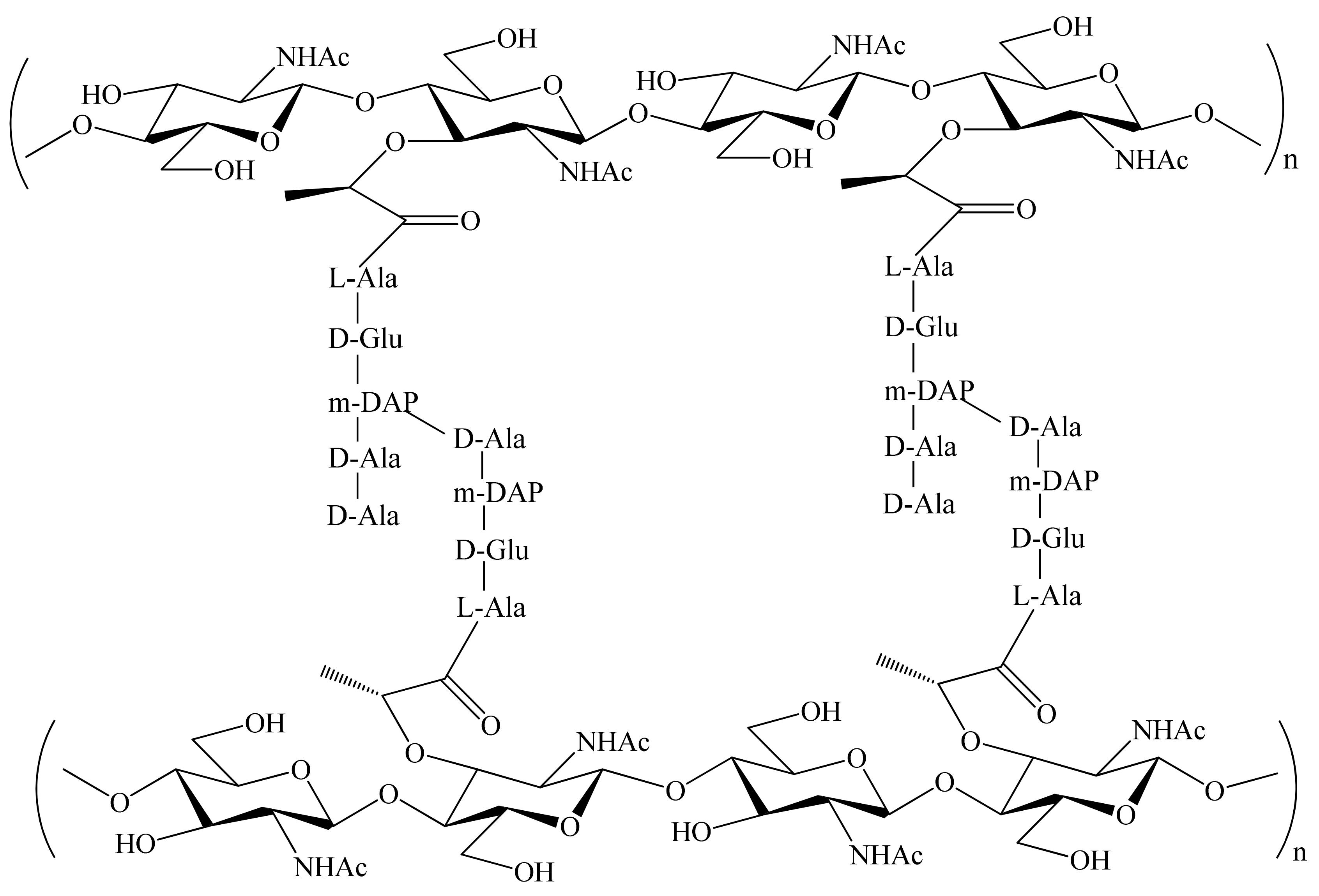
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. In Silico Toxicity Studies
3.2. ADME Parameter’s Estimation
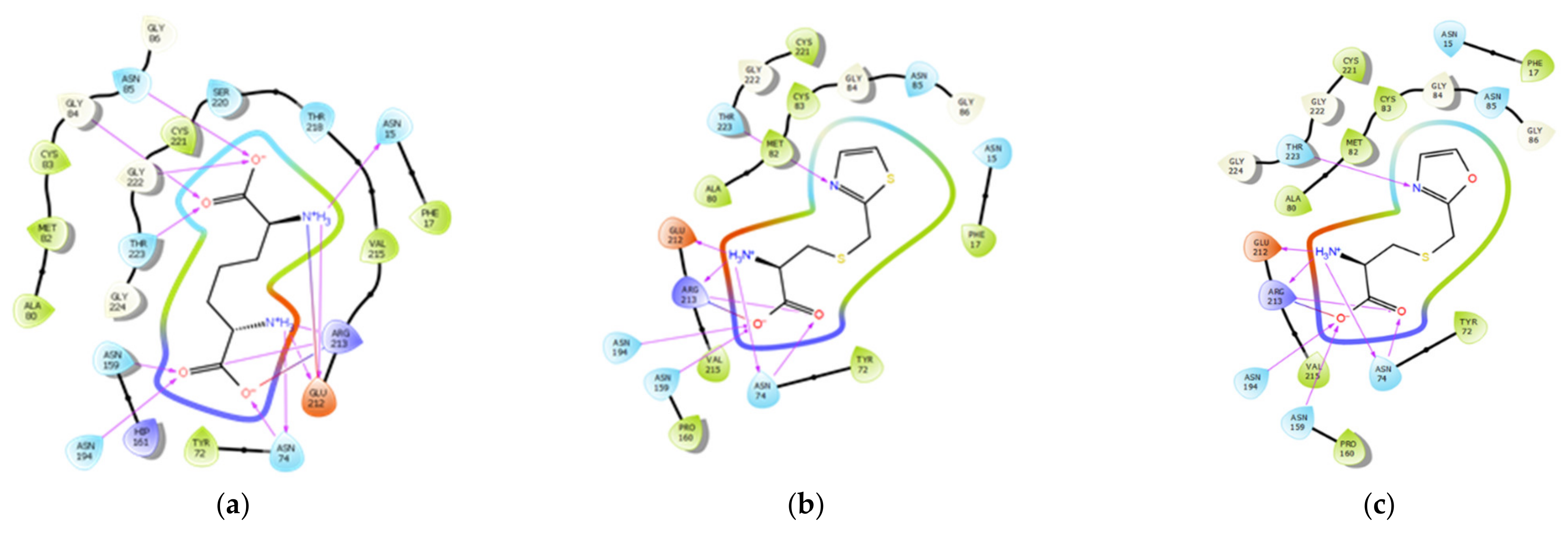
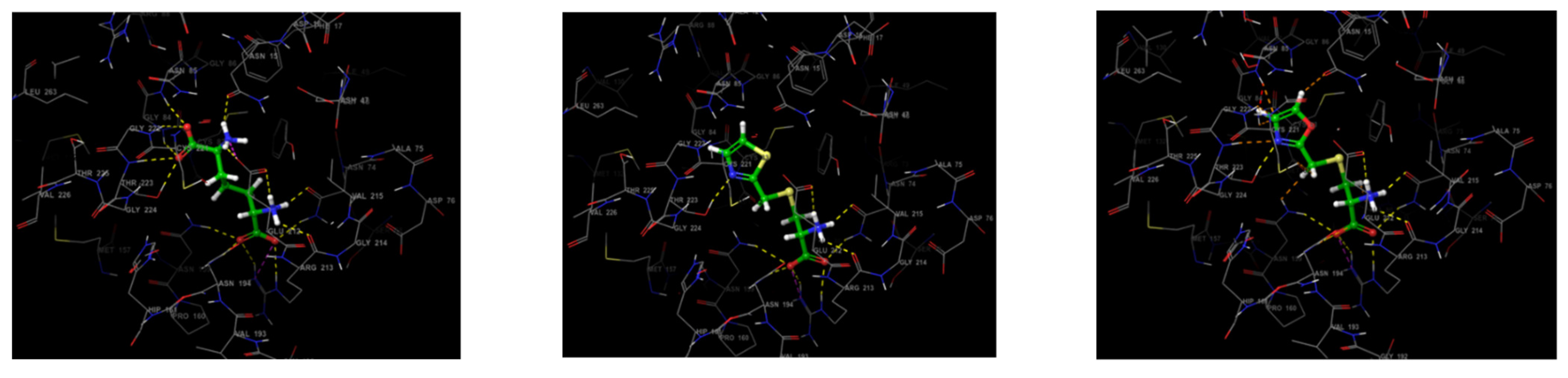
3.3. Molecular Docking Study
- (a)
- Protein structure designing
- (b) Ligand structure design
- (c) Docking with thio-DAP
- (d) Docking with oxa-DAP
- (e) Docking with LL-DAP
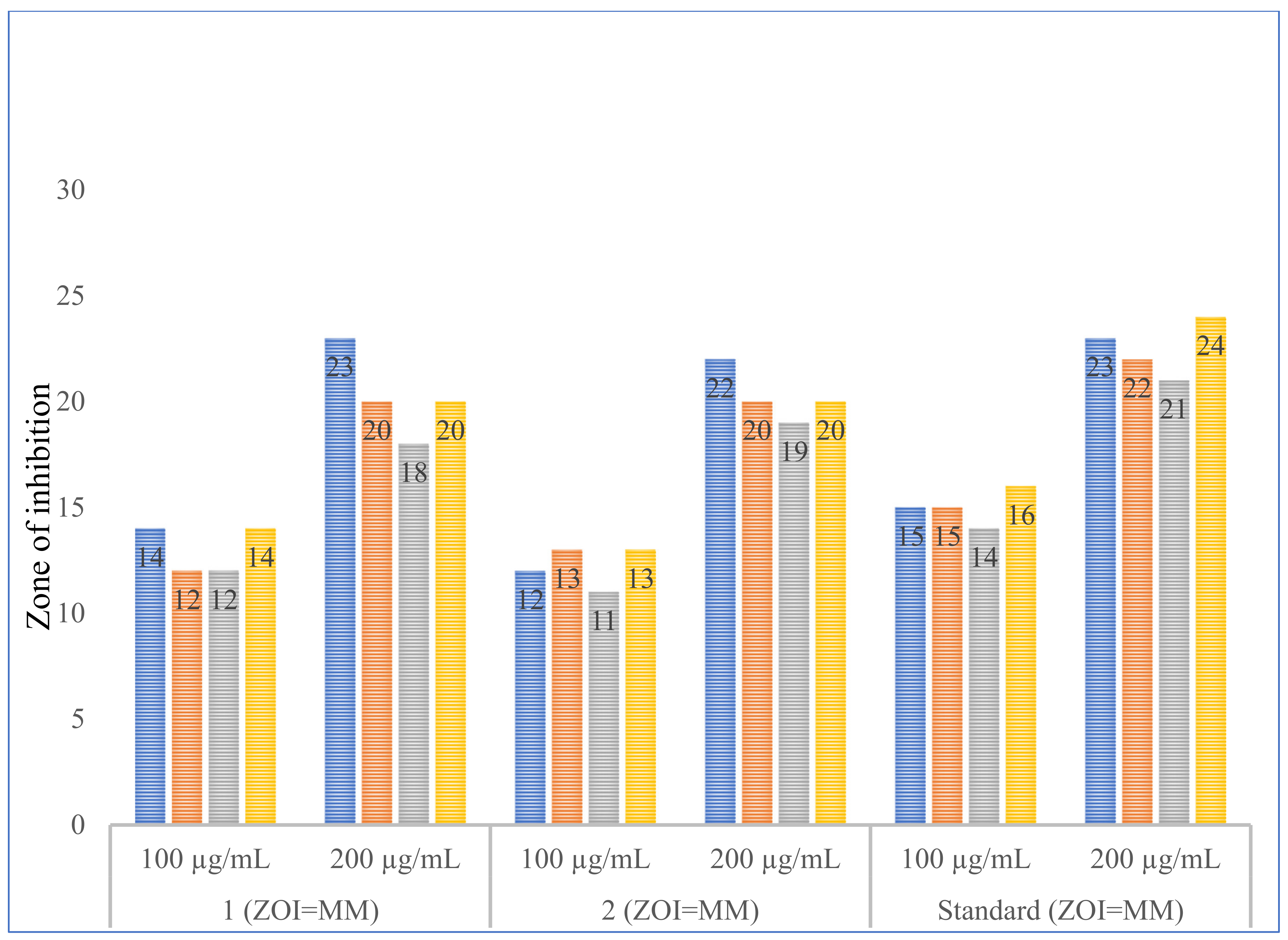
3.4. Antimicrobial Studies
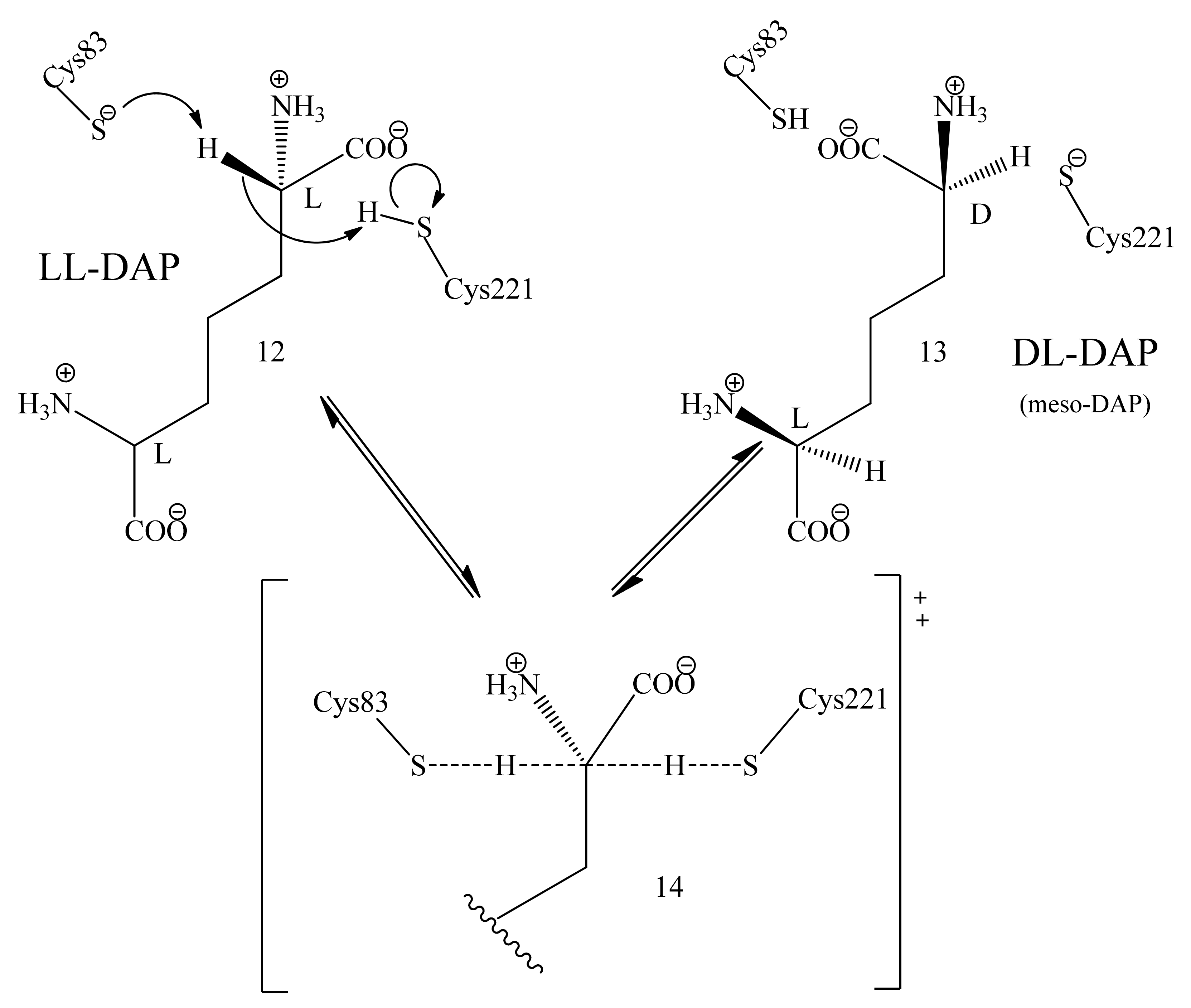
3.5. Mechanism of Epimerization by dapF
3.6. SAR for Designing the DAP Analogues
4. Conclusions
5. Chemistry
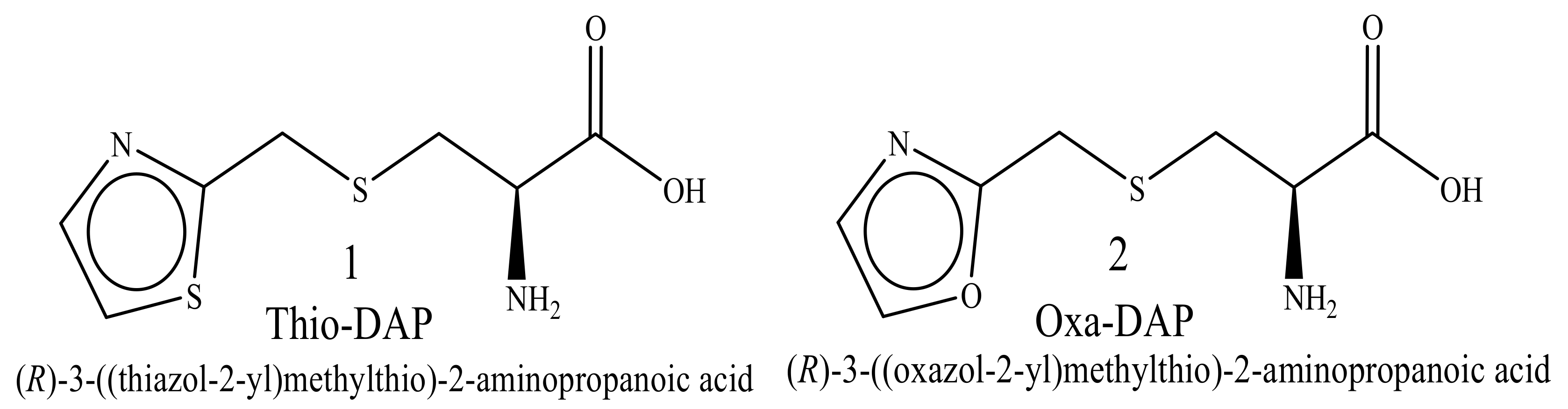
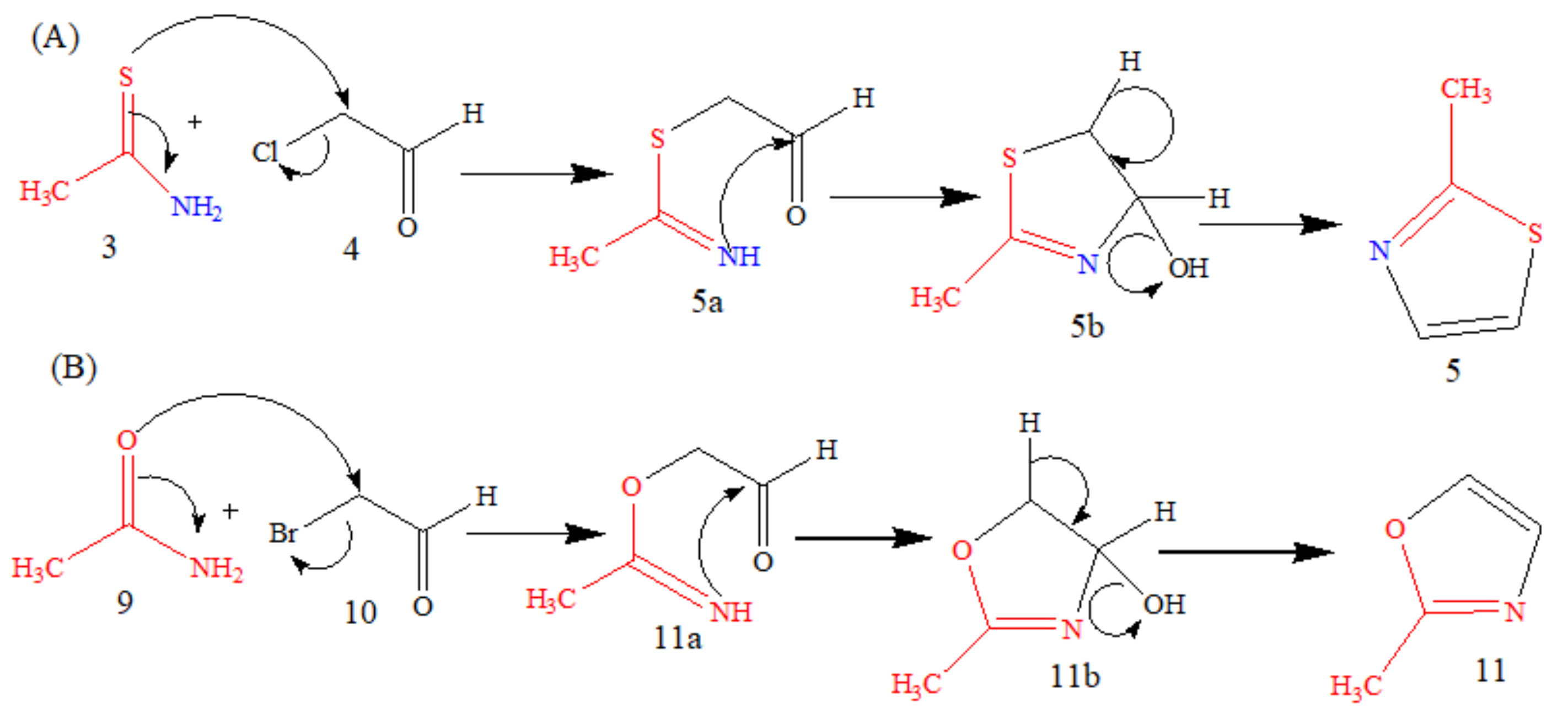
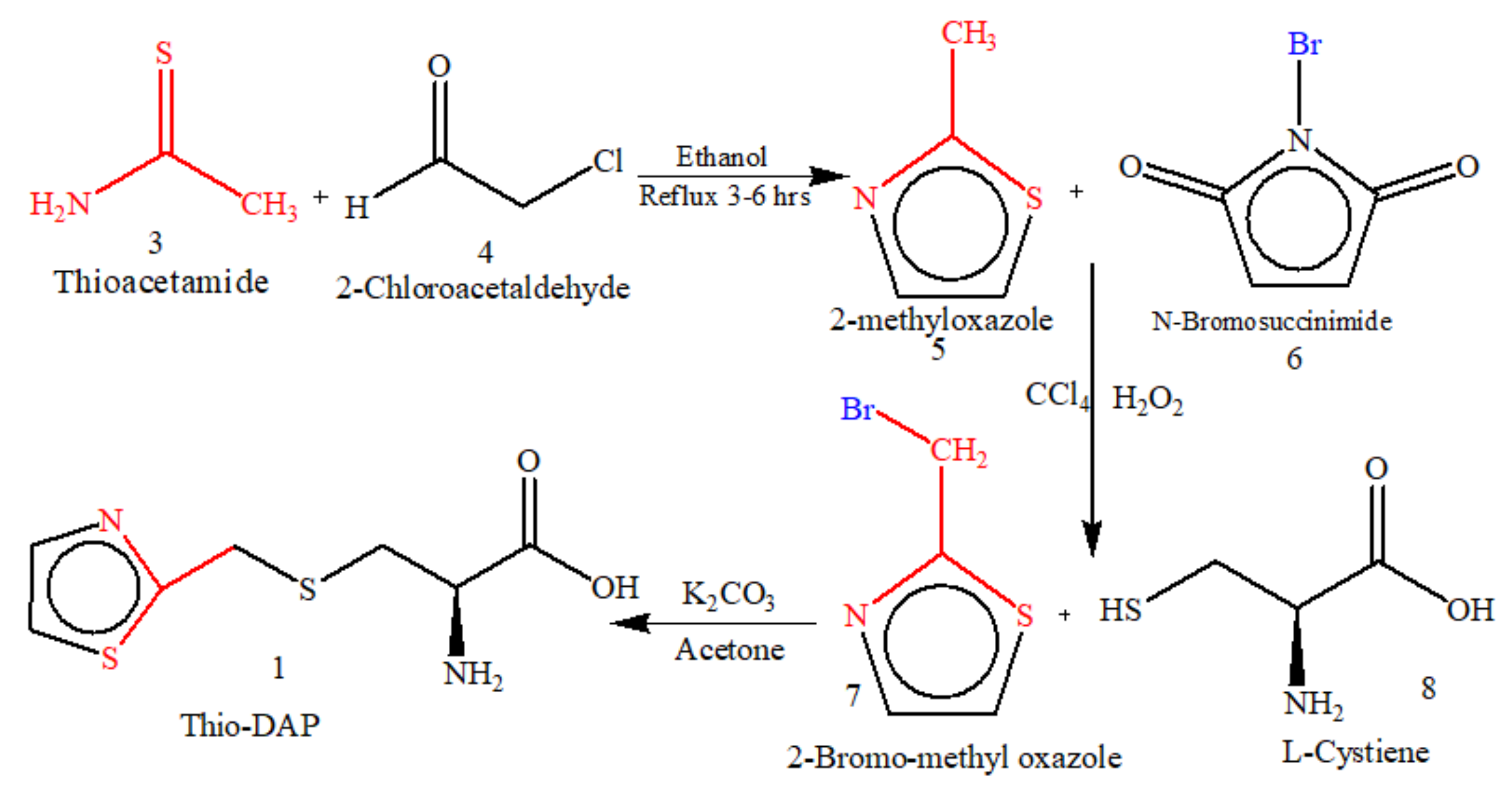
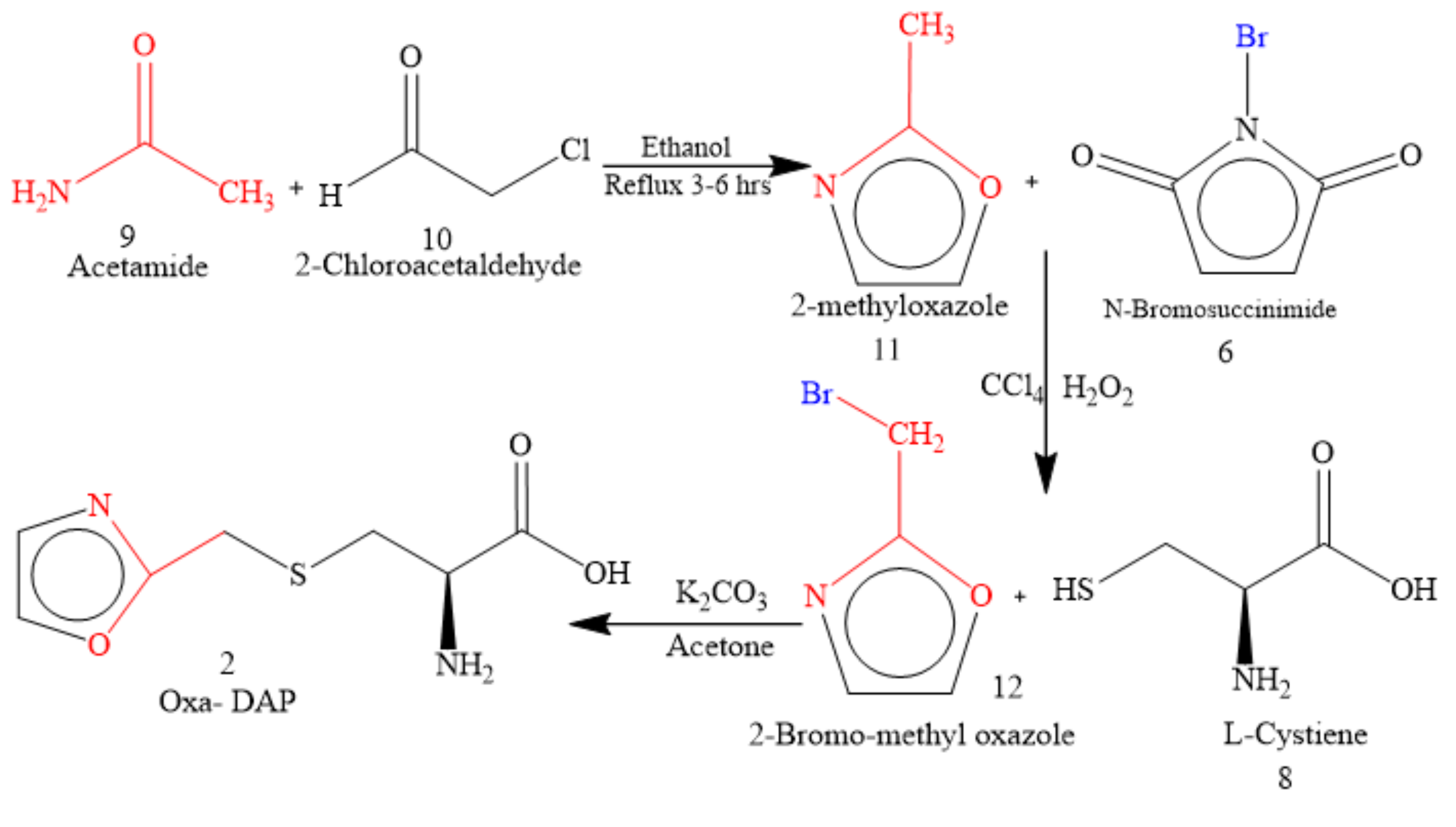
5.1. Microwave-Assisted Synthesis of Thio-DAP (01) and Oxa-DAP (02)
5.1.1. Thio-DAP (01): (R)-3-((thiazol-2-yl)methylthio)-2-aminopropanoic Acid
5.1.2. Oxa-DAP (02): (R)-3-((oxazol-2-yl)methylthio)-2-aminopropanoic Acid
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kale, M.; Shaikh, M.S. Exploration of Lysine Pathway for Developing Newer Anti-Microbial Analogs through Enzyme Inhibition Approach. Int. J. Pharm. Sci. Rev. Res. 2014, 25, 221–230. [Google Scholar]
- Hutton, C.A.; Perugini, M.A.; Gerrard, J.A. Inhibition of Lysine Biosynthesis: An Evolving Antibiotic Strategy. Mol. Biosyst. 2007, 3, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Swelum, A.A.; Elbestawy, A.R.; El-Saadony, M.T.; Hussein, E.O.S.; Alhotan, R.; Suliman, G.M.; Taha, A.E.; Ba-Awadh, H.; El-Tarabily, K.A.; Abd El-Hack, M.E. Ways to Minimize Bacterial Infections, with Special Reference to Escherichia Coli, to Cope with the First-Week Mortality in Chicks: An Updated Overview. Poult. Sci. 2021, 100, 101039. [Google Scholar] [CrossRef] [PubMed]
- Donovan, C.; Blewitt, J. Signs, Symptoms and Management of Bacterial Meningitis. Paediatr. Nurs. 2010, 22, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; Misba, L.; Khan, A.U. Nano-Therapeutics: A Revolution in Infection Control in Post Antibiotic Era. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 2281–2301. [Google Scholar] [CrossRef] [PubMed]
