
A Metabolic Model of Intestinal Secretions: The Link between Human Microbiota and Colorectal Cancer Progression

 ,
, 
Abstract
:1. Introduction
2. Results
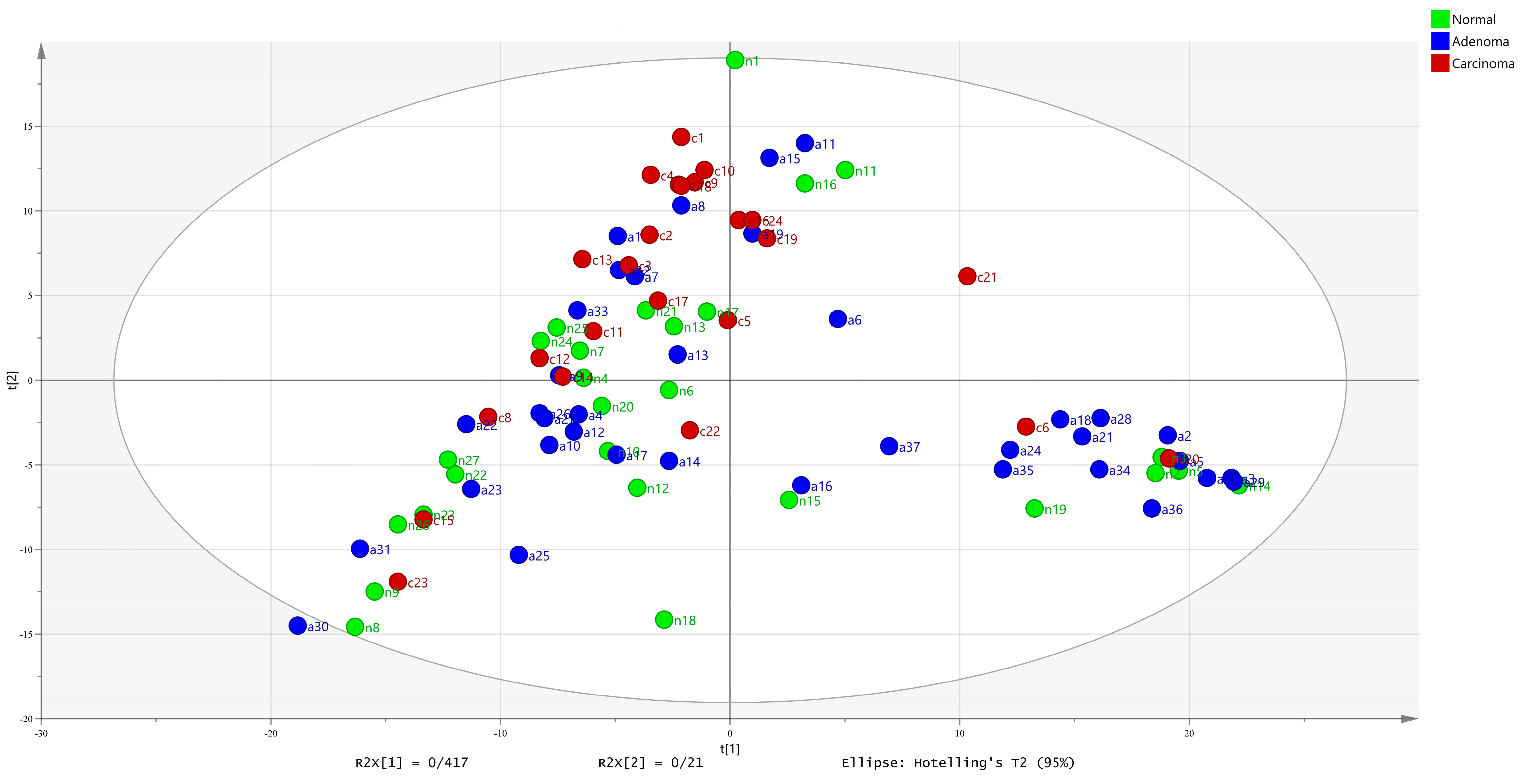
2.1. Community Microbiome Metabolic Models of CRC in Different Tumors
2.2. Meta-Model Selection and Data Analysis for Simulated Metabolism of CRC Microbiome
2.3. Meta-Models Reveal Different Patterns Among CRC Tumors
