
Advances in Huntington’s Disease Biomarkers: A 10-Year Bibliometric Analysis and a Comprehensive Review

 ,
,  and
and 
Simple Summary
Abstract
1. Introduction
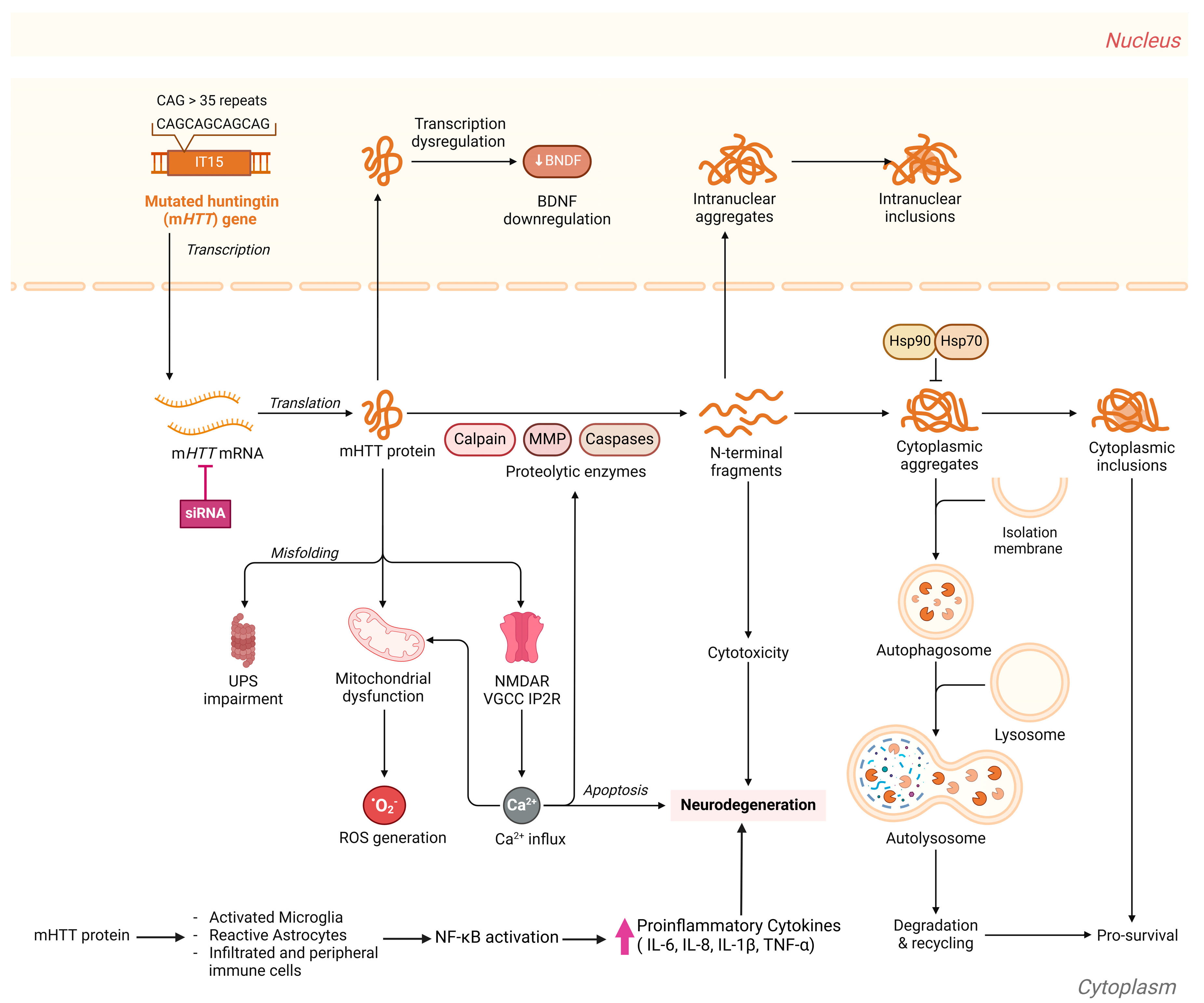
1.1. Huntington’s Disease: An Overview
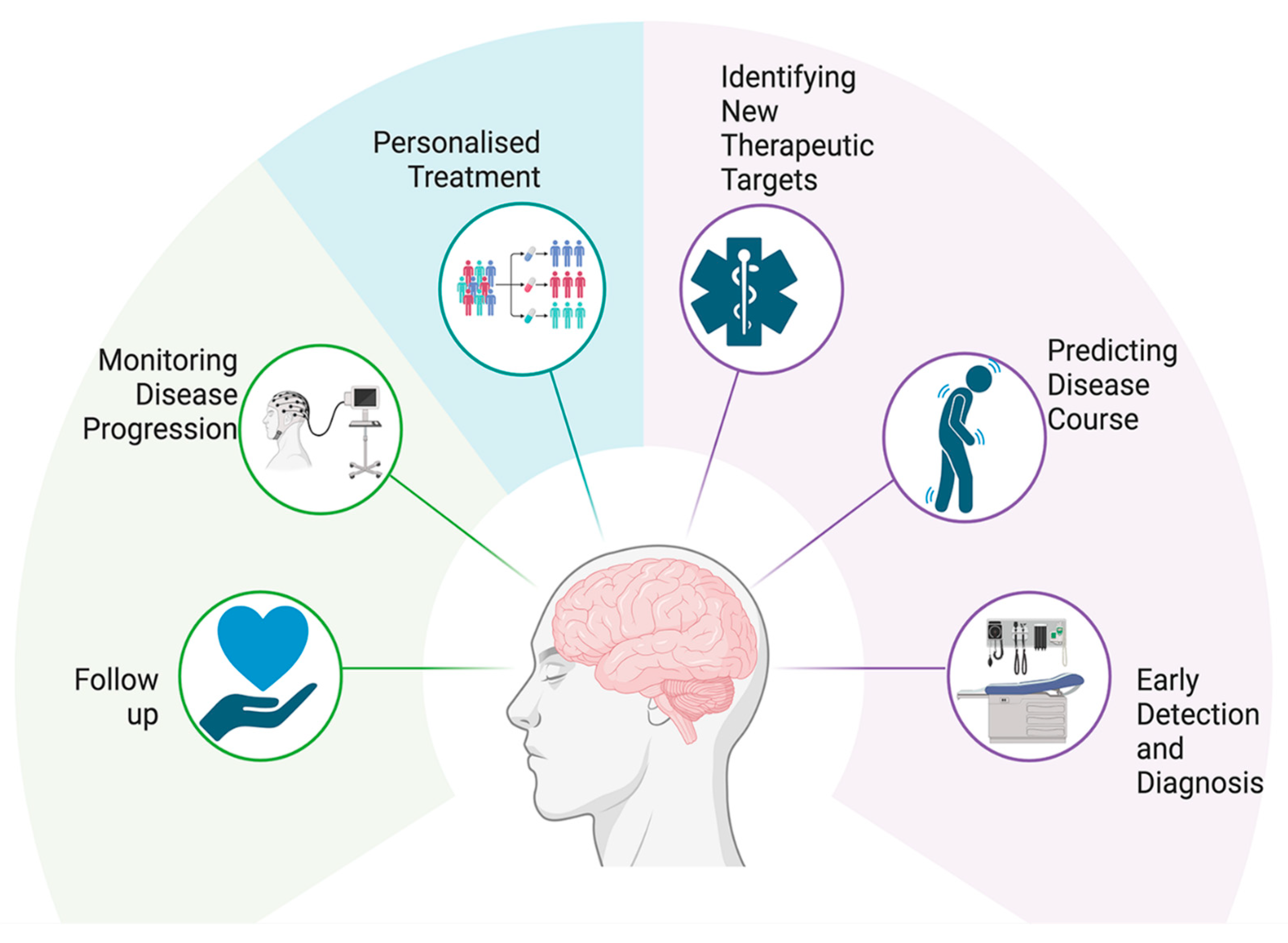
1.2. Challenges in HD Diagnosis and Treatment
2. Methods
2.1. Bibliometric Analysis
2.2. Literature Review
3. Results
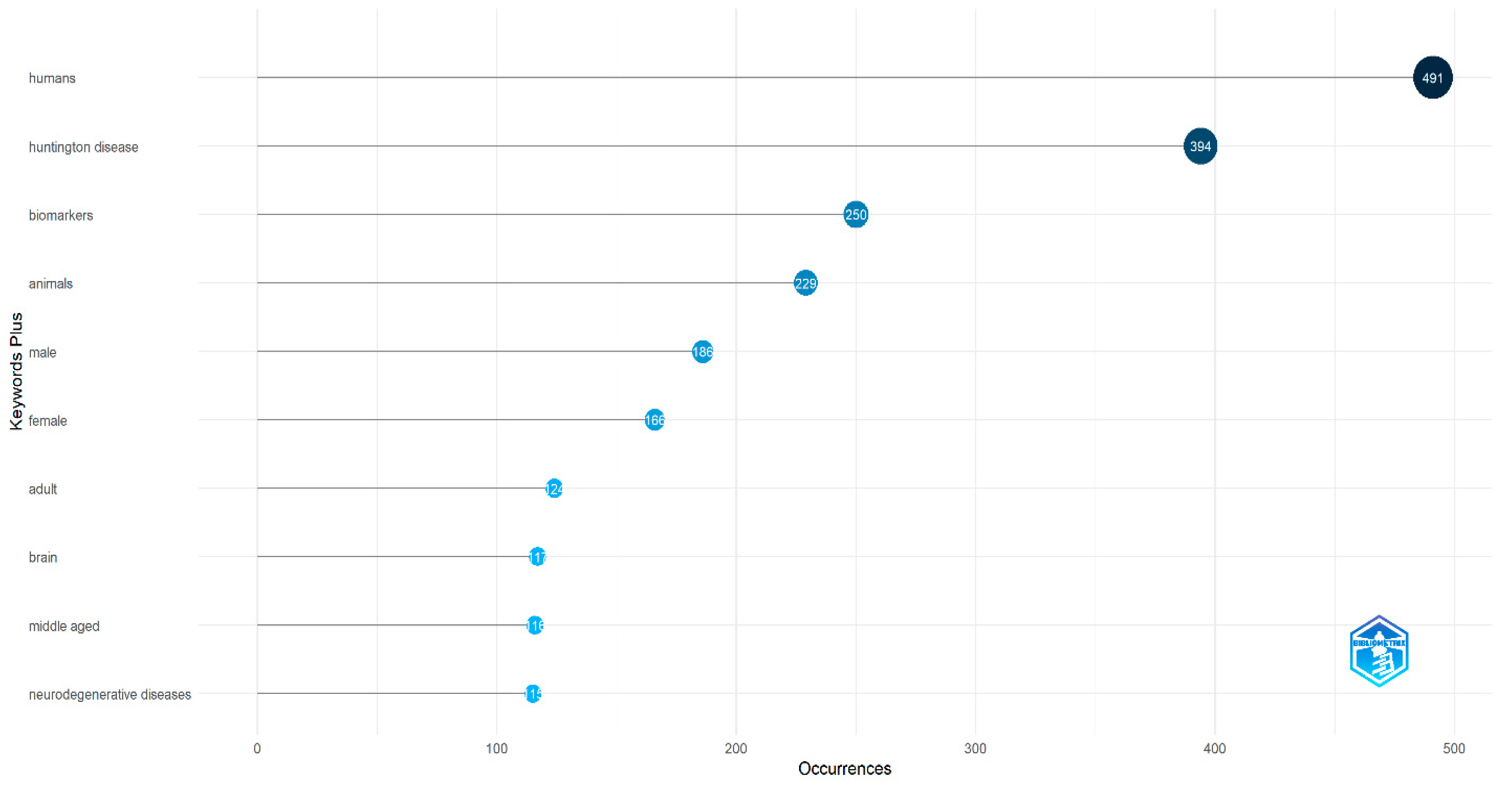
3.1. Bibliometric Analysis
3.2. Huntington’s Diseases Biomarkers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarker | Source | Clinical Utility | Advantages and Limitations | References | |
|---|---|---|---|---|---|
| Genomic Biomarkers | HTT gene mutation/CAG repeat expansion | Blood, Saliva | Disease diagnosis; predictive testing; prenatal testing |
| [64,65,66,67,68] |
| Protein Biomarkers | Mutant huntingtin (mHTT) protein | Blood, CSF | Disease progression monitoring, therapeutic targeting |
| [65,69,70,71,72,73,74] |
| Neurofilament light chain (NfL) | Blood, CSF | Neurodegeneration marker; disease progression marker; Can be used in preHD |
| [75,76,77,78,79,80,81,82] | |
| Brain-derived neurotrophic factor (BDNF) | Blood, salvia, CSF | Potential early disease marker |
| [44,83,84,85,86,87,88,89] | |
| Tau protein | Blood, CSF, skin tissue | Elevated levels of phosphorylated tau (p-tau) in plasma correlate with HD severity, aiding in staging the disease, abnormal tau accumulation in skin tissue may serve as an early indicator of HD. |
| [90,91,92,93] | |
| IL-6 | Blood, CSF | Inflammation monitoring, disease progression |
| [94,95,96,97] | |
| Non-coding RNA Biomarkers | microRNAs (miRNAs) | Blood, CSF, brain tissue | Gene expression regulation, disease state indicators |
| [98,99,100,101,102,103] |
| Metabolic Biomarkers | Uric acid | Blood, saliva | Potential predictor of disease progression in HD |
| [104,105,106,107] |
| 24S-Hydroxycholesterol (24S-OHC) | Brain-derived; measurable in plasma | Reduced plasma levels of 24S-OHC correlate with disease progression, Alterations in 24S-OHC levels may help identify premanifest HD individuals |
| [44,108,109,110,111,112] | |
| Neurophysiological Tasks Biomarkers | Motor tapping | Speeded tapping tasks measuring the number and rhythm of taps within a set time frame. | Identifies subtle motor deficits in premanifest HD individuals, Tracks motor decline over time. |
| [113,114,115,116,117] |
| Speech biomarkers | Acoustic analysis of speech patterns, including articulation rate, pause duration, and prosody. | Tracks progression of speech impairments correlating with motor and cognitive decline, Associates speech changes with genetic markers like CAG repeat length. |
| [118,119,120,121,122,123] | |
| Event-related potentials (ERPs) | Measurement of brain’s electrical response to specific sensory, cognitive, or motor events using EEG | Cognitive function assessment, disease progression monitoring |
| [124,125,126,127,128,129,130] | |
| Electroencephalography (EEG) | Brain | Identifies neural activity alterations in premanifest and manifest HD stages. |
| [131,132,133,134] | |
| Imaging Biomarkers | MRI | Brain | Structural changes, disease progression |
| [135,136,137,138] |
| PET scan | Brain | Functional brain imaging, neurotransmitter activity |
| [139,140,141,142] | |
| Diffusion tensor imaging (DTI) | Brain | Early detection, disease progression monitoring |
| [143,144,145,146,147] |
3.2.1. Genomic Biomarkers
HTT Gene Mutations/CAG Repeat Expansion
3.2.2. Wet HD Biomarkers
Non-Coding RNA Biomarkers
MicroRNAs (miRNAs)
Protein Biomarkers
- Mutant Huntingtin (mHTT) Protein
Neurofilament Light Chain (NfL)
Brain-Derived Neurotrophic Factor (BDNF)
- Tau Protein
- Inflammatory Biomarkers
- Metabolic Biomarkers
- Uric Acid (UA)
- 24 (S) Hydroxycholesterol (24OHC)
3.2.3. Imaging Biomarkers
Magnetic Resonance Imaging (MRI)
Positron Emission Tomography (PET)
Diffusion Tensor Imaging (DTI)
3.2.4. Neuropsychological Tasks-Related HD Biomarkers
Motor Tapping
Speech Biomarker
Digital Biomarker
EEG and fMRI
Event Related Potentials (ERPs)
4. Discussion
5. Study Limitations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stoker, T.B.; Mason, S.L.; Greenland, J.C.; Holden, S.T.; Santini, H.; Barker, R.A. Huntington’s disease: Diagnosis and management. Pract. Neurol. 2022, 22, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Daldin, M.; Fodale, V.; Cariulo, C.; Azzollini, L.; Verani, M.; Martufi, P.; Spiezia, M.C.; Deguire, S.M.; Cherubini, M.; Macdonald, D.; et al. Polyglutamine expansion affects huntingtin conformation in multiple Huntington’s disease models. Sci. Rep. 2017, 7, 5070. [Google Scholar] [CrossRef]
- Medina, A.; Mahjoub, Y.; Shaver, L.; Pringsheim, T. Prevalence and Incidence of Huntington’s Disease: An Updated Systematic Review and Meta-Analysis. Mov. Disord. Off. J. Mov. Disord. Soc. 2022, 37, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Papanna, B.; Lazzari, C.; Rabottini, M. Huntington’s disease prevalence in Asia: A systematic review and meta-analysis. Riv. Psichiatr. 2024, 59, 4–12. [Google Scholar]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Rawlins, M.D.; Wexler, N.S.; Wexler, A.R.; Tabrizi, S.J.; Douglas, I.; Evans, S.J.; Smeeth, L. The Prevalence of Huntington’s Disease. Neuroepidemiology 2016, 46, 144–153. [Google Scholar] [CrossRef]
- Gillis, C.; Montenigro, P.; Nejati, M.; Maserejian, N. Estimating prevalence of early Alzheimer’s disease in the United States, accounting for racial and ethnic diversity. Alzheimer’s Dement. 2023, 19, 1841–1848. [Google Scholar] [CrossRef]
- Rodríguez-Santana, I.; Mestre, T.; Squitieri, F.; Willock, R.; Arnesen, A.; Clarke, A.; D’Alessio, B.; Fisher, A.; Fuller, R.; Hamilton, J.L.; et al. Economic burden of Huntington disease in Europe and the USA: Results from the Huntington’s Disease Burden of Illness study. Eur. J. Neurol. 2023, 30, 1109–1117. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Schobel, S.; Gantman, E.C.; Mansbach, A.; Borowsky, B.; Konstantinova, P.; Mestre, T.A.; Panagoulias, J.; Ross, C.A.; Zauderer, M.; et al. A biological classification of Huntington’s disease: The Integrated Staging System. Lancet Neurol. 2022, 21, 632–644. [Google Scholar] [CrossRef]
- MacLeod, R.; Tibben, A.; Frontali, M.; Evers-Kiebooms, G.; Jones, A.; Martinez-Descales, A.; Roos, R.A. Recommendations for the predictive genetic test in Huntington’s disease. Clin. Genet. 2013, 83, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Wild, E.; Tabrizi, S. Premanifest and Early Huntington’s Disease; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- Voysey, Z.J.; Owen, N.E.; Holbrook, J.A.; Malpetti, M.; Le Draoulec, C.; Spindler, L.R.B.; Goodman, A.O.G.; Lazar, A.S.; Barker, R.A. A 14-year longitudinal study of neurofilament light chain dynamics in premanifest and transitional Huntington’s disease. J. Neurol. 2024, 271, 7572–7582. [Google Scholar] [CrossRef]
- Paulsen, J.S.; Long, J.D.; Johnson, H.J.; Aylward, E.H.; Ross, C.A.; Williams, J.K.; Nance, M.A.; Erwin, C.J.; Westervelt, H.J.; Harrington, D.L.; et al. Clinical and Biomarker Changes in Premanifest Huntington Disease Show Trial Feasibility: A Decade of the PREDICT-HD Study. Front. Aging Neurosci. 2014, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Mehanna, R.; Jankovic, J. Systemic Symptoms in Huntington’s Disease: A Comprehensive Review. Mov. Disord. Clin. Pract. 2024, 11, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Ajitkumar; Jesus, D. Huntington Disease. Available online: https://www.ncbi.nlm.nih.gov/books/NBK559166/ (accessed on 19 January 2025).
- Roth, J. Clinical Symptomatology of Huntington’s Disease. In Pathology, Prevention and Therapeutics of Neurodegenerative Disease; Singh, S., Joshi, N., Eds.; Springer: Singapore, 2019; pp. 117–131. [Google Scholar]
- Shafie, A.; Ashour, A.A.; Anwar, S.; Anjum, F.; Hassan, M.I. Exploring molecular mechanisms, therapeutic strategies, and clinical manifestations of Huntington’s disease. Arch. Pharmacal Res. 2024, 47, 571–595. [Google Scholar] [CrossRef] [PubMed]
- Krause, A.; Anderson, D.G.; Ferreira-Correia, A.; Dawson, J.; Baine-Savanhu, F.; Li, P.P.; Margolis, R.L. Huntington disease-like 2: Insight into neurodegeneration from an African disease. Nat. Rev. Neurol. 2024, 20, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Assaye, M.A.; Gizaw, S.T. Chaperone-Mediated Autophagy and Its Implications for Neurodegeneration and Cancer. Int. J. Gen. Med. 2022, 15, 5635–5649. [Google Scholar] [CrossRef]
- Jodeiri Farshbaf, M.; Ghaedi, K. Huntington’s Disease and Mitochondria. Neurotox. Res. 2017, 32, 518–529. [Google Scholar] [CrossRef]
- Srinivasan, E.; Ram, V.; Rajasekaran, R. A Review On Huntington Protein: Insight Into Protein Aggregation and Therapeutic Interventions. Curr. Drug Metab. 2022, 23, 260–282. [Google Scholar] [CrossRef] [PubMed]