- Kale, M.; Shaikh, M.S. Drug Discovery of Newer Analogs of Anti-Microbials through Enzyme-Inhibition: A Review. Int. J. Pharm. Pharm. Sci. 2014, 6, 27–35. [Google Scholar]
- Shaikh, M.S.; Kale, M.A.; Sharuk, K. Discovery of Heterocyclic Analogs of Diaminopimelic Acid as Promising Antibacterial Agents Through Enzyme Targeted Inhibition of Lysine Biosynthesis. Graph. Abstr. Curr. Enzym. Inhib. 2018, 14, 120–130. [Google Scholar] [CrossRef]
- Shaikh, M.S.; Kale, M.A.; Mahaparle, P.R.; Rajput, H.; Karkhele, S.M. Development and Validation of UV Spectrophotometric Method for the Estimation of Luliconazole in Bulk, Marketed Formulations. Curr. Pharma Res. 2020, 10, 3759–3770. [Google Scholar]
- Baltora, E.; Soubieux-Bourbon, A.; Kamami, V.; Tiret, I.; Varin, R. État Des Lieux Des Prescriptions de Daptomycine Au Sein d’un Établissement de Santé Hospitalo-Universitaire. Infect. Dis. Now 2021, 51, S48. [Google Scholar] [CrossRef]
- Durand, A.; Cayeux, S.; Adehossi, A.; Richecoeur, J.; Schmit, B. Troubles Neurologiques Sous Daptomycine En Réanimation: Effet Indésirable Ou Mésusage? Cas Clinique. Le Pharm. Hosp. Clin. 2021, 56, 87–90. [Google Scholar] [CrossRef]
- Poissy, J.; Senneville, E. New Antibiotics for Severe ICU-Aquired Bacterial Infections. Infect. Disord.-Drug Targets 2012, 11, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Crank, C.W.; Scheetz, M.H.; Brielmaier, B.; Rose, W.E.; Patel, G.P.; Ritchie, D.J.; Segreti, J. Comparison of Outcomes from Daptomycin or Linezolid Treatment for Vancomycin-Resistant Enterococcal Bloodstream Infection: A Retrospective, Multicenter, Cohort Study. Clin. Ther. 2010, 32, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Whang, D.W.; Miller, L.G.; Partain, N.M.; McKinnell, J.A. Systematic Review and Meta-Analysis of Linezolid and Daptomycin for Treatment of Vancomycin-Resistant Enterococcal Bloodstream Infections. Antimicrob. Agents Chemother. 2013, 57, 5013–5018. [Google Scholar] [CrossRef]
- Diaper, C.M.; Sutherland, A.; Pillai, B.; James, M.N.G.; Semchuk, P.; Blanchard, J.S.; Vederas, J.C. The Stereoselective Synthesis of Aziridine Analogues of Diaminopimelic Acid (DAP) and Their Interaction with Dap Epimerase. Org. Biomol. Chem. 2005, 3, 4402–4411. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.J. The DAP Pathway to Lysine as a Target for Antimicrobial Agents. Nat. Prod. Rep. 1996, 13, 29–43. [Google Scholar] [CrossRef]