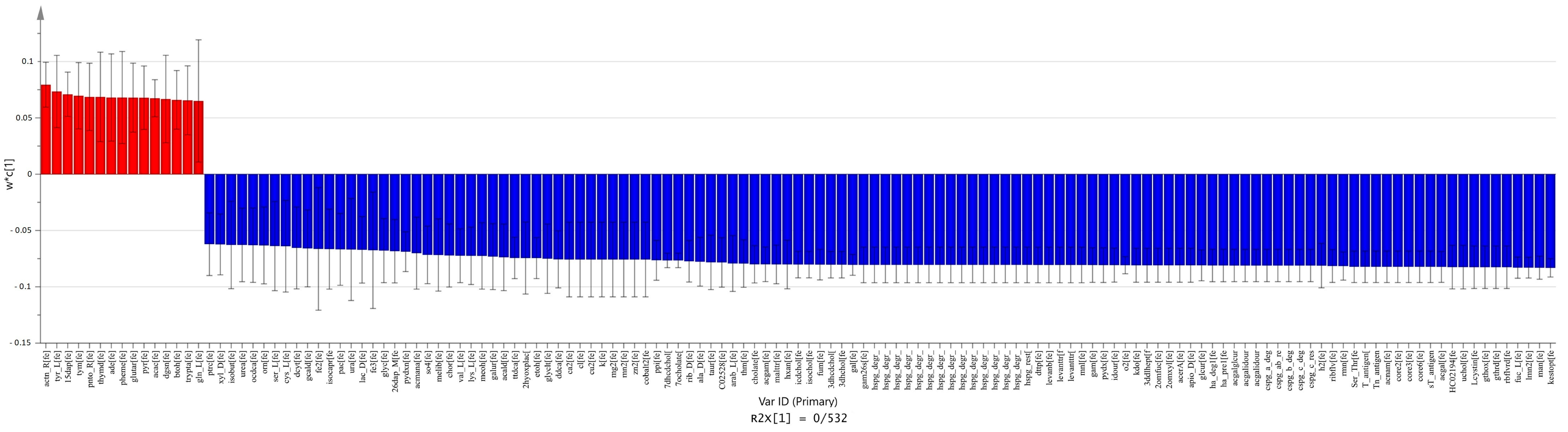
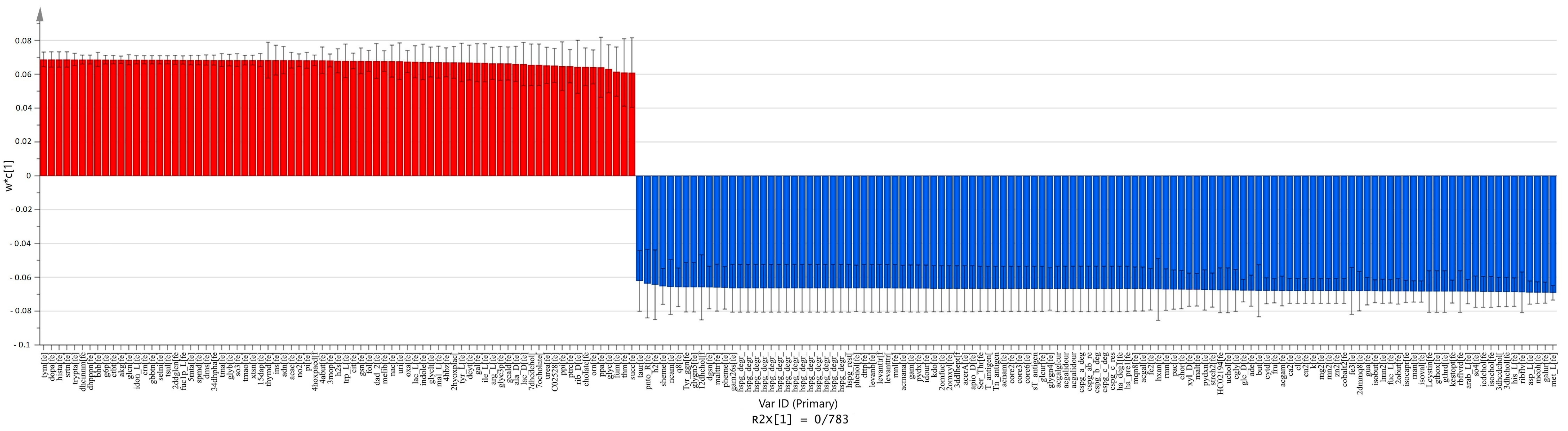
2.4. Comparison between Carcinoma and Normal Meta-Models
2.5. Comparison between Adenoma and Normal Meta-Models
2.6. Comparison between Carcinoma and Adenoma Meta-Models
3. Discussion
- The adenoma microbiome plays an important role in the mutagenesis and the progression of the adenoma to carcinoma.
