
Genetically Modified Mesenchymal Stromal/Stem Cells as a Delivery Platform for SE-33, a Cathelicidin LL-37 Analogue: Preclinical Pharmacokinetics and Tissue Distribution in C57BL/6 Mice
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
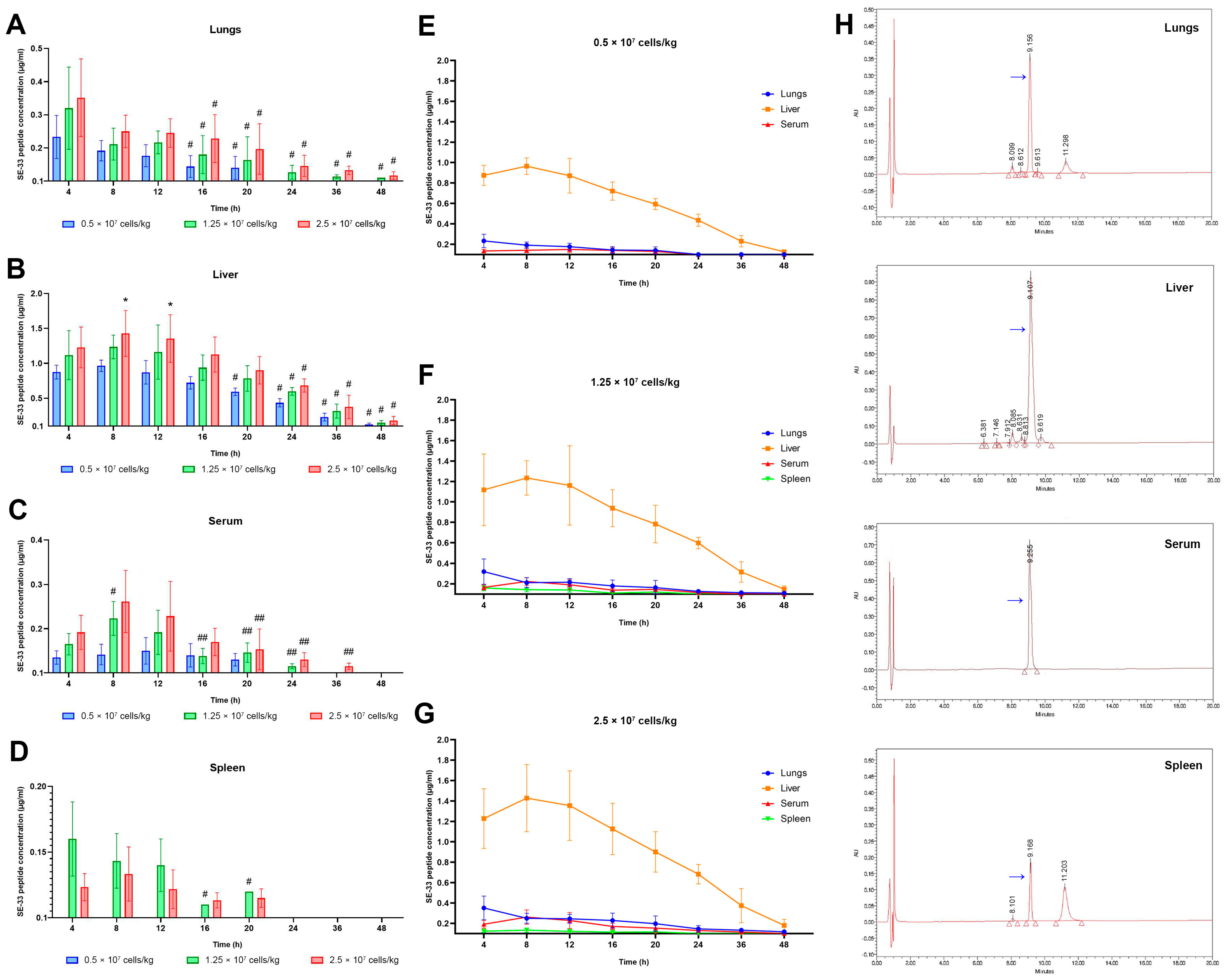
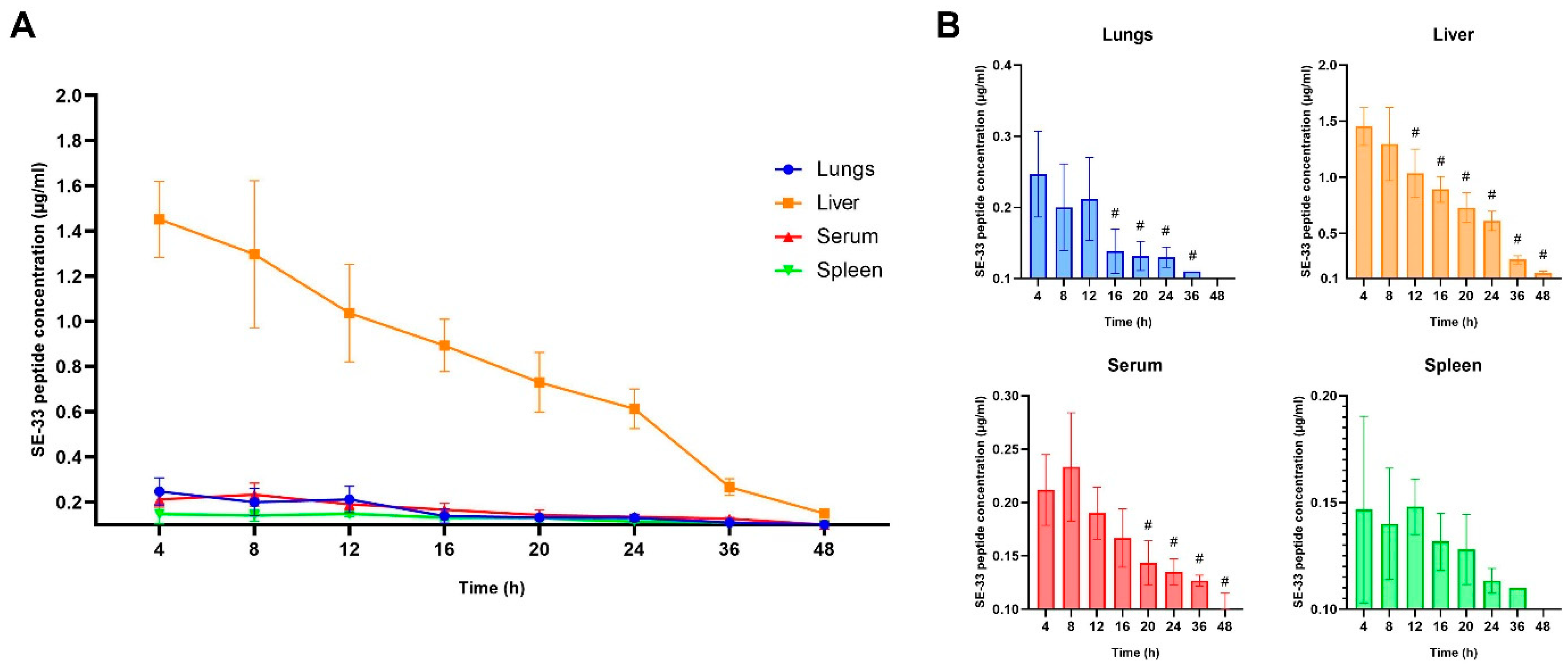
2.1. Pharmacokinetics of SE-33 Peptide in Mice Following the Single and Repeated Intravenous Administration of WJ-MSC-SE33
2.2. Linear Regression Analysis of the SE-33 Peptide Concentration in Mice Following the Intravenous Administration of WJ-MSC-SE33
2.3. The Apparent Distribution and Tissue Penetration of SE-33 Peptide in Mice Following an Intravenous Administration of WJ-MSC-SE33
3. Discussion
4. Materials and Methods
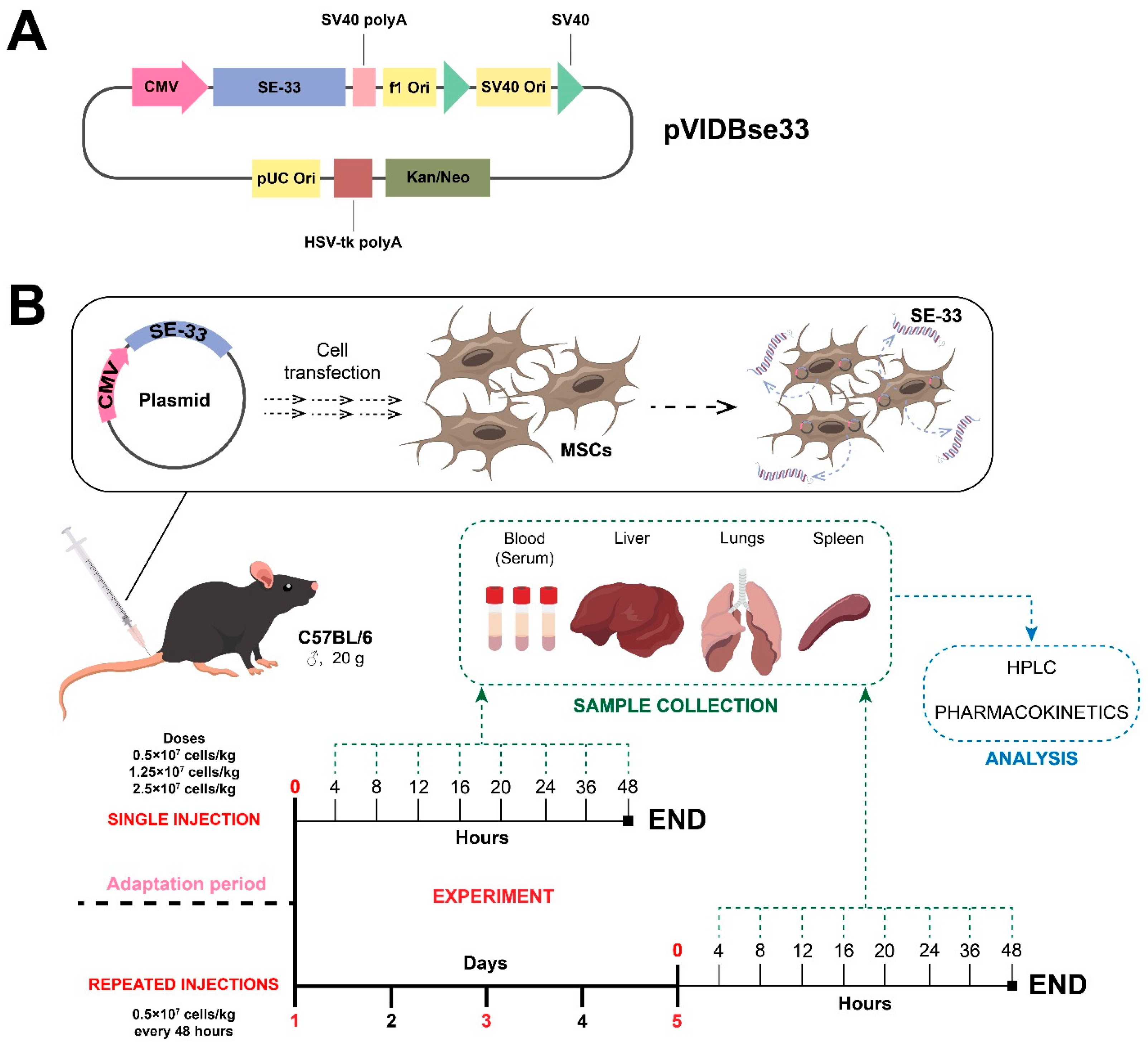
4.1. Plasmid Vector Construction and Mesenchymal Stromal/Stem Cell Transfection
4.2. Animal Studies
4.3. The Administration of Genetically Modified WJ-MSCs
4.4. Biomaterial Collection
4.5. The Determination of the Antimicrobial Peptide SE-33 in Animal Tissue and Serum Samples
4.5.1. Sample Preparation for Chromatographic Analysis
4.5.2. The Preparation of an Affinity Column for SE-33 Peptide Binding
4.5.3. Affinity Chromatography
4.5.4. Solid-Phase Extraction
4.5.5. High-Performance Liquid Chromatography
4.6. Non-Compartmental Analysis of Pharmacokinetic Data
4.7. Tissue Penetration and Distribution Analysis
4.8. Linear Regression Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Okeke, I.N.; de Kraker, M.E.A.; Van Boeckel, T.P.; Kumar, C.K.; Schmitt, H.; Gales, A.C.; Bertagnolio, S.; Sharland, M.; Laxminarayan, R. The Scope of the Antimicrobial Resistance Challenge. Lancet 2024, 403, 2426–2438. [Google Scholar] [CrossRef]
- Salam, M.A.; Al-Amin, M.Y.; Salam, M.T.; Pawar, J.S.; Akhter, N.; Rabaan, A.A.; Alqumber, M.A.A. Antimicrobial Resistance: A Growing Serious Threat for Global Public Health. Healthcare 2023, 11, 1946. [Google Scholar] [CrossRef]
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Chung, H.; Merakou, C.; Schaefers, M.M.; Flett, K.B.; Martini, S.; Lu, R.; Blumenthal, J.A.; Webster, S.S.; Cross, A.R.; Al Ahmar, R.; et al. Rapid Expansion and Extinction of Antibiotic Resistance Mutations during Treatment of Acute Bacterial Respiratory Infections. Nat. Commun. 2022, 13, 1231. [Google Scholar] [CrossRef]
- Sfeir, M.M. Diagnosis of Multidrug-Resistant Pathogens of Pneumonia. Diagnostics 2021, 11, 2287. [Google Scholar] [CrossRef] [PubMed]
- Kato, H. Antibiotic Therapy for Bacterial Pneumonia. J. Pharm. Health Care Sci. 2024, 10, 45. [Google Scholar] [CrossRef]
- Chu, V.T.; Tsitsiklis, A.; Mick, E.; Ambroggio, L.; Kalantar, K.L.; Glascock, A.; Osborne, C.M.; Wagner, B.D.; Matthay, M.A.; DeRisi, J.L.; et al. The Antibiotic Resistance Reservoir of the Lung Microbiome Expands with Age in a Population of Critically Ill Patients. Nat. Commun. 2024, 15, 92. [Google Scholar] [CrossRef]
- Jian, Z.; Zeng, L.; Xu, T.; Sun, S.; Yan, S.; Yang, L.; Huang, Y.; Jia, J.; Dou, T. Antibiotic Resistance Genes in Bacteria: Occurrence, Spread, and Control. J. Basic Microbiol. 2021, 61, 1049–1070. [Google Scholar] [CrossRef]
- Chavada, J.; Muneshwar, K.N.; Ghulaxe, Y.; Wani, M.; Sarda, P.P.; Huse, S. Antibiotic Resistance: Challenges and Strategies in Combating Infections. Cureus 2023, 15, e46013. [Google Scholar] [CrossRef]
- Peterson, E.; Kaur, P. Antibiotic Resistance Mechanisms in Bacteria: Relationships Between Resistance Determinants of Antibiotic Producers, Environmental Bacteria, and Clinical Pathogens. Front. Microbiol. 2018, 9, 2928. [Google Scholar] [CrossRef]
- de la Fuente-Nunez, C.; Cesaro, A.; Hancock, R.E.W. Antibiotic Failure: Beyond Antimicrobial Resistance. Drug Resist. Updat. 2023, 71, 101012. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, T.R.; Wells, T.J.; Souza-Fonseca-Guimaraes, F. Towards Efficient Immunotherapy for Bacterial Infection. Trends Microbiol. 2021, 30, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Laroye, C.; Gibot, S.; Reppel, L.; Bensoussan, D. Concise Review: Mesenchymal Stromal/Stem Cells: A New Treatment for Sepsis and Septic Shock? Stem Cells 2017, 35, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Yudintceva, N.; Mikhailova, N.; Fedorov, V.; Samochernych, K.; Vinogradova, T.; Muraviov, A.; Shevtsov, M. Mesenchymal Stem Cells and MSCs-Derived Extracellular Vesicles in Infectious Diseases: From Basic Research to Clinical Practice. Bioengineering 2022, 9, 662. [Google Scholar] [CrossRef]
