
Recent Advances in Pretreatment Methods and Detection Techniques for Veterinary Drug Residues in Animal-Derived Foods


Abstract
:1. Introduction
2. Pretreatment Methods
2.1. Liquid–Liquid Extraction (LLE) Technology
2.2. SPE
2.3. Immunoaffinity Chromatography (IAC)
2.4. QuEChERS
2.5. Molecular Imprinting Technology (MIT)
3. Chromatographic Detection Techniques
3.1. GC-MS
3.2. Liquid Chromatography Quadrupole-Time-of-Flight Mass Spectrometry (LC-QTOF-MS)
3.3. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)
3.4. Liquid Chromatography Coupled to Ion Trap Mass Spectrometry (LC-IT-MS)
3.5. CE-MS
4. Rapid Detection Techniques
4.1. Immunoassay Analysis Techniques
4.1.1. GICA
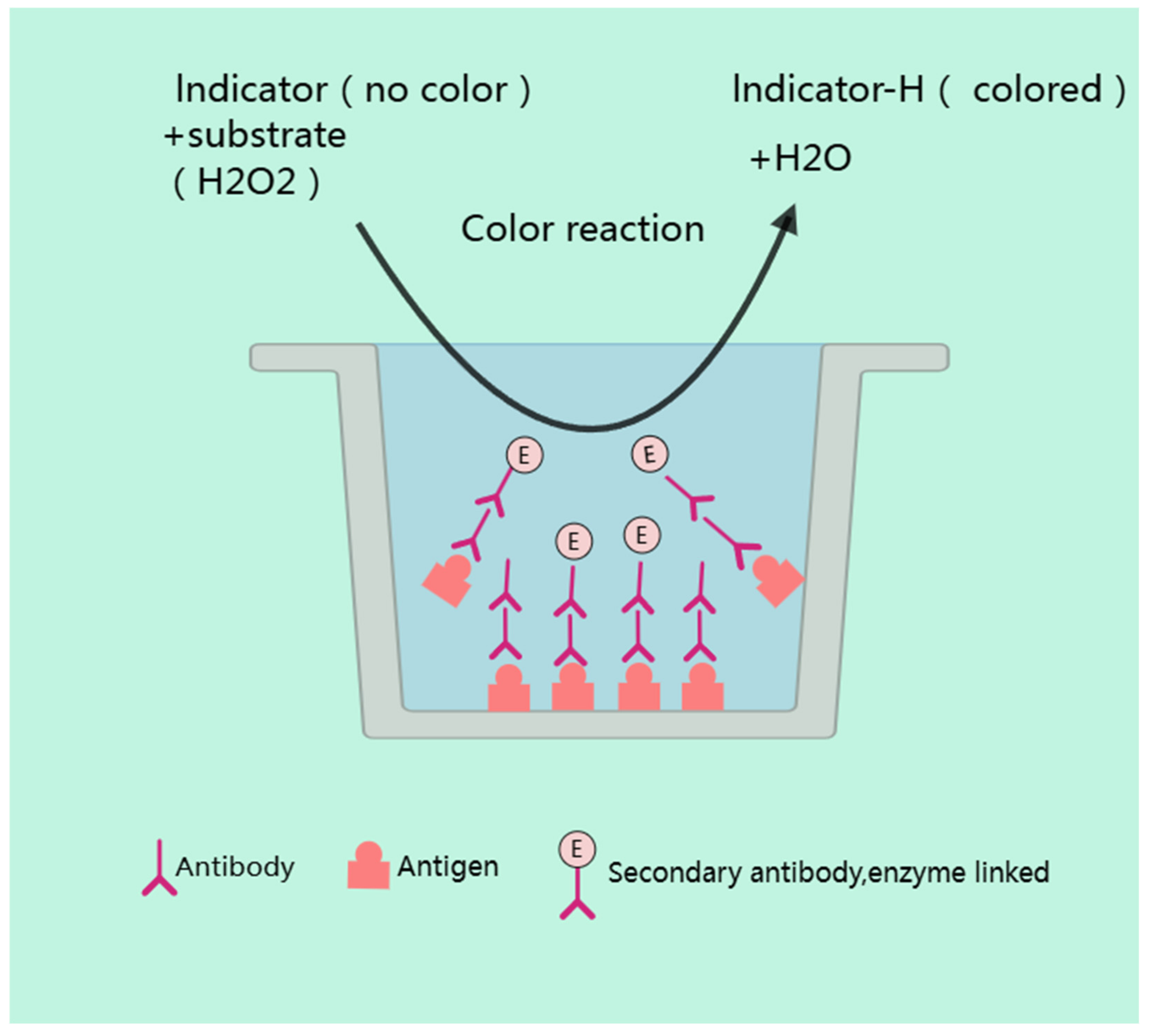
4.1.2. ELISA
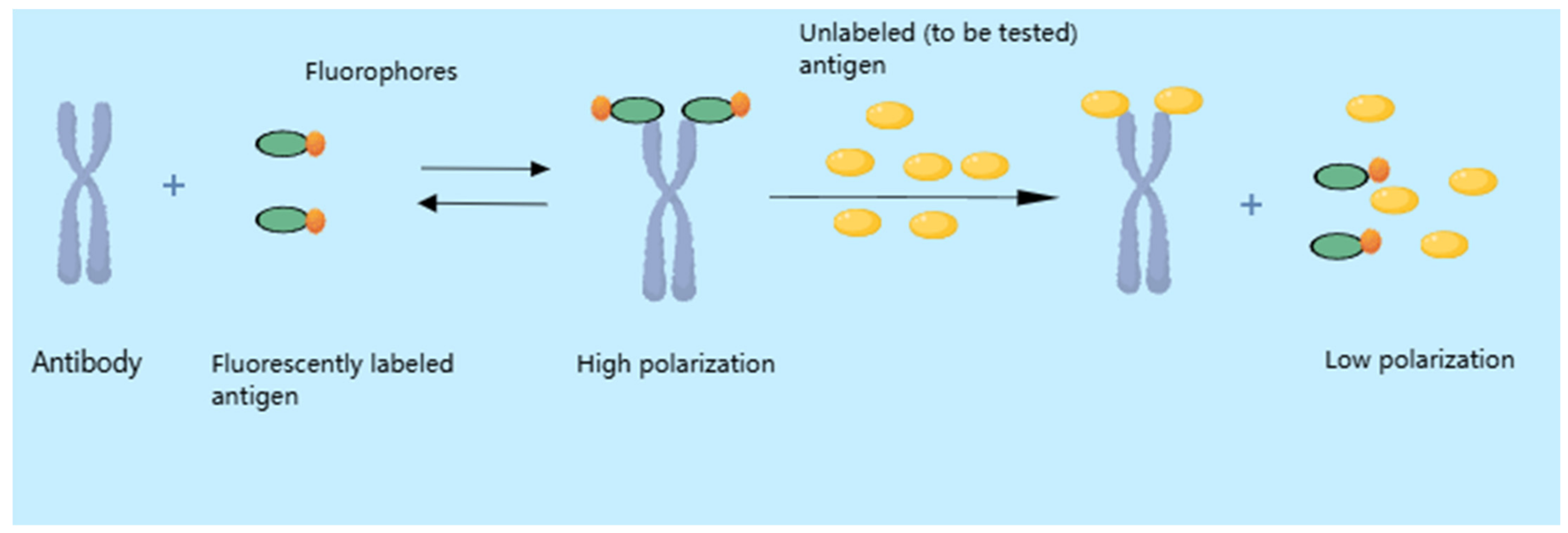
4.2. Fluorescence Polarization Immunoassay (FPIA)
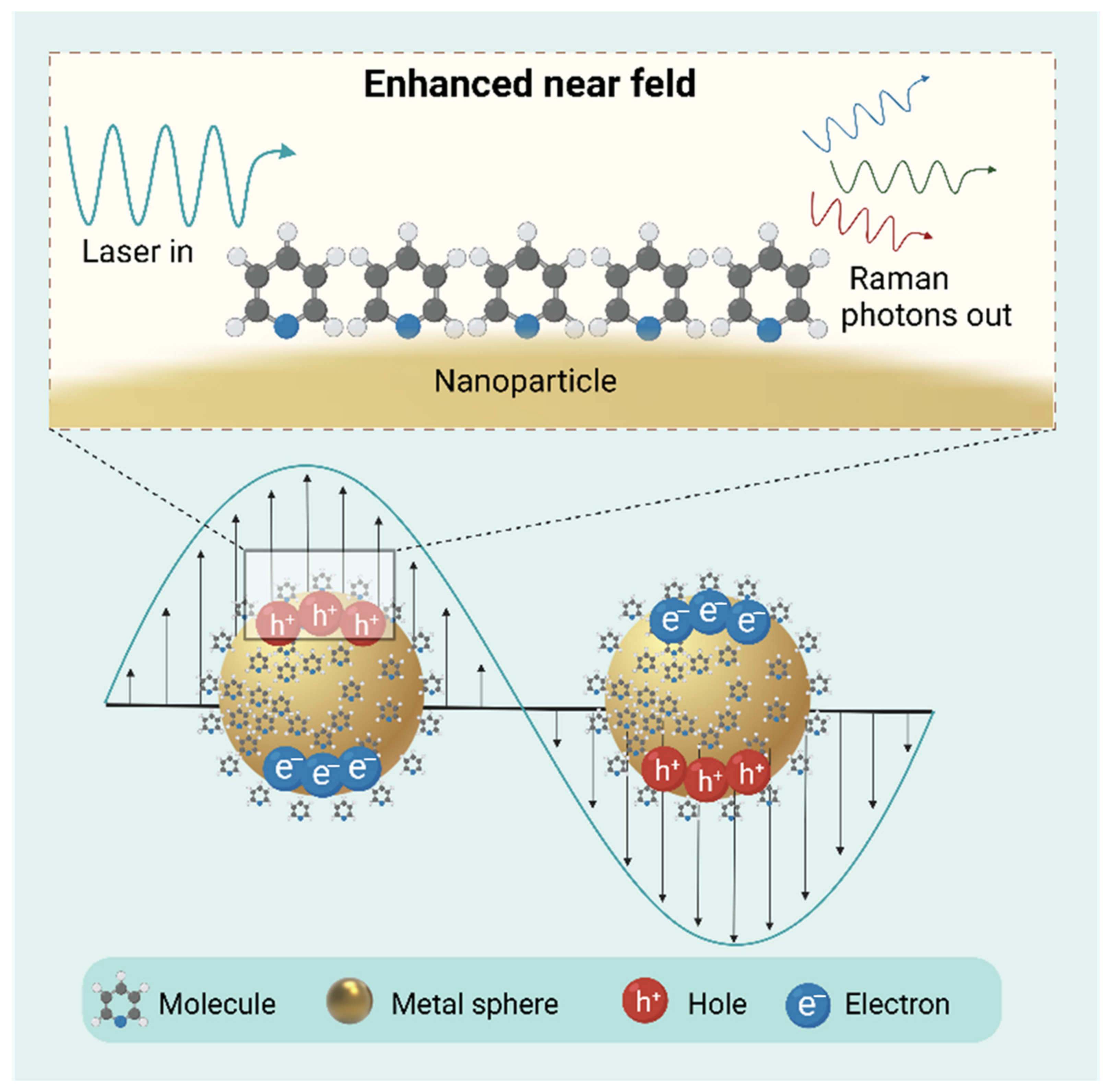
4.3. Surface-Enhanced Raman Scattering (SERS)
5. Comparative Analysis of Merits and Limitations in Contemporary Veterinary Drug Residue Analytical Techniques for Animal-Derived Food Products
6. Conclusions and Further Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, B.; Xie, K.; Lee, K. Veterinary drug residues in animal-derived foods: Sample preparation and analytical methods. Foods 2021, 10, 555. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, H.O.; Shikoray, L.; Mohamed, M.I.; Habib, I.; Matsumoto, T. Veterinary drug residues in the food chain as an emerging public health threat: Sources, analytical methods, health impacts, and preventive measures. Foods 2024, 13, 1629. [Google Scholar] [CrossRef] [PubMed]
- Pratiwi, R.; Ramadhanti, S.P.; Amatulloh, A.; Megantara, S.; Subra, L. Recent advances in the determination of veterinary drug residues in food. Foods 2023, 12, 3422. [Google Scholar] [CrossRef]
- Mesfin, Y.M.; Mitiku, B.A.; Tamrat Admasu, H. Veterinary drug residues in food products of animal origin and their public health consequences: A review. Vet. Med. Sci. 2024, 10, e70049. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Q.; Niu, B. Risk assessment of veterinary drug residues in meat products. Curr. Drug Metab. 2020, 21, 779–789. [Google Scholar] [CrossRef]
- Bacanlı, M.; Başaran, N. Importance of antibiotic residues in animal food. Food Chem. Toxicol. 2019, 125, 462–466. [Google Scholar] [CrossRef]
- Parmar, J.K.; Chaubey, K.K.; Gupta, V.; Bharath, M.N. Assessment of various veterinary drug residues in animal originated food products. Vet. World 2021, 14, 1650–1664. [Google Scholar] [CrossRef] [PubMed]