- The metabolic changes in the adenoma microbiota increase inflammation and regulate the immune system.
- The metabolites of the CRC microbiota contribute to the growth and proliferation of cancer cells in both adenoma and carcinoma tumors.
- Microbial metabolites of adenomas and carcinomas are involved in the progression of CRC, for example, (the inhibition of) apoptosis and invasion.
3.1. Adenoma Microbiota Plays an Important Role in Mutagenesis and Progression of Adenoma to Carcinoma
3.2. Metabolic Alterations in the Adenoma Microbiota Increase Inflammation and Regulate the Immune System
3.3. CRC Microbiota Contribute to the Growth and Proliferation of Cancer Cells in Both Tumors
3.4. Microbiome Metabolites in Adenoma and Carcinoma Are Involved in the Development of Colorectal Cancer, such as through (the Inhibition of) Apoptosis and Invasion
4. Materials and Methods
4.1. Data Collection and Preprocessing
4.1.1. Taxonomy Assignment Data
4.1.2. Data Preprocessing
4.2. Microbiome Metabolic Modeling
4.3. Data Analysis
4.3.1. Multivariate Analysis
4.3.2. Metabolic Set Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dalton-Griffin, L.; Kellam, P. Infectious causes of cancer and their detection. J. Biol. 2009, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Claesson, M.J.; Cusack, S.; O’Sullivan, O.; Greene-Diniz, R.; de Weerd, H.; Flannery, E.; Marchesi, J.R.; Falush, D.; Dinan, T.; Fitzgerald, G.; et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4586–4591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dethlefsen, L.; Eckburg, P.B.; Bik, E.M.; Relman, D.A. Assembly of the human intestinal microbiota. Trends Ecol. Evol. 2006, 21, 517–523. [Google Scholar] [CrossRef]
- Marchesi, J.R. Human distal gut microbiome. Environ. Microbiol. 2011, 13, 3088–3102. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin. 2009, 59, 225–249. [Google Scholar] [CrossRef]
- Proctor, L.M. The Human Microbiome Project in 2011 and beyond. Cell Host Microbe 2011, 10, 287–291. [Google Scholar] [CrossRef] [Green Version]
- Griffin, J.L. Metabolic profiles to define the genome: Can we hear the phenotypes? Philos. Trans. R. Soc. L. B Biol. Sci. 2004, 359, 857–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, J.L.; Shockcor, J.P. Metabolic profiles of cancer cells. Nat. Rev. Cancer 2004, 4, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Patton, A.L.; Seely, K.A.; Chimalakonda, K.C.; Tran, J.P.; Trass, M.; Miranda, A.; Fantegrossi, W.E.; Kennedy, P.D.; Dobrowolski, P.; Radominska-Pandya, A.; et al. Targeted metabolomic approach for assessing human synthetic cannabinoid exposure and pharmacology. Anal. Chem. 2013, 85, 9390–9399. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.G.; Watkins, P.B.; Reily, M.D. Metabolomics in toxicology: Preclinical and clinical applications. Toxicol. Sci. 2011, 120 (Suppl. 