- Varkouhi, A.K.; Monteiro, A.P.T.; Tsoporis, J.N.; Mei, S.H.J.; Stewart, D.J.; dos Santos, C.C. Genetically Modified Mesenchymal Stromal/Stem Cells: Application in Critical Illness. Stem Cell Rev. Rep. 2020, 16, 812–827. [Google Scholar] [CrossRef]
- Matthay, M.A. Therapeutic Potential of Mesenchymal Stromal Cells for Acute Respiratory Distress Syndrome. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. 1), S54–S57. [Google Scholar] [CrossRef]
- Lee, J.W.; Fang, X.; Krasnodembskaya, A.; Howard, J.P.; Matthay, M.A. Concise Review: Mesenchymal Stem Cells for Acute Lung Injury: Role of Paracrine Soluble Factors. Stem Cells 2011, 29, 913–919. [Google Scholar] [CrossRef]
- Harrell, C.R.; Sadikot, R.; Pascual, J.; Fellabaum, C.; Jankovic, M.G.; Jovicic, N.; Djonov, V.; Arsenijevic, N.; Volarevic, V. Mesenchymal Stem Cell-Based Therapy of Inflammatory Lung Diseases: Current Understanding and Future Perspectives. Stem Cells Int. 2019, 2019, 4236973. [Google Scholar] [CrossRef]
- Behnke, J.; Kremer, S.; Shahzad, T.; Chao, C.M.; Böttcher-Friebertshäuser, E.; Morty, R.E.; Bellusci, S.; Ehrhardt, H. MSC Based Therapies—New Perspectives for the Injured Lung. J. Clin. Med. 2020, 9, 682. [Google Scholar] [CrossRef]
- Le Blanc, K.; Mougiakakos, D. Multipotent Mesenchymal Stromal Cells and the Innate Immune System. Nat. Rev. Immunol. 2012, 12, 383–396. [Google Scholar] [CrossRef]
- Song, N.; Scholtemeijer, M.; Shah, K. Mesenchymal Stem Cell Immunomodulation: Mechanisms and Therapeutic Potential. Trends Pharmacol. Sci. 2020, 41, 653. [Google Scholar] [CrossRef]
- Sharma, A.; Chakraborty, A.; Jaganathan, B.G. Review of the Potential of Mesenchymal Stem Cells for the Treatment of Infectious Diseases. World J. Stem Cells 2021, 13, 568. [Google Scholar] [CrossRef] [PubMed]
- Guerra, A.D.; Rose, W.E.; Hematti, P.; Kao, W.J. Minocycline Modulates NFκB Phosphorylation and Enhances Antimicrobial Activity against Staphylococcus Aureus in Mesenchymal Stromal/Stem Cells. Stem Cell Res. Ther. 2017, 8, 171. [Google Scholar] [CrossRef] [PubMed]
- Devaney, J.; Horie, S.; Masterson, C.; Elliman, S.; Barry, F.; O’Brien, T.; Curley, G.F.; O’Toole, D.; Laffey, J.G. Human Mesenchymal Stromal Cells Decrease the Severity of Acute Lung Injury Induced by E. Coli in the Rat. Thorax 2015, 70, 625–635. [Google Scholar] [CrossRef]
- Zhu, Y.G.; Feng, X.M.; Abbott, J.; Fang, X.H.; Hao, Q.; Monsel, A.; Qu, J.M.; Matthay, M.A. Human Mesenchymal Stem Cell Microvesicles for Treatment of Escherichia Coli Endotoxin-Induced Acute Lung Injury in Mice. Stem Cells 2014, 32, 116–125. [Google Scholar] [CrossRef]
- Monsel, A.; Zhu, Y.G.; Gennai, S.; Hao, Q.; Hu, S.; Rouby, J.J.; Rosenzwajg, M.; Matthay, M.A.; Lee, J.W. Therapeutic Effects of Human Mesenchymal Stem Cell-Derived Microvesicles in Severe Pneumonia in Mice. Am. J. Respir. Crit. Care Med. 2015, 192, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Sutton, M.T.; Fletcher, D.; Ghosh, S.K.; Weinberg, A.; Van Heeckeren, R.; Kaur, S.; Sadeghi, Z.; Hijaz, A.; Reese, J.; Lazarus, H.M.; et al. Antimicrobial Properties of Mesenchymal Stem Cells: Therapeutic Potential for Cystic Fibrosis Infection, and Treatment. Stem Cells Int. 2016, 2016, 5303048. [Google Scholar] [CrossRef]
- Yagi, H.; Chen, A.F.; Hirsch, D.; Rothenberg, A.C.; Tan, J.; Alexander, P.G.; Tuan, R.S. Antimicrobial Activity of Mesenchymal Stem Cells against Staphylococcus Aureus. Stem Cell Res. Ther. 2020, 11, 293. [Google Scholar] [CrossRef]
- Ward, C.L.; Sanchez, C.J.; Pollot, B.E.; Romano, D.R.; Hardy, S.K.; Becerra, S.C.; Rathbone, C.R.; Wenke, J.C. Soluble Factors from Biofilms of Wound Pathogens Modulate Human Bone Marrow-Derived Stromal Cell Differentiation, Migration, Angiogenesis, and Cytokine Secretion. BMC Microbiol. 2015, 15, 75. [Google Scholar] [CrossRef]