- Song, Y.; Niederer, D.; Lane-Bell, P.M.; Lam, L.K.; Crawley, S.; Palcic, M.M.; Pickard, A.A.; Pruess, D.L.; Vederas, J.C. Stereospecific Synthesis of Phosphonate Analogues of Diaminopimelic Acid (DAP), Their Interaction with DAP Enzymes, and Antibacterial Activity of Peptide Derivatives. J. Org. Chem. 1994, 59, 5784–5793. [Google Scholar] [CrossRef]
- Abbott, S.D.; Lane-Bell, P.; Sidhu, K.P.S.; Vederas, J.C. Synthesis and Testing of Heterocyclic Analogues of Diaminopimelic Acid (DAP) as Inhibitors of DAP Dehydrogenase and DAP Epimerase. J. Am. Chem. Soc. 1994, 116, 6513–6520. [Google Scholar] [CrossRef]
- Kesicki, E.A.; Bailey, M.A.; Ovechkina, Y.; Early, J.V.; Alling, T.; Bowman, J.; Zuniga, E.S.; Dalai, S.; Kumar, N.; Masquelin, T.; et al. Synthesis and Evaluation of the 2-Aminothiazoles as Anti-Tubercular Agents. PLoS ONE 2016, 11, e0155209. [Google Scholar] [CrossRef]
- Kumar, S.V.; Parameshwarappa, G.; Ila, H. Synthesis of 2,4,5-Trisubstituted Thiazoles via Lawesson’s Reagent-Mediated Chemoselective Thionation-Cyclization of Functionalized Enamides. J. Org. Chem. 2013, 78, 7362–7369. [Google Scholar] [CrossRef]
- Shaw, A.Y.; Xu, Z.; Hulme, C. Ugi/Robinson-Gabriel Reactions Directed toward the Synthesis of 2,4,5-Trisubstituted Oxazoles. Tetrahedron Lett. 2012, 53, 1998–2000. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, W.; Hu, M.; Zou, H.; Yu, Y. Synthesis of Polysubstituted 5-Aminooxazoles from α- Diazocarbonyl Esters and α-Isocyanoacetamides. Org. Lett. 2010, 12, 616–618. [Google Scholar] [CrossRef] [PubMed]
- Lodh, R.S.; Borah, A.J.; Phukan, P. Synthesis of Bromohydrins Using NBS in Presence of Iodine as Catalyst. Indian J. Chem.-Sect. B Org. Med. Chem. 2014, 53B, 1425–1429. [Google Scholar]
- Das, B.; Venkateswarlu, K.; Holla, H.; Krishnaiah, M. Sulfonic Acid Functionalized Silica: A Remarkably Efficient Heterogeneous Reusable Catalyst for α-Monobromination of Carbonyl Compounds Using N-Bromosuccinimide. J. Mol. Catal. A Chem. 2006, 253, 107–111. [Google Scholar] [CrossRef]