1), S146–S170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roessner, U.; Luedemann, A.; Brust, D.; Fiehn, O.; Linke, T.; Willmitzer, L.; Fernie, A. Metabolic profiling allows comprehensive phenotyping of genetically or environmentally modified plant systems. Plant Cell 2001, 13, 11–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazzaniga, P.; Damiani, C.; Besozzi, D.; Colombo, R.; Nobile, M.S.; Gaglio, D.; Pescini, D.; Molinari, S.; Mauri, G.; Alberghina, L.; et al. Computational strategies for a system-level understanding of metabolism. Metabolites 2014, 4, 1034. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.U.; Kim, T.Y.; Lee, S.Y. Metabolic flux analysis and metabolic engineering of microorganisms. Mol. Biosyst. 2008, 4, 113–120. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Shoaie, S.; Nielsen, J. Elucidating the interactions between the human gut microbiota and its host through metabolic modeling. Front. Genet. 2014, 5, 86. [Google Scholar] [CrossRef] [Green Version]
- Stolyar, S.; Van Dien, S.; Hillesland, K.L.; Pinel, N.; Lie, T.J.; Leigh, J.A.; Stahl, D.A. Metabolic modeling of a mutualistic microbial community. Mol. Syst. Biol. 2007, 3, 92. [Google Scholar] [CrossRef]
- Mendes-Soares, H.; Chia, N. Community metabolic modeling approaches to understanding the gut microbiome: Bridging biochemistry and ecology. Free Radic. Biol. Med. 2017, 105, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Ji, B.; Babaei, P.; Das, P.; Lappa, D.; Ramakrishnan, G.; Fox, T.E.; Haque, R.; Petri, W.A.; Bäckhed, F.; et al. Gut microbiota dysbiosis is associated with malnutrition and reduced plasma amino acid levels: Lessons from genome-scale metabolic modeling. Metab. Eng. 2018, 49, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Rosario, D.; Benfeitas, R.; Bidkhori, G.; Zhang, C.; Uhlen, M.; Shoaie, S.; Mardinoglu, A. Understanding the Representative Gut Microbiota Dysbiosis in Metformin-Treated Type 2 Diabetes Patients Using Genome-Scale Metabolic Modeling. Front. Physiol. 2018, 9, 775. [Google Scholar] [CrossRef]
- Nakatsu, G.; Li, X.; Zhou, H.; Sheng, J.; Wong, S.H.; Wu, W.K.K.; Ng, S.C.; Tsoi, H.; Dong, Y.; Zhang, N.; et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat. Commun. 2015, 6, 8727. [Google Scholar] [CrossRef]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A bacterial driver–passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Jiang, B. Analysis of Mucosa-Associated Microbiota in Colorectal Cancer. Med. Sci. Monit. 2017, 23, 4422–4430. [Google Scholar] [CrossRef] [Green Version]
- Hertel, J.; Heinken, A.; Martinelli, F.; Thiele, I. Integration of constraint-based modeling with fecal metabolomics reveals large deleterious effects of Fusobacterium spp. on community butyrate production. Gut Microbes 2021, 13, 1915673. [Google Scholar] [CrossRef]
- MGnify. Human Gut Environment Targeted Loci Environmental. Available online: https://www.ebi.ac.uk/metagenomics/studies/MGYS00001248 (accessed on 23 September 2020).