- Intihar, T.A.; Martinez, E.A.; Gomez-Pastor, R. Mitochondrial Dysfunction in Huntington’s Disease; Interplay Between HSF1, p53 and PGC-1α Transcription Factors. Front. Cell Neurosci. 2019, 13, 103. [Google Scholar] [CrossRef]
- Bañez-Coronel, M.; Porta, S.; Kagerbauer, B.; Mateu-Huertas, E.; Pantano, L.; Ferrer, I.; Guzmán, M.; Estivill, X.; Martí, E. A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet. 2012, 8, e1002481. [Google Scholar] [CrossRef]
- Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium, CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset. Cell 2019, 178, 887–900.e14. [CrossRef]
- Wright, G.E.B.; Collins, J.A.; Kay, C.; McDonald, C.; Dolzhenko, E.; Xia, Q.; Bečanović, K.; Drögemöller, B.I.; Semaka, A.; Nguyen, C.M.; et al. Length of Uninterrupted CAG, Independent of Polyglutamine Size, Results in Increased Somatic Instability, Hastening Onset of Huntington Disease. Am. J. Hum. Genet. 2019, 104, 1116–1126. [Google Scholar] [CrossRef]
- Gu, X.; Richman, J.; Langfelder, P.; Wang, N.; Zhang, S.; Bañez-Coronel, M.; Wang, H.B.; Yang, L.; Ramanathan, L.; Deng, L.; et al. Uninterrupted CAG repeat drives striatum-selective transcriptionopathy and nuclear pathogenesis in human Huntingtin BAC mice. Neuron 2022, 110, 1173–1192.e7. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, M.; Sciandra, F. Molecular Mechanisms Underlying Muscle Wasting in Huntington’s Disease. Int. J. Mol. Sci. 2020, 21, 8314. [Google Scholar] [CrossRef] [PubMed]
- Andrich, J.; Schmitz, T.; Saft, C.; Postert, T.; Kraus, P.; Epplen, J.T.; Przuntek, H.; Agelink, M.W. Autonomic nervous system function in Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2002, 72, 726–731. [Google Scholar] [CrossRef]
- Carroll, J.B.; Bates, G.P.; Steffan, J.; Saft, C.; Tabrizi, S.J. Treating the whole body in Huntington’s disease. Lancet Neurol. 2015, 14, 1135–1142. [Google Scholar] [CrossRef]
- Rook, M.E.; Southwell, A.L. Antisense Oligonucleotide Therapy: From Design to the Huntington Disease Clinic. BioDrugs 2022, 36, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Morelli, K.H.; Wu, Q.; Gosztyla, M.L.; Liu, H.; Yao, M.; Zhang, C.; Chen, J.; Marina, R.J.; Lee, K.; Jones, K.L.; et al. An RNA-targeting CRISPR–Cas13d system alleviates disease-related phenotypes in Huntington’s disease models. Nat. Neurosci. 2023, 26, 27–38. [Google Scholar] [CrossRef]
- Boak, L.; McColgan, P. Understanding the treatment and post-treatment effects of tominersen in the Phase III GENERATION HD1 study. In Proceedings of the CHDI Foundation Annual Therapeutics Conference, Palm Springs, CA, USA, 28 February–3 March 2022. [Google Scholar]
- Sledzinski, P.; Nowaczyk, M.; Smielowska, M.I.; Olejniczak, M. CRISPR/Cas9-induced double-strand breaks in the huntingtin locus lead to CAG repeat contraction through DNA end resection and homology-mediated repair. BMC Biol. 2024, 22, 282. [Google Scholar] [CrossRef]
- Saft, C.; Burgunder, J.M.; Dose, M.; Jung, H.H.; Katzenschlager, R.; Priller, J.; Nguyen, H.P.; Reetz, K.; Reilmann, R.; Seppi, K.; et al. Symptomatic treatment options for Huntington’s disease (guidelines of the German Neurological Society). Neurol. Res. Pract. 2023, 5, 61. [Google Scholar] [CrossRef]
- Sampaio, C. Huntington disease—Update on ongoing therapeutic developments and a look toward the future. Park. Relat. Disord. 2024, 122, 106049. [Google Scholar] [CrossRef]
- Tippett, L.J.; Waldvogel, H.J.; Snell, R.G.; Vonsattel, J.P.; Young, A.B.; Faull, R.L.M. The Complexity of Clinical Huntington’s Disease: Developments in Molecular Genetics, Neuropathology and Neuroimaging Biomarkers. Adv. Neurobiol. 2017, 15, 129–161. [Google Scholar]
- Martí-Martínez, S.; Valor, L.M. A Glimpse of Molecular Biomarkers in Huntington’s Disease. Int. J. Mol. Sci. 2022, 23, 5411. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-M.; Wheeler, V.C.; Chao, M.J.; Vonsattel, J.P.G.; Pinto, R.M.; Lucente, D.; Abu-Elneel, K.; Ramos, E.M.; Mysore, J.S.; Gillis, T.J.C. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 2015, 162, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Mathews, E.W.; Coffey, S.R.; Gaertner, A.; Belgrad, J.; Bragg, R.M.; O’Reilly, D.; Cantle, J.P.; McHugh, C.; Summers, A.; Fentz, J.B. Suppression of Huntington’s Disease Somatic Instability by Transcriptional Repression and Direct CAG Repeat Binding. bioRxiv 2024. [Google Scholar]
- Lee, J.-M.; Huang, Y.; Orth, M.; Gillis, T.; Siciliano, J.; Hong, E.; Mysore, J.S.; Lucente, D.; Wheeler, V.C.; Seong, I.S. Genetic modifiers of Huntington disease differentially influence motor and cognitive domains. Am. J. Hum. Genet. 2022, 109, 885–899. [Google Scholar] [CrossRef]
- Mouro Pinto, R.; Arning, L.; Giordano, J.V.; Razghandi, P.; Andrew, M.A.; Gillis, T.; Correia, K.; Mysore, J.S.; Grote Urtubey, D.-M.; Parwez, C.R.J. Patterns of CAG repeat instability in the central nervous system and periphery in Huntington’s disease and in spinocerebellar ataxia type 1. Hum. Mol. Genet. 2020, 29, 2551–2567. [Google Scholar] [CrossRef]
- Moss, D.J.H.; Pardiñas, A.F.; Langbehn, D.; Lo, K.; Leavitt, B.R.; Roos, R.; Durr, A.; Mead, S.; Holmans, P.; Jones, L.; et al. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. 2017, 16, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cheng, Y.; Shang, H. The updated development of blood-based biomarkers for Huntington’s disease. J. Neurol. 2023, 270, 2483–2503. [Google Scholar] [CrossRef]
- Wei, W.; Jiang, Z. A bibliometrix-based visualization analysis of international studies on conversations of people with aphasia: Present and prospects. Heliyon 2023, 9, e16839. [Google Scholar] [CrossRef] [PubMed]
- Campra, M.; Riva, P.; Oricchio, G.; Brescia, V. Bibliometrix analysis of medical tourism. Health Serv. Manag. Res. 2022, 35, 172–188. [Google Scholar] [CrossRef]
- Wu, J.; Möhle, L.; Brüning, T.; Eiriz, I.; Rafehi, M.; Stefan, K.; Stefan, S.M.; Pahnke, J. A Novel Huntington’s Disease Assessment Platform to Support Future Drug Discovery and Development. Int. J. Mol. Sci. 2022, 23, 14763. [Google Scholar] [CrossRef] [PubMed]
- Koval, I.; Dighiero-Brecht, T.; Tobin, A.J.; Tabrizi, S.J.; Scahill, R.I.; Tezenas du Montcel, S.; Durrleman, S.; Durr, A. Forecasting individual progression trajectories in Huntington disease enables more powered clinical trials. Sci. Rep. 2022, 12, 18928. [Google Scholar] [CrossRef]
- Tang, H.; van Eimeren, T.; Sampaio, C.; Mestre, T.A. Validation of biomarkers in Huntington disease to support the development of disease-modifying therapies: A systematic review and critical appraisal scheme. Park. Relat. Disord. 2021, 93, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Przybyl, L.; Wozna-Wysocka, M.; Kozlowska, E.; Fiszer, A. What, When and How to Measure—Peripheral Biomarkers in Therapy of Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 1561. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.B.; Byrne, L.M.; Lowe, A.J.; Tortelli, R.; Heins, M.; Flik, G.; Johnson, E.B.; De Vita, E.; Scahill, R.I.; Giorgini, F.; et al. Kynurenine pathway metabolites in cerebrospinal fluid and blood as potential biomarkers in Huntington’s disease. J. Neurochem. 2021, 158, 539–553. [Google Scholar] [CrossRef]
- Scarabino, D.; Veneziano, L.; Peconi, M.; Frontali, M.; Mantuano, E.; Corbo, R.M. Leukocyte telomere shortening in Huntington’s disease. J. Neurol. Sci. 2019, 396, 25–29. [Google Scholar] [CrossRef]
- Barschke, P.; Abu-Rumeileh, S.; Al Shweiki, M.H.D.R.; Barba, L.; Paolini Paoletti, F.; Oeckl, P.; Steinacker, P.; Halbgebauer, S.; Gaetani, L.; Lewerenz, J.; et al. Cerebrospinal fluid levels of proenkephalin and prodynorphin are differentially altered in Huntington’s and Parkinson’s disease. J. Neurol. 2022, 269, 5136–5143. [Google Scholar] [CrossRef]
- Castaldo, I.; De Rosa, M.; Romano, A.; Zuchegna, C.; Squitieri, F.; Mechelli, R.; Peluso, S.; Borrelli, C.; Del Mondo, A.; Salvatore, E.; et al. DNA damage signatures in peripheral blood cells as biomarkers in prodromal huntington disease. Ann. Neurol. 2019, 85, 296–301. [Google Scholar] [CrossRef]
- Kalliolia, E.; Silajdžić, E.; Nambron, R.; Hill, N.R.; Doshi, A.; Frost, C.; Watt, H.; Hindmarsh, P.; Björkqvist, M.; Warner, T.T. Plasma melatonin is reduced in Huntington’s disease. Mov. Disord. 2014, 29, 1511–1515. [Google Scholar] [CrossRef]
- Caron, N.S.; Haqqani, A.S.; Sandhu, A.; Aly, A.E.; Findlay Black, H.; Bone, J.N.; McBride, J.L.; Abulrob, A.; Stanimirovic, D.; Leavitt, B.R.; et al. Cerebrospinal fluid biomarkers for assessing Huntington disease onset and severity. Brain Commun. 2022, 4, fcac309s. [Google Scholar] [CrossRef] [PubMed]