- Tanemura, K.; Nishida, Y.; Satsumabayashi, K.; Horaguchi, T. A Mild and Efficient Procedure for α-Bromination of Ketones Using N-Bromosuccinimide Catalysed by Ammonium Acetate. Chem. Commun. 2004, 4, 470–471. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Togo, H. Facile Oxidative Conversion of Alcohols to Esters Using Molecular Iodine. Tetrahedron 2005, 61, 5915–5925. [Google Scholar] [CrossRef]
- Gogoi, P.; Konwar, D. Transition-Metal- and Organic-Solvent-Free: A Highly Efficient Anaerobic Process for Selective Oxidation of Alcohols to Aldehydes and Ketones in Water. Org. Biomol. Chem. 2005, 3, 3473–3475. [Google Scholar] [CrossRef]
- Paiva, A.M.; Vanderwall, D.E.; Blanchard, J.S.; Kozarich, J.W.; Williamson, J.M.; Kelly, T.M. Inhibitors of Dihydrodipicolinate Reductase, a Key Enzyme of the Diaminopimelate Pathway of Mycobacterium Tuberculosis. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 2001, 1545, 67–77. [Google Scholar] [CrossRef]
- Graham, T.H. A Direct Synthesis of Oxazoles from Aldehydes. Org. Lett. 2010, 12, 3614–3617. [Google Scholar] [CrossRef]
- Mukherjee, A.; Singh, B. Binding Interaction of Pharmaceutical Drug Captopril with Calf Thymus DNA: A Multispectroscopic and Molecular Docking Study. J. Lumin. 2017, 190, 319–327. [Google Scholar] [CrossRef]
- Song, Y.; Niederer, D.; Lane-BELL, P.M.; Lam, L.K.P.; Crawley, S.; Palcic, M.M.; Pickard, M.A.; Pruess, D.L.; Vederas, J.C. ChemInform Abstract: Stereospecific Synthesis of Phosphonate Analogues of Diaminopimelic Acid (DAP), Their Interaction with DAP Enzymes, and Antibacterial Activity of Peptide Derivatives. ChemInform 2010, 26, 5784–5793. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Ghosh, S.; Nath Talapatra, S. Bioavailability Prediction of Phytochemicals Present in Calotropis Procera (Aiton) R. Br. by Using Swiss-ADME Tool. World Sci. News 2019, 131, 147–163. [Google Scholar]
- Shntaif, A.H.; Khan, S.; Tapadiya, G.; Chettupalli, A.; Saboo, S.; Shaikh, M.S.; Siddiqui, F.; Amara, R.R. Rational Drug Design, Synthesis, and Biological Evaluation of Novel N-(2-Arylaminophenyl)-2,3-Diphenylquinoxaline-6-Sulfonamides as Potential Antimalarial, Antifungal, and Antibacterial Agents. Digit. Chin. Med. 2021, 4, 290–304. [Google Scholar] [CrossRef]