- Alcayaga-Miranda, F.; Cuenca, J.; Martin, A.; Contreras, L.; Figueroa, F.E.; Khoury, M. Combination Therapy of Menstrual Derived Mesenchymal Stem Cells and Antibiotics Ameliorates Survival in Sepsis. Stem Cell Res. Ther. 2015, 6, 199. [Google Scholar] [CrossRef]
- Brandau, S.; Jakob, M.; Bruderek, K.; Bootz, F.; Giebel, B.; Radtke, S.; Mauel, K.; Jäger, M.; Flohé, S.B.; Lang, S. Mesenchymal Stem Cells Augment the Anti-Bacterial Activity of Neutrophil Granulocytes. PLoS ONE 2014, 9, e106903. [Google Scholar] [CrossRef]
- Krasnodembskaya, A.; Song, Y.; Fang, X.; Gupta, N.; Serikov, V.; Lee, J.W.; Matthay, M.A. Antibacterial Effect of Human Mesenchymal Stem Cells Is Mediated in Part from Secretion of the Antimicrobial Peptide LL-37. Stem Cells 2010, 28, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Krasnodembskaya, A.; Kapetanaki, M.; Mouded, M.; Tan, X.; Serikov, V.; Matthay, M.A. Mesenchymal Stem Cells Enhance Survival and Bacterial Clearance in Murine Escherichia Coli Pneumonia. Thorax 2012, 67, 533–539. [Google Scholar] [CrossRef]
- Sung, D.K.; Chang, Y.S.; Sung, S.I.; Yoo, H.S.; Ahn, S.Y.; Park, W.S. Antibacterial Effect of Mesenchymal Stem Cells against Escherichia Coli Is Mediated by Secretion of Beta—Defensin-2 via Toll-like Receptor 4 Signalling. Cell. Microbiol. 2016, 18, 424. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; van der Does, A.M.; Tang, X.; Lindbom, L.; Agerberth, B.; Haeggström, J.Z. Antimicrobial Peptide LL-37 Promotes Bacterial Phagocytosis by Human Macrophages. J. Leukoc. Biol. 2014, 95, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, D.; Landuyt, B.; Luyten, W.; Schoofs, L. A Comprehensive Summary of LL-37, the Factotum Human Cathelicidin Peptide. Cell. Immunol. 2012, 280, 22–35. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Kaplan, M.J. Little Peptide, Big Effects: The Role of LL-37 in Inflammation and Autoimmune Disease. J. Immunol. 2013, 191, 4895–4901. [Google Scholar] [CrossRef]
- Gad, G.I.; Abushady, N.M.; Fathi, M.S.; Elsaadany, W. Diagnostic Value of Anti-Microbial Peptide, Cathelicidin in Congenital Pneumonia. J. Matern. Fetal Neonatal Med. 2015, 28, 2197–2200. [Google Scholar] [CrossRef]
- Liu, W.; Dong, S.L.; Xu, F.; Wang, X.Q.; Withers, T.R.; Yu, H.D.; Wang, X. Effect of Intracellular Expression of Antimicrobial Peptide LL-37 on Growth of Escherichia Coli Strain TOP10 under Aerobic and Anaerobic Conditions. Antimicrob. Agents Chemother. 2013, 57, 4707–4716. [Google Scholar] [CrossRef]
- Tjabringa, G.S.; Ninaber, D.K.; Drijfhout, J.W.; Rabe, K.F.; Hiemstra, P.S. Human Cathelicidin LL-37 Is a Chemoattractant for Eosinophils and Neutrophils That Acts via Formyl-Peptide Receptors. Int. Arch. Allergy Immunol. 2006, 140, 103–112. [Google Scholar] [CrossRef]
- De Yang, B.; Chen, Q.; Schmidt, A.P.; Anderson, G.M.; Wang, J.M.; Wooters, J.; Oppenheim, J.J.; Chertov, O. LL-37, the Neutrophil Granule- and Epithelial Cell-Derived Cathelicidin, Utilizes Formyl Peptide Receptor-like 1 (FPRL1) as a Receptor to Chemoattract Human Peripheral Blood Neutrophils, Monocytes, and T Cells. J. Exp. Med. 2000, 192, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Babolewska, E.; Brzezińska-Błaszczyk, E. Human-Derived Cathelicidin LL-37 Directly Activates Mast Cells to Proinflammatory Mediator Synthesis and Migratory Response. Cell. Immunol. 2015, 293, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.; Zhang, N.; Yang, J.; Meng, X.; Yang, R.; Li, J.; Sun, T. Antimicrobial Peptide LL-37 and IDR-1 Ameliorate MRSA Pneumonia in Vivo. Cell. Physiol. Biochem. 2013, 32, 614–623. [Google Scholar] [CrossRef]
- Zhou, Y.; Shi, Y.; Yang, L.; Sun, Y.; Han, Y.; Zhao, Z.; Wang, Y.; Liu, Y.; Ma, Y.; Zhang, T.; et al. Genetically Engineered Distal Airway Stem Cell Transplantation Protects Mice from Pulmonary Infection. EMBO Mol. Med. 2020, 12, e10233. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Lu, T.K. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics 2020, 9, 24. [Google Scholar] [CrossRef]