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018, 46, 7542–7553. [Google Scholar] [CrossRef] [PubMed]
- Janssens, Y.; Nielandt, J.; Bronselaer, A.; Debunne, N.; Verbeke, F.; Wynendaele, E.; Van Immerseel, F.; Vandewynckel, Y.P.; De Tré, G.; De Spiegeleer, B. Disbiome database: Linking the microbiome to disease. BMC Microbiol. 2018, 18, 50. [Google Scholar] [CrossRef]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [Green Version]
- Calle, M.L. Statistical Analysis of Metagenomics Data. Genom. Inf. 2019, 17, e6. [Google Scholar] [CrossRef]
- Navas-Molina, J.A.; Peralta-Sánchez, J.M.; González, A.; McMurdie, P.J.; Vázquez-Baeza, Y.; Xu, Z.; Ursell, L.K.; Lauber, C.; Zhou, H.; Song, S.J.; et al. Advancing our understanding of the human microbiome using QIIME. Methods Enzymol. 2013, 531, 371–444. [Google Scholar] [CrossRef] [Green Version]
- Worley, B.; Powers, R. Multivariate Analysis in Metabolomics. Curr. Metab. 2013, 1, 92–107. [Google Scholar] [CrossRef]
- Chong, I.-G.; Jun, C.-H. Performance of some variable selection methods when multicollinearity is present. Chemom. Intell. Lab. Syst. 2005, 78, 103–112. [Google Scholar] [CrossRef]
- Sun, X.-M.; Yu, X.-P.; Liu, Y.; Xu, L.; Di, D.-L. Combining bootstrap and uninformative variable elimination: Chemometric identification of metabonomic biomarkers by nonparametric analysis of discriminant partial least squares. Chemom. Intell. Lab. Syst. 2012, 115, 37–43. [Google Scholar] [CrossRef]
- Adewiah, S.; Abubakar, A.; Yusuf, F. IDDF2018-ABS-0023 Butyrate acid as a potential marker for the diversity of gut microbiota in colorectal cancer patients. Gut 2018, 67, A1–A118. [Google Scholar] [CrossRef]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Nava, G.M.; Carbonero, F.; Croix, J.A.; Greenberg, E.; Gaskins, R. Abundance and diversity of mucosa-associated hydrogenotrophic microbes in the healthy human colon Subject Category: Microbe-microbe and microbe-host interactions. ISME J. 2012, 6, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Feng, Z.; Chen, N. Spermidine as a target for cancer therapy. Pharmacol. Res. 2020, 159, 104943. [Google Scholar] [CrossRef]
- De Mey, E.; De Maere, H.; Paelinck, H.; Fraeye, I. Volatile N-nitrosamines in meat products: Potential precursors, influence of processing, and mitigation strategies. Crit. Rev. Food Sci. Nutr. 2017, 57, 2909–2923. [Google Scholar] [CrossRef]
- Chan, C.W.H.; Law, B.M.H.; Waye, M.M.Y.; Chan, J.Y.W.; So, W.K.W.; Chow, K.M. Trimethylamine-N-oxide as One Hypothetical Link for the Relationship between Intestinal Microbiota and Cancer—Where We Are and Where Shall We Go? J. Cancer 2019, 10, 5874–5882. [Google Scholar] [CrossRef]
- Keshet, R.; Szlosarek, P.; Carracedo, A.; Erez, A. Rewiring urea cycle metabolism in cancer to support anabolism. Nat. Rev. Cancer 2018, 18, 634–645. [Google Scholar] [CrossRef]
- Shaffer, R.E. Multi- and Megavariate Data Analysis. Principles and Applications, I. Eriksson, E. Johansson, N. Kettaneh-Wold and S. Wold, Umetrics Academy, Umeå, 2001, ISBN 91-973730-1-X, 533pp. J. Chemom. 2002, 16, 261–262. [Google Scholar] [CrossRef]
- Del Rio, B.; Redruello, B.; Ladero, V.; Cal, S.; Obaya, A.J.; Alvarez, M.A. An altered gene expression profile in tyramine-exposed intestinal cell cultures supports the genotoxicity of this biogenic amine at dietary concentrations. Sci. Rep. 2018, 8, 17038. [Google Scholar] [CrossRef] [PubMed]