- Khan, S.; Kale, M.; Siddiqui, F.; Nema, N. Novel Pyrimidine-Benzimidazole Hybrids with Antibacterial and Antifungal Properties and Potential Inhibition of SARS-CoV-2 Main Protease and Spike Glycoprotein. Digit. Chin. Med. 2021, 4, 102–119. [Google Scholar] [CrossRef]
- Khan, A.; Unnisa, A.; Sohel, M.; Date, M.; Panpaliya, N.; Saboo, S.G.; Siddiqui, F.; Khan, S. Investigation of Phytoconstituents of Enicostemma Littorale as Potential Glucokinase Activators through Molecular Docking for the Treatment of Type 2 Diabetes Mellitus. Silico Pharmacol. 2022, 10, 1. [Google Scholar] [CrossRef]
- Siddiqui, F.A.; Khan, S.L.; Marathe, R.P.; Nema, N.V. Design, Synthesis, and In Silico Studies of Novel N-(2-Aminophenyl)-2,3- Diphenylquinoxaline-6-Sulfonamide Derivatives Targeting Receptor- Binding Domain (RBD) of SARS-CoV-2 Spike Glycoprotein and Their Evaluation as Antimicrobial and Antimalarial Agents. Lett. Drug Des. Discov. 2021, 18, 915–931. [Google Scholar] [CrossRef]
- Shaikh, M.S.; Kale, M.A. Formulation and Molecular Docking Simulation Study of Luliconazole Nanosuspension e Based Nanogel for Transdermal Drug Delivery Using Modi Fi Ed Polymer. Mater. Today Chem. 2020, 18, 100364. [Google Scholar] [CrossRef]
- Shaikh, M.S.; Kale, M.A.; Shaikh, M.M.; Mahaparale, P.R. Formulation, Characterization and Antimicrobial Studies of Lyophilized Luliconazole Nanosuspension for Enhancing Solubility Using Modified Polymer. Int. J. Polym. Mater. Polym. Biomater. 2022, 71, 692–706. [Google Scholar] [CrossRef]
- Khan, S.L.; Siddiui, F.A. Beta-Sitosterol: As Immunostimulant, Antioxidant and Inhibitor of SARS-CoV-2 Spike Glycoprotein. Arch. Pharmacol. Ther. 2020, 2, 12–16. [Google Scholar] [CrossRef]
- Chaudhari, R.N.; Khan, S.L.; Chaudhary, R.S.; Jain, S.P.; Siddiqui, F.A. Β-Sitosterol: Isolation from Muntingia Calabura Linn Bark Extract, Structural Elucidation And Molecular Docking Studies As Potential Inhibitor of SARS-CoV-2 Mpro (COVID-19). Asian J. Pharm. Clin. Res. 2020, 13, 204–209. [Google Scholar] [CrossRef]