- Niemela, V.; Landtblom, A.M.; Nyholm, D.; Kneider, M.; Constantinescu, R.; Paucar, M.; Svenningsson, P.; Abujrais, S.; Burman, J.; Shevchenko, G.; et al. Proenkephalin Decreases in Cerebrospinal Fluid with Symptom Progression of Huntington’s Disease. Mov. Disord. 2021, 36, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Al Shweiki, M.H.D.R.; Oeckl, P.; Pachollek, A.; Steinacker, P.; Barschke, P.; Halbgebauer, S.; Anderl-Straub, S.; Lewerenz, J.; Ludolph, A.C.; Bernhard Landwehrmeyer, G.; et al. Cerebrospinal Fluid Levels of Prodynorphin-Derived Peptides are Decreased in Huntington’s Disease. Mov. Disord. 2021, 36, 492–497. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Wu, T.; Du, G.; Huang, Y.; Zeng, Y.; Lin, L.; Chen, D.; Wu, C.; Li, X.; Burgunder, J.M.; et al. Evaluation of Blood Glial Fibrillary Acidic Protein as a Potential Marker in Huntington’s Disease. Front. Neurol. 2021, 12, 779890. [Google Scholar] [CrossRef]
- Rosseto, S.M.; Alarcon, T.A.; Rocha, D.M.C.; Ribeiro, F.M.; Ferguson, S.S.G.; Martins-Silva, C.; Muniz, M.R.; Costa, P.F.; Guimarães, D.A.; Pires, R.G.W. DYNLT1 gene expression is downregulated in whole blood of patients at different Huntington’s disease stages. Neurol. Sci. 2021, 42, 1963–1967. [Google Scholar] [CrossRef]
- Rosas, H.D.; Doros, G.; Bhasin, S.; Thomas, B.; Gevorkian, S.; Malarick, K.; Matson, W.; Hersch, S.M. A systems-level “misunderstanding”: The plasma metabolome in Huntington’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 756–768. [Google Scholar] [CrossRef]
- Kong, G.; Cao, K.-A.L.; Judd, L.M.; Li, S.; Renoir, T.; Hannan, A.J. Microbiome profiling reveals gut dysbiosis in a transgenic mouse model of Huntington’s disease. Neurobiol. Dis. 2020, 135, 104268. [Google Scholar] [CrossRef]
- Borowsky, B.; Warner, J.; Leavitt, B.R.; Tabrizi, S.J.; Roos, R.A.; Durr, A.; Becker, C.; Sampaio, C.; Tobin, A.J.; Schulman, H. 8OHdG is not a biomarker for Huntington disease state or progression. Neurology 2013, 80, 1934–1941. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-M.; Ramos, E.; Lee, J.-H.; Gillis, T.; Mysore, J.; Hayden, M.; Warby, S.; Morrison, P.; Nance, M.; Ross, C.J. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology 2012, 78, 690–695. [Google Scholar] [CrossRef]
- Craufurd, D.; MacLeod, R.; Frontali, M.; Quarrell, O.; Bijlsma, E.K.; Davis, M.; Hjermind, L.E.; Lahiri, N.; Mandich, P.; Martinez, A.; et al. Diagnostic genetic testing for Huntington’s disease. Pract. Neurol. 2015, 15, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Zubkova, A.E.; Yudkin, D.V. Regulation of HTT mRNA Biogenesis: The Norm and Pathology. Int. J. Mol. Sci. 2024, 25, 11493. [Google Scholar] [CrossRef]
- Faquih, T.O.; Aziz, N.A.; Gardiner, S.L.; Li-Gao, R.; de Mutsert, R.; Milaneschi, Y.; Trompet, S.; Jukema, J.W.; Rosendaal, F.R.; van Hylckama Vlieg, A.; et al. Normal range CAG repeat size variations in the HTT gene are associated with an adverse lipoprotein profile partially mediated by body mass index. Hum. Mol. Genet. 2023, 32, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, E.; Scalzo, D.; Zobel, M.; Iennaco, R.; Maffezzini, C.; Besusso, D.; Maestri, S. When repetita no-longer iuvant: Somatic instability of the CAG triplet in Huntington’s disease. Nucleic Acids Res. 2024, 53, gkae1204. [Google Scholar] [CrossRef] [PubMed]
- Eshraghi, M.; Karunadharma, P.P.; Blin, J.; Shahani, N.; Ricci, E.P.; Michel, A.; Urban, N.T.; Galli, N.; Sharma, M.; Ramírez-Jarquín, U.N.; et al. Mutant Huntingtin stalls ribosomes and represses protein synthesis in a cellular model of Huntington disease. Nat. Commun. 2021, 12, 1461. [Google Scholar] [CrossRef]
- Agnello, L.; Gambino, C.M.; Ciaccio, A.M.; Masucci, A.; Vassallo, R.; Tamburello, M.; Scazzone, C.; Lo Sasso, B.; Ciaccio, M. Molecular Biomarkers of Neurodegenerative Disorders: A Practical Guide to Their Appropriate Use and Interpretation in Clinical Practice. Int. J. Mol. Sci. 2024, 25, 4323. [Google Scholar] [CrossRef]
- Byrne, L.M.; Rodrigues, F.B.; Johnson, E.B.; Wijeratne, P.A.; De Vita, E.; Alexander, D.C.; Palermo, G.; Czech, C.; Schobel, S.; Scahill, R.I.; et al. Evaluation of mutant huntingtin and neurofilament proteins as potential markers in Huntington’s disease. Sci. Transl. Med. 2018, 10, eaat7108. [Google Scholar] [CrossRef] [PubMed]
- Wild, E.J.; Boggio, R.; Langbehn, D.; Robertson, N.; Haider, S.; Miller, J.R.; Zetterberg, H.; Leavitt, B.R.; Kuhn, R.; Tabrizi, S.J.; et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington’s disease patients. J. Clin. Investig. 2015, 125, 1979–1986. [Google Scholar] [CrossRef]
- Southwell, A.L.; Smith, S.E.; Davis, T.R.; Caron, N.S.; Villanueva, E.B.; Xie, Y.; Collins, J.A.; Ye, M.L.; Sturrock, A.; Leavitt, B.R.; et al. Ultrasensitive measurement of huntingtin protein in cerebrospinal fluid demonstrates increase with Huntington disease stage and decrease following brain huntingtin suppression. Sci. Rep. 2015, 5, 12166. [Google Scholar] [CrossRef] [PubMed]
- Hensman Moss, D.J.; Robertson, N.; Farmer, R.; Scahill, R.I.; Haider, S.; Tessari, M.A.; Flynn, G.; Fischer, D.F.; Wild, E.J.; Macdonald, D.; et al. Quantification of huntingtin protein species in Huntington’s disease patient leukocytes using optimised electrochemiluminescence immunoassays. PLoS ONE 2017, 12, e0189891. [Google Scholar] [CrossRef]
- Kuhle, J.; Barro, C.; Andreasson, U.; Derfuss, T.; Lindberg, R.; Sandelius, Å.; Liman, V.; Norgren, N.; Blennow, K.; Zetterberg, H. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: ELISA, electrochemiluminescence immunoassay and Simoa. Clin. Chem. Lab. Med. 2016, 54, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, L.; Blennow, K.; Calabresi, P.; Di Filippo, M.; Parnetti, L.; Zetterberg, H. Neurofilament light chain as a biomarker in neurological disorders. J. Neurol. Neurosurg. Psychiatry 2019, 90, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.B.; Byrne, L.M.; Wild, E.J. Biofluid Biomarkers in Huntington’s Disease. Methods Mol. Biol. 2018, 1780, 329–396. [Google Scholar]
- Schumacher-Schuh, A.; Bieger, A.; Borelli, W.V.; Portley, M.K.; Awad, P.S.; Bandres-Ciga, S. Advances in Proteomic and Metabolomic Profiling of Neurodegenerative Diseases. Front. Neurol. 2021, 12, 792227. [Google Scholar] [CrossRef] [PubMed]
- Byrne, L.M.; Rodrigues, F.B.; Blennow, K.; Durr, A.; Leavitt, B.R.; Roos, R.A.C.; Scahill, R.I.; Tabrizi, S.J.; Zetterberg, H.; Langbehn, D.; et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington’s disease: A retrospective cohort analysis. Lancet Neurol. 2017, 16, 601–609. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Byrne, L.M.; Tortelli, R.; Johnson, E.B.; Wijeratne, P.A.; Arridge, M.; De Vita, E.; Ghazaleh, N.; Houghton, R.; Furby, H.; et al. Mutant huntingtin and neurofilament light have distinct longitudinal dynamics in Huntington’s disease. Sci. Transl. Med. 2020, 12, eabc2888s. [Google Scholar] [CrossRef]
- Zeun, P.; Scahill, R.I.; Tabrizi, S.J.; Wild, E.J. Fluid and imaging biomarkers for Huntington’s disease. Mol. Cell. Neurosci. 2019, 97, 67–80. [Google Scholar] [CrossRef]
- Morena, E.; Romano, C.; Marconi, M.; Diamant, S.; Buscarinu, M.C.; Bellucci, G.; Romano, S.; Scarabino, D.; Salvetti, M.; Ristori, G. Peripheral Biomarkers in Manifest and Premanifest Huntington’s Disease. Int. J. Mol. Sci. 2023, 24, 6051. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Marullo, M.; Vitali, B.; Tarditi, A.; Mariotti, C.; Valenza, M.; Lahiri, N.; Wild, E.J.; Sassone, J.; Ciammola, A.; et al. Brain-Derived Neurotrophic Factor in Patients with Huntington’s Disease. PLoS ONE 2011, 6, e22966. [Google Scholar] [CrossRef]
- Baydyuk, M.; Xu, B. BDNF in Huntington’s disease: Role in pathogenesis and treatment. In Huntington’s Disease-Core Concepts and Current Advances; Intechopen: Rijeka, Croatia, 2012. [Google Scholar]
- Gutierrez, A.; Corey-Bloom, J.; Thomas, E.A.; Desplats, P. Evaluation of Biochemical and Epigenetic Measures of Peripheral Brain-Derived Neurotrophic Factor (BDNF) as a Biomarker in Huntington’s Disease Patients. Front. Mol. Neurosci. 2019, 12, 335. [Google Scholar] [CrossRef]
- Ciammola, A.; Sassone, J.; Cannella, M.; Calza, S.; Poletti, B.; Frati, L.; Squitieri, F.; Silani, V. Low brain-derived neurotrophic factor (BDNF) levels in serum of Huntington’s disease patients. Am. J. Med. Genet. Part. B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2007, 144b, 574–577. [Google Scholar] [CrossRef]
- Tasset, I.; Sánchez-López, F.; Agüera, E.; Fernández-Bolaños, R.; Sánchez, F.M.; Cruz-Guerrero, A.; Gascón-Luna, F.; Túnez, I. NGF and nitrosative stress in patients with Huntington’s disease. J. Neurol. Sci. 2012, 315, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ross, C.A.; Cai, H.; Cong, W.N.; Daimon, C.M.; Carlson, O.D.; Egan, J.M.; Siddiqui, S.; Maudsley, S.; Martin, B. Metabolic and hormonal signatures in pre-manifest and manifest Huntington’s disease patients. Front. Physiol. 2014, 5, 231. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.A.; Byrne, L.M.; Rodrigues, F.B.; Tortelli, R.; Johnson, E.B.; Foiani, M.S.; Arridge, M.; De Vita, E.; Scahill, R.I.; Heslegrave, A.; et al. Brain-derived neurotrophic factor in cerebrospinal fluid and plasma is not a biomarker for Huntington’s disease. Sci. Rep. 2021, 11, 3481. [Google Scholar] [CrossRef] [PubMed]