- Lysenko, E.S.; Gould, J.; Bals, R.; Wilson, J.M.; Weiser, J.N. Bacterial Phosphorylcholine Decreases Susceptibility to the Antimicrobial Peptide LL-37/HCAP18 Expressed in the Upper Respiratory Tract. Infect. Immun. 2000, 68, 1664. [Google Scholar] [CrossRef]
- Koprivnjak, T.; Peschel, A. Bacterial Resistance Mechanisms against Host Defense Peptides. Cell. Mol. Life Sci. 2011, 68, 2243–2254. [Google Scholar] [CrossRef]
- Ridyard, K.E.; Overhage, J. The Potential of Human Peptide Ll-37 as an Antimicrobial and Anti-Biofilm Agent. Antibiotics 2021, 10, 650. [Google Scholar] [CrossRef]
- Takahashi, T.; Kulkarni, N.N.; Lee, E.Y.; Zhang, L.J.; Wong, G.C.L.; Gallo, R.L. Cathelicidin Promotes Inflammation by Enabling Binding of Self-RNA to Cell Surface Scavenger Receptors. Sci. Rep. 2018, 8, 4032. [Google Scholar] [CrossRef]
- Chen, K.; Gong, W.; Huang, J.; Yoshimura, T.; Wang, J.M. The Potentials of Short Fragments of Human Anti-Microbial Peptide LL-37 as a Novel Therapeutic Modality for Diseases. Front. Biosci.—Landmark 2021, 26, 1362–1372. [Google Scholar] [CrossRef]
- Zhan, N.; Zhang, L.; Yang, H.; Zheng, Y.; Wei, X.; Wang, J.; Shan, A. Design and Heterologous Expression of a Novel Dimeric LL37 Variant in Pichia Pastoris. Microb. Cell Fact. 2021, 20, 143. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Rajasekaran, G.; Shin, S.Y. LL-37-Derived Short Antimicrobial Peptide KR-12-A5 and Its d-Amino Acid Substituted Analogs with Cell Selectivity, Anti-Biofilm Activity, Synergistic Effect with Conventional Antibiotics, and Anti-Inflammatory Activity. Eur. J. Med. Chem. 2017, 136, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Gasanov, V.; Vorotelyak, E.; Vasiliev, A. Expression of the Antimicrobial Peptide SE-33-A2P, a Modified Analog of Cathelicidin, and an Analysis of Its Properties. Antibiotics 2024, 13, 190. [Google Scholar] [CrossRef] [PubMed]
- Gasanov, V.A.; Vorotelyak, E.A.; Vasiliev, A.V. Production of Antimicrobial Peptides (Cathelicidin Analogues) and Evaluation of Their Biological Properties. Biol. Bull. 2022, 49, S148–S151. [Google Scholar] [CrossRef]
- Trenin, A.S.; Arzumanian, V.G.; Zhmak, M.N.; Shelukhina, I.V.; Makarova, Y.V.; Ivanov, I.A.; Bychkova, O.P.; Budikhina, A.S.; Balyasova, L.S.; Tsetlin, V.I. Synthesis and Antimicrobial Activity of a New Drug Based on Retro-Analog Cathelicidin-Polypeptide SE-33. Russ. J. Bioorg. Chem. 2019, 45, 252–264. [Google Scholar] [CrossRef]
- oglu Gasanov, V.A.; Kashirskikh, D.A.; Khotina, V.A.; Kuzmina, D.M.; Nikitochkina, S.Y.; Mukhina, I.V.; Vorotelyak, E.A.; Vasiliev, A.V. Preclinical Evaluation of the Safety, Toxicity and Efficacy of Genetically Modified Wharton’s Jelly Mesenchymal Stem/Stromal Cells Expressing the Antimicrobial Peptide SE-33. Cells 2025, 14, 341. [Google Scholar] [CrossRef]
- Cheng, Y.; Cao, X.; Qin, L. Mesenchymal Stem Cell-Derived Extracellular Vesicles: A Novel Cell-Free Therapy for Sepsis. Front. Immunol. 2020, 11, 526156. [Google Scholar] [CrossRef]
- Daniel, M.; Bedoui, Y.; Vagner, D.; Raffray, L.; Ah-Pine, F.; Doray, B.; Gasque, P. Pathophysiology of Sepsis and Genesis of Septic Shock: The Critical Role of Mesenchymal Stem Cells (MSCs). Int. J. Mol. Sci. 2022, 23, 9274. [Google Scholar] [CrossRef]
- Javaregowda, P.K.; Yoon, J.W.; Jang, G. Roles of Mesenchymal Stem Cells (MSCs) in Bacterial Diseases. J. Biomed. Res. 2013, 14, 184–194. [Google Scholar] [CrossRef]
- Yang, H.; Xu, F.; Zheng, X.; Yang, S.; Ren, Z.; Yang, J. Human Umbilical Cord Mesenchymal Stem Cells Prevent Bacterial Biofilm Formation. BioMed Res. Int. 2022, 2022, 1530525. [Google Scholar] [CrossRef]
- Pezzanite, L.M.; Chow, L.; Phillips, J.; Griffenhagen, G.M.; Moore, A.R.; Schaer, T.P.; Engiles, J.B.; Werpy, N.; Gilbertie, J.; Schnabel, L.V.; et al. TLR-Activated Mesenchymal Stromal Cell Therapy and Antibiotics to Treat Multi-Drug Resistant Staphylococcal Septic Arthritis in an Equine Model. Ann. Transl. Med. 2022, 10, 1157. [Google Scholar] [CrossRef]