- Van Assche, I.; Soroka, M.; Haemers, A.; Hooper, M.; Blanot, D.; van Heijenoort, J. Synthesis and Antibacterial Evaluation of Phosphonic Acid Analogues of Diaminopimelic Acid. Eur. J. Med. Chem. 1991, 26, 505–515. [Google Scholar] [CrossRef]
- Van Assche, I.; Soroka, M.; Haemers, A.; Hooper, M.; Blanot, D.; van Heijenoort, J. Cheminform Abstract: Synthesis and Antibacterial Evaluation of Phosphonic Acid Analogues of Diaminopimelic Acid. ChemInform 2010, 22. [Google Scholar] [CrossRef]
- Khan, S.L.; Siddiqui, F.A.; Shaikh, M.S.; Nema, N.V.; Shaikh, A.A. Discovery of Potential Inhibitors of the Receptor-Binding Domain (RBD) of Pandemic Disease-Causing SARS-CoV-2 Spike Glycoprotein from Triphala through Molecular Docking. Curr. Chinese Chem. 2022, 2, 1. [Google Scholar] [CrossRef]
- Chatterjee, B.; Mondal, D.; Bera, S. Diaminopimelic Acid and Its Analogues: Synthesis and Biological Perspective. Tetrahedron 2021, 100. [Google Scholar] [CrossRef]
- Dzierzbicka, K. Synthesis of 2,6-Diaminopimelic Acid (DAP) and Its Analogues. Pol. J. Chem. 2007, 81, 455–473. [Google Scholar] [CrossRef]
- Liu, H.; Pattabiraman, V.R.; Vederas, J.C. Stereoselective Syntheses of 4-Oxa Diaminopimelic Acid and Its Protected Derivatives via Aziridine Ring Opening. Org. Lett. 2007, 9, 4211–4214. [Google Scholar] [CrossRef]
- Brunetti, L.; Galeazzi, R.; Orena, M.; Bottoni, A. Catalytic Mechanism of l,l-Diaminopimelic Acid with Diaminopimelate Epimerase by Molecular Docking Simulations. J. Mol. Graph. Model. 2008, 26, 1082–1090. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Predicted LD50 Value (mg/Kg) | Hepatotoxicity | Carcinogenicity | Immunotoxicity | Cytotoxicity | Nuclear Receptor Signaling Pathways Active | ||
|---|---|---|---|---|---|---|---|---|
| Androgen Receptor (AR) | Aromatase Active | Estrogen Receptor Alpha (ER) | ||||||
| LL-DAP | 366 | None | None | None | None | None | None | None |
| Thio-DAP | 1630 | None | None | None | None | None | None | None |
| Oxa-DAP | 4000 | None | None | None | None | None | None | None |
| Compound | Pharmacokinetics | Drug-Likeness | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GI * Absorption | BBB * Permeability | P-gp * Substrate | CYP1A2 | CYP219 | CYP2C9 | CYP2D6 | CYP3A4 | Log Kp (Skin Permeation | Ghose | Egan | Muegge | Bioavailability | |