- Niemelä, V.; Landtblom, A.-M.; Blennow, K.; Sundblom, J. Tau or neurofilament light—Which is the more suitable biomarker for Huntington’s disease? PLoS ONE 2017, 12, e0172762. [Google Scholar] [CrossRef]
- Vuono, R.; Winder-Rhodes, S.; de Silva, R.; Cisbani, G.; Drouin-Ouellet, J.; Spillantini, M.G.; Cicchetti, F.; Barker, R.A. The role of tau in the pathological process and clinical expression of Huntington’s disease. Brain A J. Neurol. 2015, 138 Pt 7, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Barrio, I.; Vázquez-Oliver, A.; Puig-Davi, A.; Rivas-Asensio, E.; Perez-Perez, J.; Fernandez-Vizuete, C.; Horta-Barba, A.; Olmedo-Saura, G.; Salvat-Rovira, N.; Sampedro, F.J. Skin Tau Quantification as a Novel Biomarker in Huntington’s Disease. Mov. Disord. 2024, 39, 2067–2074. [Google Scholar] [CrossRef] [PubMed]
- Lepinay, E.; Cicchetti, F. Tau: A biomarker of Huntington’s disease. Mol. Psychiatry 2023, 28, 4070–4083. [Google Scholar] [CrossRef]
- Dalrymple, A.; Wild, E.J.; Joubert, R.; Sathasivam, K.; Björkqvist, M.; Petersén, A.; Jackson, G.S.; Isaacs, J.D.; Kristiansen, M.; Bates, G.P.; et al. Proteomic profiling of plasma in Huntington’s disease reveals neuroinflammatory activation and biomarker candidates. J. Proteome Res. 2007, 6, 2833–2840. [Google Scholar] [CrossRef]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Eide, S.; Misztal, M.; Feng, Z.P. Interleukin-6 as a marker of Huntington’s disease progression: Systematic review and meta-analysis. Brain Behav. Immun.-Health 2023, 30, 100635. [Google Scholar] [CrossRef]
- Wild, E.; Magnusson, A.; Lahiri, N.; Krus, U.; Orth, M.; Tabrizi, S.J.; Björkqvist, M. Abnormal peripheral chemokine profile in Huntington’s disease. PLoS Curr. 2011, 3, Rrn1231. [Google Scholar] [CrossRef] [PubMed]
- Walter, N.G. Are non-protein coding RNAs junk or treasure? BioEssays 2024, 46, 2300201. [Google Scholar] [CrossRef] [PubMed]
- Juźwik, C.A.; S, S.D.; Zhang, Y.; Paradis-Isler, N.; Sylvester, A.; Amar-Zifkin, A.; Douglas, C.; Morquette, B.; Moore, C.S.; Fournier, A.E. microRNA dysregulation in neurodegenerative diseases: A systematic review. Prog. Neurobiol. 2019, 182, 101664. [Google Scholar] [CrossRef] [PubMed]
- Azam, H.M.H.; Rößling, R.I.; Geithe, C.; Khan, M.M.; Dinter, F.; Hanack, K.; Prüß, H.; Husse, B.; Roggenbuck, D.; Schierack, P.; et al. MicroRNA biomarkers as next-generation diagnostic tools for neurodegenerative diseases: A comprehensive review. Front. Mol. Neurosci. 2024, 17, 1386735. [Google Scholar] [CrossRef] [PubMed]
- Reed, E.R.; Latourelle, J.C.; Bockholt, J.H.; Bregu, J.; Smock, J.; Paulsen, J.S.; Myers, R.H. MicroRNAs in CSF as prodromal biomarkers for Huntington disease in the PREDICT-HD study. Neurology 2018, 90, e264–e272. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Gao, F.; Wang, N.; Howland, D.; Kwak, S.; Vogt, T.F.; Aaronson, J.S.; Rosinski, J.; Coppola, G.; Horvath, S.; et al. MicroRNA signatures of endogenous Huntingtin CAG repeat expansion in mice. PLoS ONE 2018, 13, e0190550. [Google Scholar] [CrossRef] [PubMed]
- Palaiogeorgou, A.M.; Papakonstantinou, E.; Golfinopoulou, R.; Sigala, M.; Mitsis, T.; Papageorgiou, L.; Diakou, I.; Pierouli, K.; Dragoumani, K.; Spandidos, D.A.; et al. Recent approaches on Huntington’s disease (Review). Biomed. Rep. 2023, 18, 5. [Google Scholar] [CrossRef]
- Fang, P.; Li, X.; Luo, J.J.; Wang, H.; Yang, X.F. A Double-edged Sword: Uric Acid and Neurological Disorders. Brain Disord. Ther. 2013, 2, 109. [Google Scholar] [PubMed]
- Wen, M.; Zhou, B.; Chen, Y.H.; Ma, Z.L.; Gou, Y.; Zhang, C.L.; Yu, W.F.; Jiao, L. Serum uric acid levels in patients with Parkinson’s disease: A meta-analysis. PLoS ONE 2017, 12, e0173731. [Google Scholar] [CrossRef] [PubMed]
- Auinger, P.; Kieburtz, K.; McDermott, M.P. The relationship between uric acid levels and Huntington’s disease progression. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, 224–228. [Google Scholar] [CrossRef]
- Corey-Bloom, J.; Haque, A.; Aboufadel, S.; Snell, C.; Fischer, R.S.; Granger, S.W.; Granger, D.A.; Thomas, E.A. Uric Acid as a Potential Peripheral Biomarker for Disease Features in Huntington’s Patients. Front. Neurosci. 2020, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Sodero, A.O. 24S-hydroxycholesterol: Cellular effects and variations in brain diseases. J. Neurochem. 2021, 157, 899–918. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Long, J.D.; Mills, J.A.; Di Donato, S.; Paulsen, J.S. Plasma 24S-hydroxycholesterol correlation with markers of Huntington disease progression. Neurobiol. Dis. 2013, 55, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Fichera, M.; Nanetti, L.; Favagrossa, M.; Castaldo, A.; Nigri, A.; Mongelli, A.; Marchini, G.; Gellera, C.; Grisoli, M.; Birolini, G. D14 Plasma Levels of 24S-Hydroxycholesterol Are Reduced in Huntington Disease Subjects: Preliminary Results of a 2-Year Longitudinal Study; BMJ Publishing Group Ltd.: London, UK, 2022. [Google Scholar]
- Leoni, V.; Mariotti, C.; Tabrizi, S.J.; Valenza, M.; Wild, E.J.; Henley, S.M.; Hobbs, N.Z.; Mandelli, M.L.; Grisoli, M.; Björkhem, I.J. Plasma 24S-hydroxycholesterol and caudate MRI in pre-manifest and early Huntington’s disease. Brain 2008, 131, 2851–2859. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Mariotti, C.; Nanetti, L.; Salvatore, E.; Squitieri, F.; Bentivoglio, A.R.; Bandettini di Poggio, M.; Piacentini, S.; Monza, D.; Valenza, M.; et al. Whole body cholesterol metabolism is impaired in Huntington’s disease. Neurosci. Lett. 2011, 494, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Michell, A.W.; Goodman, A.O.; Silva, A.H.; Lazic, S.E.; Morton, A.J.; Barker, R.A. Hand tapping: A simple, reproducible, objective marker of motor dysfunction in Huntington’s disease. J. Neurol. 2008, 255, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, N.; Scahill, R.I.; Rosas, H.D.; Acharya, T.; van den Bogaard, S.J.; Jauffret, C.; Say, M.J.; Sturrock, A.; Johnson, H.; Onorato, C.E.; et al. Tapping linked to function and structure in premanifest and symptomatic Huntington disease. Neurology 2010, 75, 2150–2160. [Google Scholar] [CrossRef]
- Collins, L.M.; Lazic, S.E.; Barker, R.A. A retrospective analysis of hand tapping as a longitudinal marker of disease progression in Huntington’s disease. BMC Neurol. 2014, 14, 35. [Google Scholar] [CrossRef]
- Mestre, T.A.; Forjaz, M.J.; Mahlknecht, P.; Cardoso, F.; Ferreira, J.J.; Reilmann, R.; Sampaio, C.; Goetz, C.G.; Cubo, E.; Martinez-Martin, P.; et al. Rating Scales for Motor Symptoms and Signs in Huntington’s Disease: Critique and Recommendations. Mov. Disord. Clin. Pract. 2018, 5, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.L.; Waddell, E.M.; Chunga, N.; Quinn, L. Digital Measures in Huntington’s Disease. In Biomarkers for Huntington’s Disease: Improving Clinical Outcomes; Thomas, E.A., Parkin, G.M., Eds.; Springer International Publishing: Cham, Switzerland, 2023; pp. 433–457. [Google Scholar]
- Nunes, A.S.; Pawlik, M.; Mishra, R.K.; Waddell, E.; Coffey, M.; Tarolli, C.G.; Schneider, R.B.; Dorsey, E.R.; Vaziri, A.; Adams, J.L. Digital assessment of speech in Huntington disease. Front. Neurol. 2024, 15, 1310548. [Google Scholar] [CrossRef]
- Saft, C.; Jessen, J.; Hoffmann, R.; Lukas, C.; Skodda, S. Speech Biomarkers in Huntington’s Disease: A Longitudinal Follow-Up Study in Premanifest Mutation Carriers. J. Huntington’s Dis. 2024, 13, 369–373. [Google Scholar] [CrossRef]
- Kouba, T.; Frank, W.; Tykalova, T.; Mühlbäck, A.; Klempíř, J.; Lindenberg, K.S.; Landwehrmeyer, G.B.; Rusz, J. Speech biomarkers in Huntington’s disease: A cross-sectional study in pre-symptomatic, prodromal and early manifest stages. Eur. J. Neurol. 2023, 30, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.C.S.; Stout, J.C.; Vogel, A.P. Speech in prodromal and symptomatic Huntington’s disease as a model of measuring onset and progression in dominantly inherited neurodegenerative diseases. Neurosci. Biobehav. Rev. 2019, 107, 450–460. [Google Scholar] [CrossRef]
- Fahed, V.S.; Doheny, E.P.; Collazo, C.; Krzysztofik, J.; Mann, E.; Morgan-Jones, P.; Mills, L.; Drew, C.; Rosser, A.E.; Cousins, R. Language-independent acoustic biomarkers for quantifying speech impairment in Huntington’s Disease. Am. J. Speech-Lang. Pathol. 2024, 33, 1390–1405. [Google Scholar] [CrossRef] [PubMed]
- Skodda, S.; Grönheit, W.; Lukas, C.; Bellenberg, B.; von Hein, S.M.; Hoffmann, R.; Saft, C. Two different phenomena in basic motor speech performance in premanifest Huntington disease. Neurology 2016, 86, 1329–1335. [Google Scholar] [CrossRef]