- Johnson, V.; Chow, L.; Harrison, J.; Soontararak, S.; Dow, S. Activated Mesenchymal Stromal Cell Therapy for Treatment of Multi-Drug Resistant Bacterial Infections in Dogs. Front. Vet. Sci. 2022, 9, 925701. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Sun, L.C.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q.Y. The Antimicrobial Peptides and Their Potential Clinical Applications. Am. J. Transl. Res. 2019, 11, 3919. [Google Scholar] [PubMed]
- Chung, P.Y.; Khanum, R. Antimicrobial Peptides as Potential Anti-Biofilm Agents against Multidrug-Resistant Bacteria. J. Microbiol. Immunol. Infect. 2017, 50, 405–410. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Yan, Z.B.; Meng, Y.M.; Hong, X.Y.; Shao, G.; Ma, J.J.; Cheng, X.R.; Liu, J.; Kang, J.; Fu, C.Y. Antimicrobial Peptides: Mechanism of Action, Activity and Clinical Potential. Mil. Med. Res. 2021, 8, 48. [Google Scholar] [CrossRef]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial Host Defence Peptides: Functions and Clinical Potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Roudi, R.; Syn, N.L.; Roudbary, M. Antimicrobial Peptides as Biologic and Immunotherapeutic Agents against Cancer: A Comprehensive Overview. Front. Immunol. 2017, 8, 1320. [Google Scholar] [CrossRef]
- Pfalzgraff, A.; Brandenburg, K.; Weindl, G. Antimicrobial Peptides and Their Therapeutic Potential for Bacterial Skin Infections and Wounds. Front. Pharmacol. 2018, 9, 352601. [Google Scholar] [CrossRef]
- Dunvald, A.C.D.; Iversen, D.B.; Svendsen, A.L.O.; Agergaard, K.; Kuhlmann, I.B.; Mortensen, C.; Andersen, N.E.; Järvinen, E.; Stage, T.B. Tutorial: Statistical Analysis and Reporting of Clinical Pharmacokinetic Studies. Clin. Transl. Sci. 2022, 15, 1856–1866. [Google Scholar] [CrossRef]
- Gabrielsson, J.; Weiner, D. Pharmacokinetic and Pharmacodynamic Data Analysis: Concepts and Applications, 5th ed.; Swedish Pharmaceutical Press: Stockholm, Sweden, 2016; ISBN 9 789 198 299 106. [Google Scholar]
- Wang, H.; Liang, X.; Xu, Z.P.; Crawford, D.H.G.; Liu, X.; Roberts, M.S. A Physiologically Based Kinetic Model for Elucidating the in Vivo Distribution of Administered Mesenchymal Stem Cells. Sci. Rep. 2016, 6, 22293. [Google Scholar] [CrossRef]
- Mould, D.R.; Upton, R.N. Basic Concepts in Population Modeling, Simulation, and Model-Based Drug Development—Part 2: Introduction to Pharmacokinetic Modeling Methods. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e38. [Google Scholar] [CrossRef]
- Kraitchman, D.L.; Tatsumi, M.; Gilson, W.D.; Ishimori, T.; Kedziorek, D.; Walczak, P.; Segars, W.P.; Chen, H.H.; Fritzges, D.; Izbudak, I.; et al. Dynamic Imaging of Allogeneic Mesenchymal Stem Cells Trafficking to Myocardial Infarction. Circulation 2005, 112, 1451. [Google Scholar] [CrossRef] [PubMed]
- Esposito, T.V.F.; Rodríguez-Rodríguez, C.; Blackadar, C.; Kłodzińska, S.; Mørck Nielsen, H.; Saatchi, K.; Häfeli, U.O. Biodistribution of the Cationic Host Defense Peptide LL-37 Using SPECT/CT. Eur. J. Pharm. Biopharm. 2024, 202, 114398. [Google Scholar] [CrossRef]
- Chin, S.P.; Marzuki, M.; Tai, L.; Mohamed Shahrehan, N.A.; Ricky, C.; Fanty, A.; Salleh, A.; Low, C.T.; Then, K.Y.; Hoe, S.L.L.; et al. Dynamic Tracking of Human Umbilical Cord Mesenchymal Stem Cells (HUC-MSCs) Following Intravenous Administration in Mice Model. Regen. Ther. 2024, 25, 273. [Google Scholar] [CrossRef] [PubMed]
- Pichardo, A.H.; Amadeo, F.; Wilm, B.; Lévy, R.; Ressel, L.; Murray, P.; Sée, V. Optical Tissue Clearing to Study the Intra-Pulmonary Biodistribution of Intravenously Delivered Mesenchymal Stromal Cells and Their Interactions with Host Lung Cells. Int. J. Mol. Sci. 2022, 23, 14171. [Google Scholar] [CrossRef]
- Han, L.; Ma, C.; Peng, H.; Wu, Z.; Xu, H.; Wu, J.; Zhang, N.; Jiang, Q.; Ma, C.; Huang, R.; et al. Define Mesenchymal Stem Cell from Its Fate: Biodisposition of Human Mesenchymal Stem Cells in Normal and Concanavalin A-Induced Liver Injury Mice. J. Pharmacol. Exp. Ther. 2021, 379, 125–133. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Liu, B.; Du, X.; Wei, Y.; Kong, D.; Feng, B.; Guo, R.; Asiamah, E.A.; Griffin, M.D.; Hynes, S.O.; et al. Amelioration of Diabetic Nephropathy in Mice by a Single Intravenous Injection of Human Mesenchymal Stromal Cells at Early and Later Disease Stages Is Associated with Restoration of Autophagy. Stem Cell Res. Ther. 2024, 15, 66. [Google Scholar] [CrossRef]