- Losurdo, G.; Principi, M.; Girardi, B.; Pricci, M.; Barone, M.; Ierardi, E.; Di Leo, A. Histamine and Histaminergic Receptors in Colorectal Cancer: From Basic Science to Evidence-based Medicine. Anticancer Agents Med. Chem. 2018, 18, 15–20. [Google Scholar] [CrossRef]
- Ibáñez-Sanz, G.; Díez-Villanueva, A.; Vilorio-Marqués, L.; Gracia, E.; Aragonés, N.; Olmedo-Requena, R.; Llorca, J.; Vidán, J.; Amiano, P.; Nos, P.; et al. Possible role of chondroitin sulphate and glucosamine for primary prevention of colorectal cancer. Results from the MCC-Spain study. Sci. Rep. 2018, 8, 2040. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Hu, Z.; Cai, L.; Li, K.; Choi, E.; Faubert, B.; Bezwada, D.; Rodriguez-Canales, J.; Villalobos, P.; Lin, Y.F.; et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 2017, 546, 168–172. [Google Scholar] [CrossRef]
- Celiktas, M.; Tanaka, I.; Tripathi, S.C.; Fahrmann, J.F.; Aguilar-Bonavides, C.; Villalobos, P.; Delgado, O.; Dhillon, D.; Dennison, J.B.; Ostrin, E.J.; et al. Role of CPS1 in cell growth, metabolism, and prognosis in LKB1-inactivated lung adenocarcinoma. J. Natl. Cancer Inst. 2017, 109, djw231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.Y.; Li, C.F.; Lin, C.Y.; Lee, S.W.; Sheu, M.J.; Lin, L.C.; Chen, T.J.; Wu, T.F.; Hsing, C.H. Overexpression of CPS1 is an independent negative prognosticator in rectal cancers receiving concurrent chemoradiotherapy. Tumor Biol. 2014, 35, 11097–11105. [Google Scholar] [CrossRef] [PubMed]
- Palaniappan, A.; Ramar, K.; Ramalingam, S. Computational identification of novel stage-specific biomarkers in colorectal cancer progression. PLoS ONE 2016, 11, e0156665. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef]
- Kodama, M.; Oshikawa, K.; Shimizu, H.; Yoshioka, S.; Takahashi, M.; Izumi, Y.; Bamba, T.; Tateishi, C.; Tomonaga, T.; Matsumoto, M.; et al. A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer. Nat. Commun. 2020, 11, 1320. [Google Scholar] [CrossRef] [PubMed]
- Caneba, C.A.; Bellance, N.; Yang, L.; Pabst, L.; Nagrath, D. Pyruvate uptake is increased in highly invasive ovarian cancer cells under anoikis conditions for anaplerosis, mitochondrial function, and migration. Am. J. Physiol. Metab. 2012, 303, E1036–E1052. [Google Scholar] [CrossRef] [Green Version]
- Diers, A.R.; Broniowska, K.A.; Chang, C.F.; Hogg, N. Pyruvate fuels mitochondrial respiration and proliferation of breast cancer cells: Effect of monocarboxylate transporter inhibition. Biochem. J. 2012, 444, 561–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohshima, K.; Nojima, S.; Tahara, S.; Kurashige, M.; Kawasaki, K.; Hori, Y.; Taniguchi, M.; Umakoshi, Y.; Okuzaki, D.; Wada, N.; et al. Serine racemase enhances growth of colorectal cancer by producing pyruvate from serine. Nat. Metab. 2020, 2, 81–96. [Google Scholar] [CrossRef]
- Krashin, E.; Piekiełko-Witkowska, A.; Ellis, M.; Ashur-Fabian, O. Thyroid Hormones and Cancer: A Comprehensive Review of Preclinical and Clinical Studies. Front. Endocrinol. 2019, 10, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirakov, M.; Plateroti, M. The thyroid hormones and their nuclear receptors in the gut: From developmental biology to cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.A.M.; Walenkamp, A.M.E.; Kema, I.P.; Meijer, C.; De Vries, E.G.E.; Oosting, S.F. Dopamine and serotonin regulate tumor behavior by affecting angiogenesis. Drug Resist. Updates 2014, 17, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Borcherding, D.C.; Tong, W.; Hugo, E.R.; Barnard, D.F.; Fox, S.; Lasance, K.; Shaughnessy, E.; Ben-Jonathan, N. Expression and therapeutic targeting of dopamine receptor-1 (D1R) in breast cancer. Oncogene 2016, 35, 3103–3113. [Google Scholar] [CrossRef]