- Josiassen, R.C.; Shagass, C.; Roemer, R.A.; Mancall, E. A sensory evoked potential comparison of persons ’at risk’ for Huntington’s disease and hospitalized neurotic patients. Int. J. Psychophysiol. Off. J. Int. Organ. Psychophysiol. 1988, 6, 281–289. [Google Scholar] [CrossRef]
- Sokhadze, E.M.; Casanova, M.F.; Casanova, E.L.; Lamina, E.; Kelly, D.P.; Khachidze, I. Event-related potentials (ERP) in cognitive neuroscience research and applications. NeuroRegulation 2017, 4, 14. [Google Scholar] [CrossRef]
- Kaan, E. Event-related potentials and language processing: A brief overview. Lang. Linguist. Compass 2007, 1, 571–591. [Google Scholar] [CrossRef]
- Hömberg, V.; Hefter, H.; Granseyer, G.; Strauss, W.; Lange, H.; Hennerici, M. Event-related potentials in patients with Huntington’s disease and relatives at risk in relation to detailed psychometry. Electroencephalogr. Clin. Neurophysiol. 1986, 63, 552–569. [Google Scholar] [CrossRef] [PubMed]
- Beste, C.; Saft, C.; Güntürkün, O.; Falkenstein, M. Increased cognitive functioning in symptomatic Huntington’s disease as revealed by behavioral and event-related potential indices of auditory sensory memory and attention. J. Neurosci. 2008, 28, 11695–11702. [Google Scholar] [CrossRef] [PubMed]
- Beste, C.; Saft, C.; Konrad, C.; Andrich, J.; Habbel, A.; Schepers, I.; Jansen, A.; Pfleiderer, B.; Falkenstein, M. Levels of error processing in Huntington’s disease: A combined study using event-related potentials and voxel-based morphometry. Hum. Brain Mapp. 2008, 29, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Beste, C.; Ness, V.; Falkenstein, M.; Saft, C. On the role of fronto-striatal neural synchronization processes for response inhibition—Evidence from ERP phase-synchronization analyses in pre-manifest Huntington’s disease gene mutation carriers. Neuropsychologia 2011, 49, 3484–3493. [Google Scholar] [CrossRef] [PubMed]
- Ponomareva, N.V.; Klyushnikov, S.A.; Abramycheva, N.; Konovalov, R.N.; Krotenkova, M.; Kolesnikova, E.; Malina, D.; Urazgildeeva, G.; Kanavets, E.; Mitrofanov, A.; et al. Neurophysiological hallmarks of Huntington’s disease progression: An EEG and fMRI connectivity study. Front. Aging Neurosci. 2023, 15, 1270226. [Google Scholar] [CrossRef] [PubMed]
- Odish, O.F.; Johnsen, K.; van Someren, P.; Roos, R.A.; van Dijk, J.G. EEG may serve as a biomarker in Huntington’s disease using machine learning automatic classification. Sci. Rep. 2018, 8, 16090. [Google Scholar] [CrossRef]
- Thomas, E.A. The Utility of Biomarkers for Huntington’s Disease. In Biomarkers for Huntington’s Disease: Improving Clinical Outcomes; Thomas, E.A., Parkin, G.M., Eds.; Springer International Publishing: Cham, Switzerland, 2023; pp. 3–15. [Google Scholar]
- Maddury, S. The performance of domain-based feature extraction on EEG, ECG, and fNIRS for Huntington’s disease diagnosis via shallow machine learning. Front. Signal Process. 2024, 4, 1321861. [Google Scholar] [CrossRef]
- Coppen, E.M.; van der Grond, J.; Hafkemeijer, A.; Rombouts, S.A.; Roos, R.A. Early grey matter changes in structural covariance networks in Huntington’s disease. NeuroImage. Clin. 2016, 12, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Király, A.; Kincses, Z.T.; Szabó, N.; Tóth, E.; Csete, G.; Faragó, P.; Vécsei, L. Gray matter atrophy in presymptomatic Huntington’s patients. Ideggyogy. Szle. 2016, 69, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.; Dervenoulas, G.; Politis, M. Structural magnetic resonance imaging in Huntington’s disease. Int. Rev. Neurobiol. 2018, 142, 335–380. [Google Scholar] [PubMed]
- Reiner, A.; Dragatsis, I.; Dietrich, P. Genetics and neuropathology of Huntington’s disease. Int. Rev. Neurobiol. 2011, 98, 325–372. [Google Scholar]
- Young, A.B.; Penney, J.B.; Starosta-Rubinstein, S.; Markel, D.S.; Berent, S.; Giordani, B.; Ehrenkaufer, R.; Jewett, D.; Hichwa, R. PET scan investigations of Huntington’s disease: Cerebral metabolic correlates of neurological features and functional decline. Ann. Neurol. 1986, 20, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Herben-Dekker, M.; van Oostrom, J.C.; Roos, R.A.; Jurgens, C.K.; Witjes-Ané, M.N.; Kremer, H.P.; Leenders, K.L.; Spikman, J.M. Striatal metabolism and psychomotor speed as predictors of motor onset in Huntington’s disease. J. Neurol. 2014, 261, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Kuwert, T.; Lange, H.W.; Boecker, H.; Titz, H.; Herzog, H.; Aulich, A.; Wang, B.C.; Nayak, U.; Feinendegen, L.E. Striatal glucose consumption in chorea-free subjects at risk of Huntington’s disease. J. Neurol. 1993, 241, 31–36. [Google Scholar] [CrossRef]
- Antonini, A.; Leenders, K.L.; Spiegel, R.; Meier, D.; Vontobel, P.; Weigell-Weber, M.; Sanchez-Pernaute, R.; de Yébenez, J.G.; Boesiger, P.; Weindl, A.; et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington’s disease. Brain A J. Neurol. 1996, 119 Pt 6, 2085–2095. [Google Scholar]
- De Natale, E.R.; Wilson, H.; Politis, M. Imaging Biomarkers in Huntington’s Disease; Humana: New York, NY, USA, 2022; p. 457. [Google Scholar]
- Magnotta, V.A.; Kim, J.; Koscik, T.; Beglinger, L.J.; Espinso, D.; Langbehn, D.; Nopoulos, P.; Paulsen, J.S. Diffusion tensor imaging in preclinical Huntington’s disease. Brain Imaging Behav. 2009, 3, 77–84. [Google Scholar] [CrossRef]
- Yin, J.-H.; Liu, Y.-O.; Li, H.-L.; Burgunder, J.M.; Huang, Y.J. White Matter Microstructure Changes Revealed by Diffusion Kurtosis and Diffusion Tensor Imaging in Mutant Huntingtin Gene Carriers. J. Huntington’s Dis. 2024, 13, 301–313. [Google Scholar] [CrossRef]
- Müller, H.-P.; Kassubek, J. Toward diffusion tensor imaging as a biomarker in neurodegenerative diseases: Technical considerations to optimize recordings and data processing. Front. Hum. Neurosci. 2024, 18, 1378896. [Google Scholar] [CrossRef] [PubMed]
- Estevez-Fraga, C.; Scahill, R.; Rees, G.; Tabrizi, S.J.; Gregory, S. Neurosurgery; Psychiatry, Diffusion imaging in Huntington’s disease: Comprehensive review. J. Neurol. Neurosurg. Psychiatry 2021, 92, 62–69. [Google Scholar] [CrossRef]
- Gusella, J.F.; MacDonald, M.E.; Lee, J.M. Genetic modifiers of Huntington’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, A.; Kumar, B.V.; Mo, A.; Welsh, C.S.; Margolis, R.L.; Ross, C.A. Age, CAG repeat length, and clinical progression in Huntington’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2012, 27, 272–276. [Google Scholar] [CrossRef]
- Ganesh, S.; Chithambaram, T.; Krishnan, N.R.; Vincent, D.R.; Kaliappan, J.; Srinivasan, K. Exploring Huntington’s Disease Diagnosis via Artificial Intelligence Models: A Comprehensive Review. Diagnostics 2023, 13, 3592. [Google Scholar] [CrossRef]
- Wexler, N.S.; Lorimer, J.; Porter, J.; Gomez, F.; Moskowitz, C.; Shackell, E.; Marder, K.; Penchaszadeh, G.; Roberts, S.A.; Gayán, J.; et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc. Natl. Acad. Sci. USA 2004, 101, 3498–3503. [Google Scholar]
- Lipe, H.; Bird, T. Late onset Huntington Disease: Clinical and genetic characteristics of 34 cases. J. Neurol. Sci. 2009, 276, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef]
- Sathe, S.; Ware, J.; Levey, J.; Neacy, E.; Blumenstein, R.; Noble, S.; Mühlbäck, A.; Rosser, A.; Landwehrmeyer, G.B.; Sampaio, C. Enroll-HD: An Integrated Clinical Research Platform and Worldwide Observational Study for Huntington’s Disease. Front. Neurol. 2021, 12, 667420. [Google Scholar] [CrossRef] [PubMed]
- Hoss, A.G.; Labadorf, A.; Latourelle, J.C.; Kartha, V.K.; Hadzi, T.C.; Gusella, J.F.; MacDonald, M.E.; Chen, J.F.; Akbarian, S.; Weng, Z.; et al. miR-10b-5p expression in Huntington’s disease brain relates to age of onset and the extent of striatal involvement. BMC Med. Genom. 2015, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Díez-Planelles, C.; Sánchez-Lozano, P.; Crespo, M.C.; Gil-Zamorano, J.; Ribacoba, R.; González, N.; Suárez, E.; Martínez-Descals, A.; Martínez-Camblor, P.; Álvarez, V.; et al. Circulating microRNAs in Huntington’s disease: Emerging mediators in metabolic impairment. Pharmacol. Res. 2016, 108, 102–110. [Google Scholar] [CrossRef]
- Chang, K.H.; Wu, Y.R.; Chen, C.M. Down-regulation of miR-9* in the peripheral leukocytes of Huntington’s disease patients. Orphanet J. Rare Dis. 2017, 12, 185. [Google Scholar] [CrossRef]
- Ferraldeschi, M.; Romano, S.; Giglio, S.; Romano, C.; Morena, E.; Mechelli, R.; Annibali, V.; Ubaldi, M.; Buscarinu, M.C.; Umeton, R.; et al. Circulating hsa-miR-323b-3p in Huntington’s Disease: A Pilot Study. Front. Neurol. 2021, 12, 657973. [Google Scholar] [CrossRef] [PubMed]