- de Witte, S.F.H.; Luk, F.; Sierra Parraga, J.M.; Gargesha, M.; Merino, A.; Korevaar, S.S.; Shankar, A.S.; O’Flynn, L.; Elliman, S.J.; Roy, D.; et al. Immunomodulation by Therapeutic Mesenchymal Stromal Cells (MSC) Is Triggered Through Phagocytosis of MSC By Monocytic Cells. Stem Cells 2018, 36, 602–615. [Google Scholar] [CrossRef]
- Gallagher, D.; Siddiqui, F.; Fish, J.; Charlat, M.; Chaudry, E.; Moolla, S.; Gauthier-Fisher, A.; Librach, C. Mesenchymal Stromal Cells Modulate Peripheral Stress-Induced Innate Immune Activation Indirectly Limiting the Emergence of Neuroinflammation-Driven Depressive and Anxiety-like Behaviors. Biol. Psychiatry 2019, 86, 712–724. [Google Scholar] [CrossRef]
- Gaafar, T.; Attia, W.; Mahmoud, S.; Sabry, D.; Aziz, O.A.; Rasheed, D.; Hamza, H. Cardioprotective Effects of Wharton Jelly Derived Mesenchymal Stem Cell Transplantation in a Rodent Model of Myocardial Injury. Int. J. Stem Cells 2017, 10, 48–59. [Google Scholar] [CrossRef]
- Mello, T.G.; Rosado-De-Castro, P.H.; Campos, R.M.P.; Vasques, J.F.; Rangel-Junior, W.S.; Mattos, R.S.D.A.R.D.; Puig-Pijuan, T.; Foerster, B.U.; Gutfilen, B.; Souza, S.A.L.; et al. Intravenous Human Umbilical Cord-Derived Mesenchymal Stromal Cell Administration in Models of Moderate and Severe Intracerebral Hemorrhage. Stem Cells Dev. 2020, 29, 586–598. [Google Scholar] [CrossRef]
- Bandekar, M.; Maurya, D.K.; Sharma, D.; Checker, R.; Gota, V.; Mishra, N.; Sandur, S.K. Xenogeneic Transplantation of Human WJ-MSCs Rescues Mice from Acute Radiation Syndrome via Nrf-2-Dependent Regeneration of Damaged Tissues. Am. J. Transplant. 2020, 20, 2044–2057. [Google Scholar] [CrossRef] [PubMed]
- Van Der Velden, V.H.J.; Hulsmann, A.R. Peptidases: Structure, Function and Modulation of Peptide-mediated Effects in the Human Lung. Clin. Exp. Allergy 1999, 29, 445. [Google Scholar] [CrossRef]
- Meyer, M.; Jaspers, I. Respiratory Protease/Antiprotease Balance Determines Susceptibility to Viral Infection and Can Be Modified by Nutritional Antioxidants. Am. J. Physiol.—Lung Cell. Mol. Physiol. 2015, 308, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Andrault, P.M.; Samsonov, S.A.; Weber, G.; Coquet, L.; Nazmi, K.; Bolscher, J.G.M.; Lalmanach, A.C.; Jouenne, T.; Brömme, D.; Pisabarro, M.T.; et al. Antimicrobial Peptide LL-37 Is Both a Substrate of Cathepsins S and K and a Selective Inhibitor of Cathepsin L. Biochemistry 2015, 54, 2785–2798. [Google Scholar] [CrossRef]
- Engelberg, Y.; Landau, M. The Human LL-37(17-29) Antimicrobial Peptide Reveals a Functional Supramolecular Structure. Nat. Commun. 2020, 11, 3894. [Google Scholar] [CrossRef] [PubMed]
- Mahlapuu, M.; Björn, C.; Ekblom, J. Antimicrobial Peptides as Therapeutic Agents: Opportunities and Challenges. Crit. Rev. Biotechnol. 2020, 40, 978–992. [Google Scholar] [CrossRef]
- Bellotti, D.; Remelli, M. Lights and Shadows on the Therapeutic Use of Antimicrobial Peptides. Molecules 2022, 27, 4584. [Google Scholar] [CrossRef]
- National Research Council (US) Committee. Guide for the Care and Use of Laboratory Animals; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Shao, J.; Xia, L.; Ye, Z.; Yang, Q.; Zhang, C.; Shi, Y.; Zhang, L.; Gu, L.; Xu, C.; Chen, Y.; et al. A Repeat-Dose Toxicity Study of Human Umbilical Cord Mesenchymal Stem Cells in NOG Mice by Intravenous Injection. Expert Opin. Drug Metab. Toxicol. 2023, 19, 857–866. [Google Scholar] [CrossRef]
- Eurasian Economic Commission Decision of the College of the Eurasian Economic Commission № 202 “On Approval of the Guidelines for Preclinical Safety Studies for the Purposes of Conducting Clinical Trials and Registration of Medicinal Products” from 26.11.2019; 2019. Available online: https://docs.eaeunion.org/documents/335/4808/ (accessed on 23 April 2025).