- Moreno-Smith, M.; Lee, S.J.; Chunhua, L.; Nagaraja, A.S.; He, G.; Rupaimoole, R.; Han, H.D.; Jennings, N.B.; Roh, J.W.; Nishimura, M.; et al. Biologic effects of dopamine on tumor vasculature in ovarian carcinoma. Neoplasia 2013, 15, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, C.; Chakroborty, D.; Chowdhury, U.R.; Dasgupta, P.S.; Basu, S. Dopamine increases the efficacy of anticancer drugs in breast and colon cancer preclinical models. Clin. Cancer Res. 2008, 14, 2502–2510. [Google Scholar] [CrossRef] [Green Version]
- Asada, M.; Ebihara, S.; Yamanda, S.; Niu, K.; Okazaki, T.; Sora, I.; Arai, H. Depletion of Serotonin and Selective Inhibition of 2B Receptor Suppressed Tumor Angiogenesis by Inhibiting Endothelial Nitric Oxide Synthase and Extracellular Signal-Regulated Kinase 1/2 Phosphorylation. Neoplasia 2009, 11, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Jonnakuty, C.; Gragnoli, C. What do we know about serotonin? J. Cell. Physiol. 2008, 217, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Zamani, A.; Qu, Z. Serotonin activates angiogenic phosphorylation signaling in human endothelial cells. FEBS Lett. 2012, 586, 2360–2365. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.A.M.; Meijer, C.; Fehrmann, R.S.N.; Walenkamp, A.M.E.; Kema, I.P.; de Vries, E.G.E.; Hollema, H.; Oosting, S.F. Serotonin and Dopamine Receptor Expression in Solid Tumours Including Rare Cancers. Pathol. Oncol. Res. 2020, 26, 1539–1547. [Google Scholar] [CrossRef] [Green Version]
- Dang, D.; Chun, S.; Burkitt, K.; Cruz-Correa, M.; Han, X.; Dang, L. HIF-αs promote mitochondrial cardiolipin synthesis and respiration efficiency. Cancer Res. 2008, 68, 4109. [Google Scholar]
- Zhang, X.; Fryknäs, M.; Hernlund, E.; Fayad, W.; De Milito, A.; Olofsson, M.H.; Gogvadze, V.; Dang, L.; Påhlman, S.; Schughart, L.A.; et al. Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat. Commun. 2014, 5, 3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabata, S.; Yamamoto, M.; Goto, H.; Hirayama, A.; Ohishi, M.; Kuramoto, T.; Mitsuhashi, A.; Ikeda, R.; Haraguchi, M.; Kawahara, K.; et al. Thymidine Catabolism as a Metabolic Strategy for Cancer Survival. Cell Rep. 2017, 19, 1313–1321. [Google Scholar] [CrossRef] [Green Version]
- Mahey, S.; Kumar, R.; Arora, R.; Mahajan, J.; Arora, S.; Bhardwaj, R.; Thukral, A.K. Effect of cobalt(II) chloride hexahydrate on some human cancer cell lines. Springerplus 2016, 5, 930. [Google Scholar] [CrossRef] [Green Version]
- Rosa, L.; Nja, S. Anticancer Properties of Phenolic Acids in Colon Cancer—A Review. J. Nutr. Food Sci. 2016, 6, 1000468. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Tu, S.; Wang, Y.; Xu, B.; Wan, F. Mechanism of taurine-induced apoptosis in human colon cancer cells. Acta Biochim. Biophys. Sin. 2014, 46, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Tu, S.; Zhang, X.-L.; Wan, H.-F.; Xia, Y.-Q.; Liu, Z.-Q.; Yang, X.-H.; Wan, F.-S. Effect of taurine on cell proliferation and apoptosis human lung cancer A549 cells. Oncol. Lett. 2018, 15, 5473–5480. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Huang, C.; Li, Z.-F.; Wang, A.-Y.; Ying, L.; Zhao, X.-G.; Luo, Y.; Ni, L.; Zhang, W.-G.; Song, T.-S. Exogenous phosphatidylethanolamine induces apoptosis of human hepatoma HepG2 cells via the bcl-2/Bax pathway. World J. Gastroenterol. 2009, 15, 1751–1758. [Google Scholar] [CrossRef]