| Inhibitors | |||||||||||||
| LL-DAP | High | None | None | None | None | None | None | None | −11.64 cm/s | No | Yes | No | 0.55 |
| Oxa-DAP | High | None | None | None | None | None | None | None | −9.50 cm/s | Yes | Yes | No | 0.55 |
| Thio-DAP | High | None | None | None | None | None | None | None | −9.17 cm/s | Yes | Yes | No | 0.55 |
| Compound | Molecular Formula | Lipinski Rule of Five | Veber’s Rule | |||||
|---|---|---|---|---|---|---|---|---|
| Molecular Weight | HBA * | HBD * | Log P | Violation | Total Polar Surface Area (TPSA) (A2) | Number of Rotatable Bonds | ||
| LL-DAP | C7H14N2O4 | 190.20 | 6 | 4 | −2.73 | 00 | 126.64 | 6 |
| Oxa-DAP | C7H10N2O3S | 202.23 | 5 | 2 | −0.99 | 00 | 114.65 | 5 |
| Thio-DAP | C7H10N2O2S2 | 218.30 | 4 | 2 | −0.33 | 00 | 129.75 | 5 |
| Structure | Docking Score/Binding Free Energy | Protein-Ligand Interactions | Glide H-Bond | ||
|---|---|---|---|---|---|
| H-Bond Interactions | H-Bond Distance (A˚) | Hydrophobic Interactions | |||
| LL-DAP | −9.426 | Asn15 | 2.14 | Phe17, Tyr72, Cys83, Met82, Ala80, Val215, Val215a, and Cys221. | −1.455 |
| Asn74 | 1.34 | ||||
| Gly84 | 3.31 | ||||
| Asn85 | 3.14 | ||||
| Asn159 | 1.21 | ||||
| Asn194 | 2.75 | ||||
| Glu212 | 1.42, 4.22 | ||||
| Arg213 | 1.13, 4.53 | ||||
| Gly222 | 2.23 | ||||
| Thr223 | 1.41 | ||||
| T-DAP (1) | −9.823 | Asn74 | 1.82, 3.47 | Phe17, Tyr72, Ala80, Met82, Cys83, Pro160, Val215, and Cys221. | −1.532 |
| Asn159 | 2.07 | ||||
| Asn194 | 2.73 | ||||
| Glu212 | 0.81 | ||||
| Arg213 | 0.98, 3.24 | ||||
| Thr223 | 2.45 | ||||
| O-DAP (2) | −10.098 | Asn74 | 1.05, 2.83 | Phe17, Tyr72, Ala80, Met82, Cys83, Pro160, Val215, and Cys221. | −1.757 |
| Asn159 | 2.09 | ||||
| Asn194 | 1.88 | ||||
| Glu212 | 0.78 | ||||
| Arg213 | 0.83, 2.26 | ||||
| Thr223 | 2.42 | ||||
| Microorganism | 1 | 2 | Standard | 1 (ZOI = MM) | 2 (ZOI = MM) | Standard (ZOI = MM) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC | MBC | MIC | MBC | MIC | MBC | 100 µg/mL | 200 µg/mL | 100 µg/mL | 200 µg/mL | 100 µg/mL | 200 µg/mL | |
| B. subtilis | 70 ± 0 | 80 ± 0 | 80 ± 0 | 90 ± 0 | 70 ± 0 | 80 + 0 | 14 ± 02 | 22.67 ± 0.58 | 11.67 ± 1.53 | 21.67 ± 0.58 | 15.67 ± 1.15 | 23.67 ± 0.58 |