- European Medicines Agency. ICH Guideline M3(R2) on Non-Clinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorisation for Pharmaceuticals; 2009; Volume 3. Available online: https://www.ema.europa.eu/en/ich-m3-r2-non-clinical-safety-studies-conduct-human-clinical-trials-pharmaceuticals-scientific-guideline (accessed on 23 April 2025).
- Wang, G.; Li, X.; Wang, Z. APD3: The Antimicrobial Peptide Database as a Tool for Research and Education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- APD3. Antimicrobial Peptide Database. Available online: https://aps.unmc.edu/ (accessed on 28 November 2024).
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 5 March 2025).
- Denney, W.; Duvvuri, S.; Buckeridge, C. Simple, Automatic Noncompartmental Analysis: The PKNCA R Package. J. Pharmacokinet. Pharmacodyn. 2015, 42, 11–107. [Google Scholar] [CrossRef]
- Santos-Borges, K.T.; Henz, P.; de Araújo, B.V. The Influence of Sepsis on Antimicrobials Tissue Penetration: The Use of Microdialysis Technique to Access Free Drug Distribution. Braz. J. Pharm. Sci. 2023, 59, e22982. [Google Scholar] [CrossRef]
- Finazzi, S.; Luci, G.; Olivieri, C.; Langer, M.; Mandelli, G.; Corona, A.; Viaggi, B.; Di Paolo, A. Tissue Penetration of Antimicrobials in Intensive Care Unit Patients: A Systematic Review—Part I. Antibiotics 2022, 11, 1164. [Google Scholar] [CrossRef] [PubMed]
- Viaggi, B.; Cangialosi, A.; Langer, M.; Olivieri, C.; Gori, A.; Corona, A.; Finazzi, S.; Di Paolo, A. Tissue Penetration of Antimicrobials in Intensive Care Unit Patients: A Systematic Review—Part II. Antibiotics 2022, 11, 1193. [Google Scholar] [CrossRef]
- Jager, N.G.L.; van Hest, R.M.; Lipman, J.; Roberts, J.A.; Cotta, M.O. Antibiotic Exposure at the Site of Infection: Principles and Assessment of Tissue Penetration. Expert Rev. Clin. Pharmacol. 2019, 12, 623–634. [Google Scholar] [CrossRef]
- Wickham, V.H.; François, R.D.; Henry, L.; Müller, K. Dplyr: A Grammar of Data Manipulation. Available online: https://dplyr.tidyverse.org/ (accessed on 4 April 2025).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis. J. Stat. Softw. 2010, 35, 245–246. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Serum | Lungs | Liver | Spleen | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | |
| Cmax (μg/mL) | 0.142 | 0.223 | 0.262 | 0.233 | 0.320 | 0.352 | 0.965 | 1.235 | 1.428 | 0.044 | 0.072 | 0.133 |
| Tmax (h) | 8 | 8 | 8 | 4 | 4 | 4 | 8 | 8 | 8 | 8 | 8 | 8 |
| λz (h−1) | 0.144 | 0.174 | 0.173 | 0.160 | 0.048 | 0.031 | 0.061 | 0.057 | 0.056 | NC | 0.124 | 0.136 |
| T1/2 (h) | 4.80 | 3.98 | 4.00 | 4.34 | 14.3 | 22.9 | 11.4 | 12.2 | 12.4 | NC | 5.59 | 5.09 |
| AUC0–24 (h×μg/mL) | 2.01 | 4.58 | 6.08 | 3.15 | 5.13 | 6.86 | 24.6 | 33.1 | 36.4 | NC | 0.999 | 1.78 |
| AUC4–24 (h×μg/mL) | 1.74 | 4.25 | 5.70 | 2.68 | 4.48 | 6.15 | 22.8 | 30.9 | 34.0 | NC | 0.889 | 1.53 |
| AUC0–∞ (h×μg/mL) | 2.04 | 3.73 | 5.17 | 3.18 | 6.50 | 9.37 | 24.0 | 32.7 | 38.0 | NC | 1.03 | 1.81 |
| AUC4–∞ (h×μg/mL) | 1.77 | 3.4 | 4.79 | 2.71 | 5.86 | 8.67 | 22.2 | 30.4 | 35.6 | NC | 0.918 | 1.56 |
| AUMC0–24 (h2×μg/mL) | 20.0 | 82.3 | 127 | 31.7 | 73.3 | 119 | 519 | 733 | 746 | NC | 10.4 | 16.9 |
| AUMC4–24 (h2×μg/mL) | 12.0 | 64.0 | 103 | 19.2 | 52.8 | 91.4 | 421 | 601 | 600 | NC | 6.41 | 9.80 |
| AUMC0–∞ (h2×μg/mL) | 21.2 | 47.8 | 79.7 | 32.9 | 148 | 292 | 489 | 702 | 833 | NC | 11.6 | 18.1 |
| AUMC4–∞ (h2×μg/mL) | 13.1 | 32.9 | 59.1 | 20.2 | 122 | 255 | 393 | 571 | 680 | NC | 7.53 | 10.9 |
| MRT0–24 (h) | 9.95 | 18.0 | 20.9 | 10.1 | 14.3 | 17.3 | 21.1 | 22.1 | 20.5 | NC | 10.4 | 9.51 |
| MRT4–24 (h) | 6.89 | 15.1 | 18.1 | 7.15 | 11.8 | 14.9 | 18.4 | 19.4 | 17.7 | NC | 7.21 | 6.41 |
| MRT0–∞ (h) | 10.4 | 12.8 | 15.4 | 10.4 | 22.7 | 31.2 | 20.4 | 21.5 | 21.9 | NC | 11.3 | 10.0 |
| MRT4–∞ (h) | 7.38 | 9.68 | 12.3 | 7.46 | 20.8 | 29.4 | 17.7 | 18.8 | 19.1 | NC | 8.20 | 6.99 |
| Parameter | Serum | Lungs | Liver | Spleen |
|---|---|---|---|---|
| Cmax (μg/mL) | 0.233 | 0.247 | 1.45 | 0.147 |