- Belkozhayev, A.; Niyazova, R.; Kamal, M.A.; Ivashchenko, A.; Sharipov, K.; Wilson, C.M. Differential microRNA expression in the SH-SY5Y human cell model as potential biomarkers for Huntington’s disease. Front. Cell. Neurosci. 2024, 18, 1399742. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Romano, C.; Peconi, M.; Fiore, A.; Bellucci, G.; Morena, E.; Troili, F.; Cipollini, V.; Annibali, V.; Giglio, S.; et al. Circulating U13 Small Nucleolar RNA as a Potential Biomarker in Huntington’s Disease: A Pilot Study. Int. J. Mol. Sci. 2022, 23, 12440. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Im, W.; Ban, J.J.; Lee, M.; Jung, K.H.; Lee, S.K.; Chu, K.; Kim, M. Exosome-Based Delivery of miR-124 in a Huntington’s Disease Model. J. Mov. Disord. 2017, 10, 45–52. [Google Scholar] [CrossRef]
- Gaughwin, P.M.; Ciesla, M.; Lahiri, N.; Tabrizi, S.J.; Brundin, P.; Björkqvist, M. Hsa-miR-34b is a plasma-stable microRNA that is elevated in pre-manifest Huntington’s disease. Hum. Mol. Genet. 2011, 20, 2225–2237. [Google Scholar] [CrossRef]
- Dhawan, A. Extracellular miRNA biomarkers in neurologic disease: Is cerebrospinal fluid helpful? Biomark. Med. 2021, 15, 1377–1388. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef] [PubMed]
- Fodale, V.; Boggio, R.; Daldin, M.; Cariulo, C.; Spiezia, M.C.; Byrne, L.M.; Leavitt, B.R.; Wild, E.J.; Macdonald, D.; Weiss, A.; et al. Validation of Ultrasensitive Mutant Huntingtin Detection in Human Cerebrospinal Fluid by Single Molecule Counting Immunoassay. J. Huntington’s Dis. 2017, 6, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Vauleon, S.; Schutz, K.; Massonnet, B.; Gruben, N.; Manchester, M.; Buehler, A.; Schick, E.; Boak, L.; Hawellek, D.J. Quantifying mutant huntingtin protein in human cerebrospinal fluid to support the development of huntingtin-lowering therapies. Sci. Rep. 2023, 13, 5332. [Google Scholar] [CrossRef]
- Fodale, V.; Pintauro, R.; Daldin, M.; Spiezia, M.C.; MacDonald, D.; Bresciani, A. Quantifying Huntingtin Protein in Human Cerebrospinal Fluid Using a Novel Polyglutamine Length-Independent Assay. J. Huntington’s Dis. 2022, 11, 291–305. [Google Scholar] [CrossRef]
- Moscovitch-Lopatin, M.; Goodman, R.E.; Eberly, S.; Ritch, J.J.; Rosas, H.D.; Matson, S.; Matson, W.; Oakes, D.; Young, A.B.; Shoulson, I.; et al. HTRF analysis of soluble huntingtin in PHAROS PBMCs. Neurology 2013, 81, 1134–1140. [Google Scholar] [CrossRef]
- Corey-Bloom, J.; Haque, A.S.; Park, S.; Nathan, A.S.; Baker, R.W.; Thomas, E.A. Salivary levels of total huntingtin are elevated in Huntington’s disease patients. Sci. Rep. 2018, 8, 7371. [Google Scholar] [CrossRef] [PubMed]
- Bridel, C.; van Wieringen, W.N.; Zetterberg, H.; Tijms, B.M.; Teunissen, C.E.; Alvarez-Cermeño, J.C.; Andreasson, U.; Axelsson, M.; Bäckström, D.C.; Bartos, A.; et al. Diagnostic Value of Cerebrospinal Fluid Neurofilament Light Protein in Neurology: A Systematic Review and Meta-analysis. JAMA Neurol. 2019, 76, 1035–1048. [Google Scholar] [CrossRef]
- Parkin, G.M.; Corey-Bloom, J.; Snell, C.; Castleton, J.; Thomas, E.A. Plasma neurofilament light in Huntington’s disease: A marker for disease onset, but not symptom progression. Park. Relat. Disord. 2021, 87, 32–38. [Google Scholar] [CrossRef]
- Scahill, R.I.; Zeun, P.; Osborne-Crowley, K.; Johnson, E.B.; Gregory, S.; Parker, C.; Lowe, J.; Nair, A.; O’Callaghan, C.; Langley, C.; et al. Biological and clinical characteristics of gene carriers far from predicted onset in the Huntington’s disease Young Adult Study (HD-YAS): A cross-sectional analysis. Lancet Neurol. 2020, 19, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.B.; Owen, G.; Sathe, S.; Pak, E.; Kaur, D.; Ehrhardt, A.G.; Lifer, S.; Townhill, J.; Schubert, K.; Leavitt, B.R.; et al. Safety and Feasibility of Research Lumbar Puncture in Huntington’s Disease: The HDClarity Cohort and Bioresource. J. Huntington’s Dis. 2022, 11, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Machacek, M.; Garcia-Montoya, E.; McColgan, P.; Sanwald-Ducray, P.; Mazer, N.A. NfL concentration in CSF is a quantitative marker of the rate of neurodegeneration in aging and Huntington’s disease: A semi-mechanistic model-based analysis. Front. Neurosci. 2024, 18, 1420198. [Google Scholar] [CrossRef] [PubMed]
- Parkin, G.M.; Corey-Bloom, J.; Long, J.D.; Snell, C.; Smith, H.; Thomas, E.A. Associations between prognostic index scores and plasma neurofilament light in Huntington’s disease. Park. Relat. Disord. 2022, 97, 25–28. [Google Scholar] [CrossRef]
- Parkin, G.M.; Thomas, E.A.; Corey-Bloom, J. Plasma NfL as a prognostic biomarker for enriching HD-ISS stage 1 categorisation: A cross-sectional study. eBioMedicine 2023, 93, 104646. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.B.; Byrne, L.M.; Gregory, S.; Rodrigues, F.B.; Blennow, K.; Durr, A.; Leavitt, B.R.; Roos, R.A.; Zetterberg, H.; Tabrizi, S.J.; et al. Neurofilament light protein in blood predicts regional atrophy in Huntington disease. Neurology 2018, 90, e717–e723. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Scahill, R.I.; Owen, G.; Durr, A.; Leavitt, B.R.; Roos, R.A.; Borowsky, B.; Landwehrmeyer, B.; Frost, C.; Johnson, H.; et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: Analysis of 36-month observational data. Lancet Neurol. 2013, 12, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.E.; Xu, B.; Lu, B.; Hempstead, B.L. New insights in the biology of BDNF synthesis and release: Implications in CNS function. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 12764–12767. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef]
- Devaux, S.; Cizkova, D.; Quanico, J.; Franck, J.; Nataf, S.; Pays, L.; Hauberg-Lotte, L.; Maass, P.; Kobarg, J.H.; Kobeissy, F.; et al. Proteomic Analysis of the Spatio-temporal Based Molecular Kinetics of Acute Spinal Cord Injury Identifies a Time- and Segment-specific Window for Effective Tissue Repair. Mol. Cell Proteom. 2016, 15, 2641–2670. [Google Scholar] [CrossRef] [PubMed]
- Mouhieddine, T.H.; Kobeissy, F.H.; Itani, M.; Nokkari, A.; Wang, K.K. Stem cells in neuroinjury and neurodegenerative disorders: Challenges and future neurotherapeutic prospects. Neural Regen. Res. 2014, 9, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Raad, M.; El Tal, T.; Gul, R.; Mondello, S.; Zhang, Z.; Boustany, R.M.; Guingab, J.; Wang, K.K.; Kobeissy, F. Neuroproteomics approach and neurosystems biology analysis: ROCK inhibitors as promising therapeutic targets in neurodegeneration and neurotrauma. Electrophoresis 2012, 33, 3659–3668. [Google Scholar] [CrossRef] [PubMed]
- Tümer, N.; Svetlov, S.; Whidden, M.; Kirichenko, N.; Prima, V.; Erdos, B.; Sherman, A.; Kobeissy, F.; Yezierski, R.; Scarpace, P.J.; et al. Overpressure blast-wave induced brain injury elevates oxidative stress in the hypothalamus and catecholamine biosynthesis in the rat adrenal medulla. Neurosci. Lett. 2013, 544, 62–67. [Google Scholar] [CrossRef]
- Masnata, M.; Salem, S.; de Rus Jacquet, A.; Anwer, M.; Cicchetti, F. Targeting Tau to Treat Clinical Features of Huntington’s Disease. Front. Neurol. 2020, 11, 580732. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.B.; Byrne, L.; McColgan, P.; Robertson, N.; Tabrizi, S.J.; Leavitt, B.R.; Zetterberg, H.; Wild, E.J. Cerebrospinal fluid total tau concentration predicts clinical phenotype in Huntington’s disease. J. Neurochem. 2016, 139, 22–25. [Google Scholar] [CrossRef] [PubMed]
- St-Amour, I.; Turgeon, A.; Goupil, C.; Planel, E.; Hébert, S.S. Co-occurrence of mixed proteinopathies in late-stage Huntington’s disease. Acta Neuropathol. 2018, 135, 249–265. [Google Scholar] [CrossRef]
- Petry, S.; Nateghi, B.; Keraudren, R.; Sergeant, N.; Planel, E.; Hébert, S.S.; St-Amour, I. Differential Regulation of Tau Exon 2 and 10 Isoforms in Huntington’s Disease Brain. Neuroscience 2023, 518, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.S.; Oh, E.; Kim, M.; Lee, C.Y.; Kim, H.S.; Chung, S.J.; Sung, Y.H.; Yoon, W.T.; Cho, J.H.; Lee, J.H.; et al. Plasma neurofilament light-chain and phosphorylated tau as biomarkers of disease severity in Huntington’s disease: Korean cohort data. J. Neurol. Sci. 2023, 452, 120744. [Google Scholar] [CrossRef]
- Andrade-Navarro, M.A.; Mühlenberg, K.; Spruth, E.J.; Mah, N.; González-López, A.; Andreani, T.; Russ, J.; Huska, M.R.; Muro, E.M.; Fontaine, J.F.; et al. RNA Sequencing of Human Peripheral Blood Cells Indicates Upregulation of Immune-Related Genes in Huntington’s Disease. Front. Neurol. 2020, 11, 573560. [Google Scholar] [CrossRef] [PubMed]
- Valadão, P.A.C.; Santos, K.B.S.; Ferreira, E.V.T.H.; Macedo, E.C.T.; Teixeira, A.L.; Guatimosim, C.; de Miranda, A.S. Inflammation in Huntington’s disease: A few new twists on an old tale. J. Neuroimmunol. 2020, 348, 577380. [Google Scholar] [CrossRef]
- Crotti, A.; Benner, C.; Kerman, B.E.; Gosselin, D.; Lagier-Tourenne, C.; Zuccato, C.; Cattaneo, E.; Gage, F.H.; Cleveland, D.W.; Glass, C.K. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat. Neurosci. 2014, 17, 513–521. [Google Scholar] [CrossRef]