- Frank, L.A.; Gazzi, R.P.; Pohlmann, A.R.; Guterres, S.S. New Pectin-based Approaches for Colon Cancer Treatment. EC Pharmacol. Toxicol. 2020, 7–9. [Google Scholar]
- Fung, K.Y.C.; Brierley, G.V.; Henderson, S.; Hoffmann, P.; McColl, S.R.; Lockett, T.; Head, R.; Cosgrove, L. Butyrate-induced apoptosis in HCT116 colorectal cancer cells includes induction of a cell stress response. J. Proteome Res. 2011, 10, 1860–1869. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Liu, C.X.; Xu, W.; Huang, L.; Zhao, J.Y.; Zhao, S.M. Butyrate induces apoptosis by activating PDC and inhibiting complex i through SIRT3 inactivation. Signal Transduct. Target. Ther. 2017, 2, 16035. [Google Scholar] [CrossRef]
- Park, H.; Ohshima, K.; Nojima, S.; Tahara, S.; Kurashige, M.; Hori, Y.; Okuzaki, D.; Wada, N.; Ikeda, J.; Morii, E. Adenylosuccinate lyase enhances aggressiveness of endometrial cancer by increasing killer cell lectin-like receptor C3 expression by fumarate. Lab. Investig. 2018, 98, 449–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noronha, A.; Modamio, J.; Jarosz, Y.; Guerard, E.; Sompairac, N.; Preciat, G.; Daníelsdóttir, A.D.; Krecke, M.; Merten, D.; Haraldsdóttir, H.S.; et al. The Virtual Metabolic Human database: Integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res. 2019, 47, D614–D624. [Google Scholar] [CrossRef] [PubMed]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J. Vegan: Ecological Diversity. R Package Version 2.4-4. 2017. Available online: https://www.researchgate.net/publication/323265820_vegan_Community_Ecology_Package_R_package_version_24-4_http_s (accessed on 23 September 2020).
- Eastment, H.T.; Krzanowski, W.J. Cross-Validatory Choice of the Number of Components from a Principal Component Analysis. Technometrics 1982, 24, 73–77. [Google Scholar] [CrossRef]
- De Jong, S. Multivariate calibration, H. Martens and T. Naes, Wiley, New York, 1989. ISBN 0 471 90979 3. 504. J. Chemom. 1990, 4, 441. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010, 38, W71–W77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normalization Input | Normalized Input for Microbiome Metabolic Modeling | Reconstructed Microbiome Metabolic Models | Data Analysis and Meta-Model Selection | |
|---|---|---|---|---|
| Adenoma | 57 | 41 | 37 | 8 |
| Carcinoma | 52 | 26 | 24 | 7 |
| Normal | 61 | 27 | 27 | 6 |
| Total | 170 | 94 | 88 | 21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salahshouri, P.; Emadi-Baygi, M.; Jalili, M.; Khan, F.M.; Wolkenhauer, O.; Salehzadeh-Yazdi, A. A Metabolic Model of Intestinal Secretions: The Link between Human Microbiota and Colorectal Cancer Progression. Metabolites 2021, 11, 456. https://doi.org/10.3390/metabo11070456
Salahshouri P, Emadi-Baygi M, Jalili M, Khan FM, Wolkenhauer O, Salehzadeh-Yazdi A. A Metabolic Model of Intestinal Secretions: The Link between Human Microbiota and Colorectal Cancer Progression. Metabolites. 2021; 11(7):456. https://doi.org/10.3390/metabo11070456
Chicago/Turabian StyleSalahshouri, Pejman, Modjtaba Emadi-Baygi, Mahdi Jalili, Faiz M. Khan, Olaf Wolkenhauer, and Ali Salehzadeh-Yazdi. 2021. "A Metabolic Model of Intestinal Secretions: The Link between Human Microbiota and Colorectal Cancer Progression" Metabolites 11, no. 7: 456. https://doi.org/10.3390/metabo11070456
APA StyleSalahshouri, P., Emadi-Baygi, M., Jalili, M., Khan, F. M., Wolkenhauer, O., & Salehzadeh-Yazdi, A. (2021). A Metabolic Model of Intestinal Secretions: The Link between Human Microbiota and Colorectal Cancer Progression. Metabolites, 11(7), 456. https://doi.org/10.3390/metabo11070456