| B. megatorium | 70 ± 0 | 90 ± 0 | 80 ± 0 | 90 ± 0 | 70 ± 0 | 90 + 0 | 12 ± 01 | 20.33 ± 0.58 | 12.67 ± 0.58 | 20 ± 1.0 | 15.67 ± 1.15 | 21.67 ± 0.58 |
| E. coli | 80 ± 0 | 100 ± 0 | 70 ± 0 | 100 ± 0 | 80 ± 0 | 90 ± 0 | 11.67 ± 0.58 | 18.33 ± 1.53 | 11.0 ± 00 | 18.67 ± 1.53 | 14.33 ± 0.58 | 21 ± 0 |
| P. aeruginosa | 80 ± 0 | 90 ± 0 | 80 ± 0 | 90 ± 0 | 70 ± 0 | 80 ± 0 | 14 ± 01 | 20.33 ± 0.58 | 12.67 ± 0.58 | 19.67 ± 0.58 | 16.33 ± 0.58 | 24.33 ± 0.58 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaikh, M.S.; Kale, M.A.; Muralidharan, V.; Venkatachalam, T.; Ali, S.S.; Islam, F.; Khan, S.L.; Siddiqui, F.A.; Urmee, H.; Tapadiya, G.G.; et al. The Design, Synthesis, and Evaluation of Diaminopimelic Acid Derivatives as Potential dapF Inhibitors Preventing Lysine Biosynthesis for Antibacterial Activity. Antibiotics 2023, 12, 47. https://doi.org/10.3390/antibiotics12010047
Shaikh MS, Kale MA, Muralidharan V, Venkatachalam T, Ali SS, Islam F, Khan SL, Siddiqui FA, Urmee H, Tapadiya GG, et al. The Design, Synthesis, and Evaluation of Diaminopimelic Acid Derivatives as Potential dapF Inhibitors Preventing Lysine Biosynthesis for Antibacterial Activity. Antibiotics. 2023; 12(1):47. https://doi.org/10.3390/antibiotics12010047
Chicago/Turabian StyleShaikh, Mohd Sayeed, Mayura A. Kale, V. Muralidharan, T. Venkatachalam, Syed Sarfaraz Ali, Fahadul Islam, Sharuk L. Khan, Falak A. Siddiqui, Humaira Urmee, Ganesh G. Tapadiya, and et al. 2023. "The Design, Synthesis, and Evaluation of Diaminopimelic Acid Derivatives as Potential dapF Inhibitors Preventing Lysine Biosynthesis for Antibacterial Activity" Antibiotics 12, no. 1: 47. https://doi.org/10.3390/antibiotics12010047
APA StyleShaikh, M. S., Kale, M. A., Muralidharan, V., Venkatachalam, T., Ali, S. S., Islam, F., Khan, S. L., Siddiqui, F. A., Urmee, H., Tapadiya, G. G., Dhawale, S. A., Ming, L. C., Ibrahim, I. A. A., Alzahrani, A. R., Sarker, M. M. R., & Azlina, M. F. N. (2023). The Design, Synthesis, and Evaluation of Diaminopimelic Acid Derivatives as Potential dapF Inhibitors Preventing Lysine Biosynthesis for Antibacterial Activity. Antibiotics, 12(1), 47. https://doi.org/10.3390/antibiotics12010047