| Tmax (h) | 8 | 4 | 4 | 4 |
| λz (h−1) | 0.030 | 0.091 | 0.055 | 0.102 |
| T1/2 (h) | 23.1 | 7.67 | 12.6 | 6.80 |
| AUC4–24 (h×μg/mL) | 8.21 | 3.25 | 33.0 | 2.94 |
| AUC4–∞ (h×μg/mL) | 8.05 | 3.36 | 30.1 | 2.99 |
| AUMC4–24 (h2×μg/mL) | 281 | 34.1 | 708 | 37.5 |
| AUMC4–∞ (h2×μg/mL) | 253 | 38.3 | 556 | 39.3 |
| MRT4–24 (h) | 34.2 | 10.5 | 21.5 | 12.8 |
| MRT4–∞ (h) | 31.4 | 11.4 | 18.5 | 13.2 |
| Administration, Dose | Coefficient of Determination (R2) | |||
|---|---|---|---|---|
| Serum | Lungs | Liver | Spleen | |
| Single, 0.5 × 107 cells/kg | 0.438344 | 0.655836 | 0.870367 | 0.069132 |
| Single, 1.25 × 107 cells/kg | 0.744957 | 0.552811 | 0.785668 | 0.199126 |
| Single, 2.5 × 107 cells/kg | 0.723056 | 0.581298 | 0.776045 | 0.496153 |
| Repeated, 0.5 × 107 cells/kg | 0.658007 | 0.567349 | 0.837087 | 0.599679 |
| Administration, Dose | Liver | Lungs | Spleen |
|---|---|---|---|
| Single, 0.5 × 107 cells/kg | 11.76 | 1.56 | NC |
| Single, 1.25 × 107 cells/kg | 8.77 | 1.74 | 0.28 |
| Single, 2.5 × 107 cells/kg | 7.35 | 1.81 | 0.35 |
| Repeated, 0.5 × 107 cells/kg | 3.74 | 0.37 | 0.42 |
| Administration, Dose | Time (h) | Liver | Lungs | Spleen |
|---|---|---|---|---|
| Single, 0.5 × 107 cells/kg | 4 | 6.48 | 1.73 | NC |
| 8 | 6.80 | 1.35 | 0.30 | |
| 12 | 6.98 | 1.42 | NC | |
| 16 | 10.31 | 1.71 | NC | |
| 20 | 13.79 | 2.16 | NC | |
| 24 | NC | NC | NC | |
| 36 | NC | NC | NC | |
| 48 | NC | NC | NC | |
| Single, 1.25 × 107 cells/kg | 4 | 6.78 | 1.94 | 0.32 |
| 8 | 5.54 | 0.95 | 0.32 | |
| 12 | 6.05 | 1.13 | 0.36 | |
| 16 | 6.80 | 1.30 | 0.27 | |
| 20 | 6.42 | 1.34 | 0.16 | |
| 24 | 7.77 | 0.82 | NC | |
| 36 | NC | NC | NC | |
| 48 | NC | NC | NC | |
| Single, 2.5 × 107 cells/kg | 4 | 6.40 | 1.83 | 0.64 |
| 8 | 5.45 | 0.95 | 0.51 | |
| 12 | 5.94 | 1.07 | 0.54 | |
| 16 | 5.93 | 1.20 | 0.30 | |
| 20 | 5.90 | 1.29 | 0.12 | |
| 24 | 6.32 | 1.13 | NC | |
| 36 | 9.87 | 2.32 | NC | |
| 48 | NC | NC | NC | |
| Repeated, 0.5 × 107 cells/kg | 4 | 1.06 | 1.17 | 0.69 |
| 8 | 1.11 | 0.86 | 0.60 | |
| 12 | 1.11 | 1.12 | 0.65 | |
| 16 | 1.09 | 0.44 | 0.79 | |
| 20 | 1.20 | 0.62 | 0.75 | |
| 24 | 1.07 | 0.32 | 0.42 | |
| 36 | 0.97 | 0.13 | 0.15 | |
| 48 | 1.36 | NC | NC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gasanov, V.A.o.; Kashirskikh, D.A.; Khotina, V.A.; Lee, A.A.; Nikitochkina, S.Y.; Kuzmina, D.M.; Mukhina, I.V.; Vorotelyak, E.A.; Vasiliev, A.V. Genetically Modified Mesenchymal Stromal/Stem Cells as a Delivery Platform for SE-33, a Cathelicidin LL-37 Analogue: Preclinical Pharmacokinetics and Tissue Distribution in C57BL/6 Mice. Antibiotics 2025, 14, 429. https://doi.org/10.3390/antibiotics14050429
Gasanov VAo, Kashirskikh DA, Khotina VA, Lee AA, Nikitochkina SY, Kuzmina DM, Mukhina IV, Vorotelyak EA, Vasiliev AV. Genetically Modified Mesenchymal Stromal/Stem Cells as a Delivery Platform for SE-33, a Cathelicidin LL-37 Analogue: Preclinical Pharmacokinetics and Tissue Distribution in C57BL/6 Mice. Antibiotics. 2025; 14(5):429. https://doi.org/10.3390/antibiotics14050429
Chicago/Turabian StyleGasanov, Vagif Ali oglu, Dmitry Alexandrovich Kashirskikh, Victoria Alexandrovna Khotina, Arthur Anatolievich Lee, Sofya Yurievna Nikitochkina, Daria Mikhailovna Kuzmina, Irina Vasilievna Mukhina, Ekaterina Andreevna Vorotelyak, and Andrey Valentinovich Vasiliev. 2025. "Genetically Modified Mesenchymal Stromal/Stem Cells as a Delivery Platform for SE-33, a Cathelicidin LL-37 Analogue: Preclinical Pharmacokinetics and Tissue Distribution in C57BL/6 Mice" Antibiotics 14, no. 5: 429. https://doi.org/10.3390/antibiotics14050429
APA StyleGasanov, V. A. o., Kashirskikh, D. A., Khotina, V. A., Lee, A. A., Nikitochkina, S. Y., Kuzmina, D. M., Mukhina, I. V., Vorotelyak, E. A., & Vasiliev, A. V. (2025). Genetically Modified Mesenchymal Stromal/Stem Cells as a Delivery Platform for SE-33, a Cathelicidin LL-37 Analogue: Preclinical Pharmacokinetics and Tissue Distribution in C57BL/6 Mice. Antibiotics, 14(5), 429. https://doi.org/10.3390/antibiotics14050429