- Ellrichmann, G.; Reick, C.; Saft, C.; Linker, R.A. The role of the immune system in Huntington’s disease. Clin. Dev. Immunol. 2013, 2013, 541259. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.-X.; Yin, J.-H.; Du, G.; Yang, Q.; Huang, Y. Identifying and verifying Huntington’s disease subtypes: Clinical features, neuroimaging, and cytokine changes. Brain Behav. 2024, 14, e3469. [Google Scholar] [CrossRef]
- Soltani Khaboushan, A.; Moeinafshar, A.; Ersi, M.H.; Teixeira, A.L.; Majidi Zolbin, M.; Kajbafzadeh, A.M. Circulating levels of inflammatory biomarkers in Huntington’s disease: A systematic review and meta-analysis. J. Neuroimmunol. 2023, 385, 578243. [Google Scholar] [CrossRef]
- Rojas, N.G.; Cesarini, M.E.; Peker, G.; Da Prat, G.A.; Etcheverry, J.L.; Gatto, E.M. Review of huntington’s disease: From basics to advances in diagnosis and treatment. J. Neurol. Res. 2022, 12, 93–113. [Google Scholar] [CrossRef]
- Hsiao, H.-Y.; Chiu, F.-L.; Chen, C.-M.; Wu, Y.-R.; Chen, H.-M.; Chen, Y.-C.; Kuo, H.-C.; Chern, Y. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum. Mol. Genet. 2014, 23, 4328–4344. [Google Scholar] [CrossRef] [PubMed]
- von Essen, M.R.; Hellem, M.N.N.; Vinther-Jensen, T.; Ammitzbøll, C.; Hansen, R.H.; Hjermind, L.E.; Nielsen, T.T.; Nielsen, J.E.; Sellebjerg, F. Early Intrathecal T Helper 17.1 Cell Activity in Huntington Disease. Ann. Neurol. 2020, 87, 246–255. [Google Scholar] [CrossRef]
- Kunwar, O.K.; Singh, S. Neuroinflammation and neurodegeneration in Huntington’s disease: Genetic hallmarks, role of metals and organophosphates. Neurogenetics 2025, 26, 21. [Google Scholar] [CrossRef]
- Chen, K.-P.; Hua, K.-F.; Tsai, F.-T.; Lin, T.-Y.; Cheng, C.-Y.; Yang, D.-I.; Hsu, H.-T.; Ju, T.-C. A selective inhibitor of the NLRP3 inflammasome as a potential therapeutic approach for neuroprotection in a transgenic mouse model of Huntington’s disease. J. Neuroinflammation 2022, 19, 56. [Google Scholar] [CrossRef]
- Plinta, K.; Plewka, A.; Wójcik-Pędziwiatr, M.; Zmarzły, N.; Rudziński, M.; Rudzińska-Bar, M. Is tgf-β1 a biomarker of huntington’s disease progression? J. Clin. Med. 2021, 10, 3001. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Wu, Y.R.; Chen, Y.C.; Chen, C.M. Plasma inflammatory biomarkers for Huntington’s disease patients and mouse model. Brain Behav. Immun. 2015, 44, 121–127. [Google Scholar] [CrossRef]
- van der Burg, J.M.M.; Gardiner, S.L.; Ludolph, A.C.; Landwehrmeyer, G.B.; Roos, R.A.C.; Aziz, N.A. Body weight is a robust predictor of clinical progression in Huntington disease. Ann. Neurol. 2017, 82, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Silajdžić, E.; Björkqvist, M. A Critical Evaluation of Wet Biomarkers for Huntington’s Disease: Current Status and Ways Forward. J. Huntington’s Dis. 2018, 7, 109–135. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.M.; Dai, J.; Smith, A.C.; Beckley, J.T.; Rahmati, N.; Lewis, M.C.; Quirk, M.C. Changes in 24(S)-Hydroxycholesterol Are Associated with Cognitive Performance in Early Huntington’s Disease: Data from the TRACK and ENROLL HD Cohorts. J. Huntington’s Dis. 2024, 13, 449–465. [Google Scholar] [CrossRef]
- Halliday, G.; McRitchie, D.; Macdonald, V.; Double, K.; Trent, R.; McCusker, E. Regional specificity of brain atrophy in Huntington’s disease. Exp. Neurol. 1998, 154, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Kinnunen, K.M.; Schwarz, A.J.; Turner, E.C.; Pustina, D.; Gantman, E.C.; Gordon, M.F.; Joules, R.; Mullin, A.P.; Scahill, R.I.; Georgiou-Karistianis, N. Volumetric MRI-based biomarkers in Huntington’s disease: An evidentiary review. Front. Neurol. 2021, 12, 712555. [Google Scholar] [CrossRef] [PubMed]
- Rees, E.M.; Farmer, R.; Cole, J.H.; Haider, S.; Durr, A.; Landwehrmeyer, B.; Scahill, R.I.; Tabrizi, S.J.; Hobbs, N.Z. Cerebellar abnormalities in Huntington’s disease: A role in motor and psychiatric impairment? Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1648–1654. [Google Scholar] [CrossRef]
- Scharmüller, W.; Ille, R.; Schienle, A. Cerebellar contribution to anger recognition deficits in Huntington’s disease. Cerebellum 2013, 12, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Wolf, R.C.; Vasic, N.; Schönfeldt-Lecuona, C.; Ecker, D.; Landwehrmeyer, G.B. Cortical dysfunction in patients with Huntington’s disease during working memory performance. Hum. Brain Mapp. 2009, 30, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, C.G. Uses of Human MR and PET Imaging in Research of Neurodegenerative Brain Diseases. Neurotherapeutics 2021, 18, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Kuwert, T.; Lange, H.W.; Langen, K.J.; Herzog, H.; Aulich, A.; Feinendegen, L.E. Cortical and subcortical glucose consumption measured by PET in patients with Huntington’s disease. Brain A J. Neurol. 1990, 113 Pt 5, 1405–1423. [Google Scholar] [CrossRef] [PubMed]
- Berent, S.; Giordani, B.; Lehtinen, S.; Markel, D.; Penney, J.B.; Buchtel, H.A.; Starosta-Rubinstein, S.; Hichwa, R.; Young, A.B. Positron emission tomographic scan investigations of Huntington’s disease: Cerebral metabolic correlates of cognitive function. Ann. Neurol. 1988, 23, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Bertoglio, D.; Weiss, A.R.; Liguore, W.; Martin, L.D.; Hobbs, T.; Templon, J.; Srinivasan, S.; Dominguez, C.; Munoz-Sanjuan, I.; Khetarpal, V.; et al. In Vivo Cerebral Imaging of Mutant Huntingtin Aggregates Using (11)C-CHDI-180R PET in a Nonhuman Primate Model of Huntington Disease. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2023, 64, 1581–1587. [Google Scholar]
- Matusch, A.; Saft, C.; Elmenhorst, D.; Kraus, P.H.; Gold, R.; Hartung, H.P.; Bauer, A. Cross sectional PET study of cerebral adenosine A1 receptors in premanifest and manifest Huntington’s disease. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 1210–1220. [Google Scholar] [CrossRef]
- Tang, C.C.; Feigin, A.; Ma, Y.; Habeck, C.; Paulsen, J.S.; Leenders, K.L.; Teune, L.K.; van Oostrom, J.C.; Guttman, M.; Dhawan, V.; et al. Metabolic network as a progression biomarker of premanifest Huntington’s disease. J. Clin. Investig. 2013, 123, 4076–4088. [Google Scholar] [CrossRef]
- Russell, D.S.; Barret, O.; Jennings, D.L.; Friedman, J.H.; Tamagnan, G.D.; Thomae, D.; Alagille, D.; Morley, T.J.; Papin, C.; Papapetropoulos, S.; et al. The phosphodiesterase 10 positron emission tomography tracer, [18F]MNI-659, as a novel biomarker for early Huntington disease. JAMA Neurol. 2014, 71, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Niccolini, F.; Haider, S.; Reis Marques, T.; Muhlert, N.; Tziortzi, A.C.; Searle, G.E.; Natesan, S.; Piccini, P.; Kapur, S.; Rabiner, E.A.J.B. Altered PDE10A expression detectable early before symptomatic onset in Huntington’s disease. Brain 2015, 138, 3016–3029. [Google Scholar] [CrossRef]
- Pavese, N.; Andrews, T.C.; Brooks, D.J.; Ho, A.K.; Rosser, A.E.; Barker, R.A.; Robbins, T.W.; Sahakian, B.J.; Dunnett, S.B.; Piccini, P. Progressive striatal and cortical dopamine receptor dysfunction in Huntington’s disease: A PET study. Brain A J. Neurol. 2003, 126, 1127–1135. [Google Scholar] [CrossRef]
- van Oostrom, J.C.; Maguire, R.P.; Verschuuren-Bemelmans, C.C.; Veenma-van der Duin, L.; Pruim, J.; Roos, R.A.; Leenders, K.L. Striatal dopamine D2 receptors, metabolism, and volume in preclinical Huntington disease. Neurology 2005, 65, 941–943. [Google Scholar] [CrossRef]
- Feigin, A.; Tang, C.; Ma, Y.; Mattis, P.; Zgaljardic, D.; Guttman, M.; Paulsen, J.S.; Dhawan, V.; Eidelberg, D. Thalamic metabolism and symptom onset in preclinical Huntington’s disease. Brain A J. Neurol. 2007, 130 Pt 11, 2858–2867. [Google Scholar]
- Andica, C.; Kamagata, K.; Hatano, T.; Saito, Y.; Ogaki, K.; Hattori, N.; Aoki, S. MR Biomarkers of Degenerative Brain Disorders Derived From Diffusion Imaging. J. Magn. Reson. Imaging JMRI 2020, 52, 1620–1636. [Google Scholar] [CrossRef]
- Liu, W.; Yang, J.; Burgunder, J.; Cheng, B.; Shang, H. Diffusion imaging studies of Huntington’s disease: A meta-analysis. Park. Relat. Disord. 2016, 32, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Weaver, K.E.; Richards, T.L.; Liang, O.; Laurino, M.Y.; Samii, A.; Aylward, E.H. Longitudinal diffusion tensor imaging in Huntington’s Disease. Exp. Neurol. 2009, 216, 525–529. [Google Scholar] [CrossRef]
- Sritharan, A.; Egan, G.F.; Johnston, L.; Horne, M.; Bradshaw, J.L.; Bohanna, I.; Asadi, H.; Cunnington, R.; Churchyard, A.J.; Chua, P.; et al. A longitudinal diffusion tensor imaging study in symptomatic Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2010, 81, 257–262. [Google Scholar] [CrossRef]
- Vandenberghe, W.; Demaerel, P.; Dom, R.; Maes, F. Diffusion-weighted versus volumetric imaging of the striatum in early symptomatic Huntington disease. J. Neurol. 2009, 256, 109–114. [Google Scholar] [CrossRef]
- Odish, O.F.; Leemans, A.; Reijntjes, R.H.; van den Bogaard, S.J.; Dumas, E.M.; Wolterbeek, R.; Tax, C.M.; Kuijf, H.J.; Vincken, K.L.; van der Grond, J.; et al. Microstructural brain abnormalities in Huntington’s disease: A two-year follow-up. Hum. Brain Mapp. 2015, 36, 2061–2074. [Google Scholar] [CrossRef] [PubMed]
- Harrington, D.L.; Smith, M.M.; Zhang, Y.; Carlozzi, N.E.; Paulsen, J.S. Cognitive domains that predict time to diagnosis in prodromal Huntington disease. J. Neurol. Neurosurg. Psychiatry 2012, 83, 612–619. [Google Scholar] [CrossRef]
- Paulsen, J.S.; Langbehn, D.R.; Stout, J.C.; Aylward, E.; Ross, C.A.; Nance, M.; Guttman, M.; Johnson, S.; MacDonald, M.; Beglinger, L.J.; et al. Detection of Huntington’s disease decades before diagnosis: The Predict-HD study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 874–880. [Google Scholar] [CrossRef]
- Stout, J.C.; Queller, S.; Baker, K.N.; Cowlishaw, S.; Sampaio, C.; Fitzer-Attas, C.; Borowsky, B. HD-CAB: A cognitive assessment battery for clinical trials in Huntington’s disease 1,2,3. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Schobel, S.A.; Palermo, G.; Auinger, P.; Long, J.D.; Ma, S.; Khwaja, O.S.; Trundell, D.; Cudkowicz, M.; Hersch, S.; Sampaio, C.; et al. Motor, cognitive, and functional declines contribute to a single progressive factor in early HD. Neurology 2017, 89, 2495–2502. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Reilmann, R.; Roos, R.A.; Durr, A.; Leavitt, B.; Owen, G.; Jones, R.; Johnson, H.; Craufurd, D.; Hicks, S.L.; et al. Potential endpoints for clinical trials in premanifest and early Huntington’s disease in the TRACK-HD study: Analysis of 24 month observational data. Lancet Neurol. 2012, 11, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Langbehn, D.R.; Leavitt, B.R.; Roos, R.A.; Durr, A.; Craufurd, D.; Kennard, C.; Hicks, S.L.; Fox, N.C.; Scahill, R.I.; et al. Biological and clinical manifestations of Huntington’s disease in the longitudinal TRACK-HD study: Cross-sectional analysis of baseline data. Lancet Neurol. 2009, 8, 791–801. [Google Scholar] [CrossRef] [PubMed]
- van der Plas, E.; Schubert, R.; Reilmann, R.; Nopoulos, P.C. A Feasibility Study of Quantitative Motor Assessments in Children Using the Q-Motor Suite. J. Huntington’s Dis. 2019, 8, 333–338. [Google Scholar] [CrossRef]
- Antoniades, C.A.; Ober, J.; Hicks, S.; Siuda, G.; Carpenter, R.H.S.; Kennard, C.; Nemeth, A.H. Statistical characteristics of finger-tapping data in Huntington’s disease. Med. Biol. Eng. Comput. 2012, 50, 341–346. [Google Scholar] [CrossRef]
- Saft, C.; Andrich, J.; Meisel, N.M.; Przuntek, H.; Müller, T. Assessment of simple movements reflects impairment in Huntington’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2006, 21, 1208–1212. [Google Scholar] [CrossRef]
- Lipsmeier, F.; Simillion, C.; Bamdadian, A.; Tortelli, R.; Byrne, L.M.; Zhang, Y.P.; Wolf, D.; Smith, A.V.; Czech, C.; Gossens, C.; et al. A Remote Digital Monitoring Platform to Assess Cognitive and Motor Symptoms in Huntington Disease: Cross-sectional Validation Study. J. Med. Internet Res. 2022, 24, e32997. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.P.; Shirbin, C.; Churchyard, A.J.; Stout, J.C. Speech acoustic markers of early stage and prodromal Huntington’s disease: A marker of disease onset? Neuropsychologia 2012, 50, 3273–3278. [Google Scholar] [CrossRef] [PubMed]
- Riad, R.; Lunven, M.; Titeux, H.; Cao, X.-N.; Hamet Bagnou, J.; Lemoine, L.; Montillot, J.; Sliwinski, A.; Youssov, K.; Cleret de Langavant, L.; et al. Predicting clinical scores in Huntington’s disease: A lightweight speech test. J. Neurol. 2022, 269, 5008–5021. [Google Scholar] [CrossRef]
- Thomas, E.A.; Parkin, G.M. Biomarkers for Huntington’s Disease: Improving Clinical Outcomes; Springer Nature: Berlin/Heidelberg, Germany, 2023. [Google Scholar]
- Tortelli, R.; Rodrigues, F.B.; Wild, E. The use of wearable/portable digital sensors in Huntington’s disease: A systematic review. Park. Relat. Disord. 2021, 83, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Leuchter, M.K.; Donzis, E.J.; Cepeda, C.; Hunter, A.M.; Estrada-Sánchez, A.M.; Cook, I.A.; Levine, M.S.; Leuchter, A.F. Quantitative Electroencephalographic Biomarkers in Preclinical and Human Studies of Huntington’s Disease: Are They Fit-for-Purpose for Treatment Development? Front. Neurol. 2017, 8, 91. [Google Scholar] [CrossRef]
- Delussi, M.; Nazzaro, V.; Ricci, K.; de Tommaso, M. EEG Functional Connectivity and Cognitive Variables in Premanifest and Manifest Huntington’s Disease: EEG Low-Resolution Brain Electromagnetic Tomography (LORETA) Study. Front. Physiol. 2020, 11, 612325. [Google Scholar] [CrossRef]
- Ponomareva, N.; Klyushnikov, S.; Abramycheva, N.; Malina, D.; Scheglova, N.; Fokin, V.; Ivanova-Smolenskaia, I.; Illarioshkin, S. Alpha-theta border EEG abnormalities in preclinical Huntington’s disease. J. Neurol. Sci. 2014, 344, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Lazar, A.S.; Panin, F.; Goodman, A.O.; Lazic, S.E.; Lazar, Z.I.; Mason, S.L.; Rogers, L.; Murgatroyd, P.R.; Watson, L.P.; Singh, P.; et al. Sleep deficits but no metabolic deficits in premanifest Huntington’s disease. Ann. Neurol. 2015, 78, 630–648. [Google Scholar] [CrossRef]
- Piano, C.; Mazzucchi, E.; Bentivoglio, A.R.; Losurdo, A.; Calandra Buonaura, G.; Imperatori, C.; Cortelli, P.; Della Marca, G. Wake and Sleep EEG in Patients With Huntington Disease: An eLORETA Study and Review of the Literature. Clin. EEG Neurosci. 2017, 48, 60–71. [Google Scholar] [CrossRef]
- Painold, A.; Anderer, P.; Holl, A.K.; Letmaier, M.; Saletu-Zyhlarz, G.M.; Saletu, B.; Bonelli, R.M. EEG low-resolution brain electromagnetic tomography (LORETA) in Huntington’s disease. J. Neurol. 2011, 258, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Beste, C.; Ness, V.; Lukas, C.; Hoffmann, R.; Stüwe, S.; Falkenstein, M.; Saft, C. Mechanisms mediating parallel action monitoring in fronto-striatal circuits. Neuroimage 2012, 62, 137–146. [Google Scholar] [CrossRef]
- Rosenberg, C.; Nudleman, K.; Starr, A. Cognitive evoked potentials (P300) in early Huntington’s disease. Arch. Neurol. 1985, 42, 984–987. [Google Scholar] [CrossRef]
- Beste, C.; Stock, A.K.; Ness, V.; Hoffmann, R.; Lukas, C.; Saft, C. A novel cognitive-neurophysiological state biomarker in premanifest Huntington’s disease validated on longitudinal data. Sci. Rep. 2013, 3, 1797. [Google Scholar] [CrossRef]
- Ibañez, K.; Jadhav, B.; Zanovello, M.; Gagliardi, D.; Clarkson, C.; Facchini, S.; Garg, P.; Martin-Trujillo, A.; Gies, S.J.; Galassi Deforie, V.; et al. Increased frequency of repeat expansion mutations across different populations. Nat. Med. 2024, 30, 3357–3368. [Google Scholar] [CrossRef]
- Berth, S.H.; Lloyd, T.E. Disruption of axonal transport in neurodegeneration. J. Clin. Investig. 2023, 133, e168554. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Scahill, R.I.; Durr, A.; Roos, R.A.; Leavitt, B.R.; Jones, R.; Landwehrmeyer, G.B.; Fox, N.C.; Johnson, H.; Hicks, S.L.; et al. Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: The 12-month longitudinal analysis. Lancet Neurol. 2011, 10, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R. Characterization of a large group of individuals with Huntington disease and their relatives enrolled in the COHORT study. PloS One 2012, 7, e29522. [Google Scholar] [CrossRef]
- Orth, M.; European Huntington’s Disease Network. Observing Huntington’s disease: The European Huntington’s disease network’s REGISTRY. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1409–1412. [Google Scholar] [CrossRef] [PubMed]



| Description | Results |
|---|---|
| Timespan | 2013:2024 |
| Sources (Journals, Books, etc.) | 319 |
| Documents | 730 |
| Annual Growth Rate % | 9.13% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aqel, S.; Ahmad, J.; Saleh, I.; Fathima, A.; Al Thani, A.A.; Mohamed, W.M.Y.; Shaito, A.A. Advances in Huntington’s Disease Biomarkers: A 10-Year Bibliometric Analysis and a Comprehensive Review. Biology 2025, 14, 129. https://doi.org/10.3390/biology14020129
Aqel S, Ahmad J, Saleh I, Fathima A, Al Thani AA, Mohamed WMY, Shaito AA. Advances in Huntington’s Disease Biomarkers: A 10-Year Bibliometric Analysis and a Comprehensive Review. Biology. 2025; 14(2):129. https://doi.org/10.3390/biology14020129
Chicago/Turabian StyleAqel, Sarah, Jamil Ahmad, Iman Saleh, Aseela Fathima, Asmaa A. Al Thani, Wael M. Y. Mohamed, and Abdullah A. Shaito. 2025. "Advances in Huntington’s Disease Biomarkers: A 10-Year Bibliometric Analysis and a Comprehensive Review" Biology 14, no. 2: 129. https://doi.org/10.3390/biology14020129
APA StyleAqel, S., Ahmad, J., Saleh, I., Fathima, A., Al Thani, A. A., Mohamed, W. M. Y., & Shaito, A. A. (2025). Advances in Huntington’s Disease Biomarkers: A 10-Year Bibliometric Analysis and a Comprehensive Review. Biology, 14(2), 129. https://doi.org/10.3390/biology14020129