- Colopi, A.; Guida, E.; Cacciotti, S.; Fuda, S.; Lampitto, M.; Onorato, A.; Zucchi, A.; Balistreri, C.R.; Grimaldi, P.; Barchi, M. Dietary exposure to pesticide and veterinary drug residues and their effects on human fertility and embryo development: A global overview. Int. J. Mol. Sci. 2024, 25, 9116. [Google Scholar] [CrossRef]
- Chicoine, A.; Erdely, H.; Fattori, V.; Finnah, A.; Fletcher, S.; Lipp, M.; Sanders, P.; Scheid, S. Assessment of veterinary drug residues in food: Considerations when dealing with sub-optimal data. Regul. Toxicol. Pharmacol. 2020, 118, 104806. [Google Scholar] [CrossRef]
- Rana, M.S.; Lee, S.Y.; Kang, H.J.; Hur, S.J. Reducing veterinary drug residues in animal products: A review. Food Sci. Anim. Resour. 2019, 39, 687–703. [Google Scholar] [CrossRef]
- Atta, A.H.; Atta, S.A.; Nasr, S.M.; Mouneir, S.M. Current perspective on veterinary drug and chemical residues in food of animal origin. Environ. Sci. Pollut. Res. Int. 2022, 29, 15282–15302. [Google Scholar] [CrossRef]
- Lozano, A.; Hernando, M.D.; Ucles, S.; Hakme, E.; Fernandez-Alba, A.R. Identification and measurement of veterinary drug residues in beehive products. Food Chem. 2019, 274, 61–70. [Google Scholar] [CrossRef]
- Xia, S.; Niu, B.; Chen, J.; Deng, X.; Chen, Q. Risk analysis of veterinary drug residues in aquatic products in the Yangtze river delta of China. J. Food Prot. 2021, 84, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Tian, H.; Yang, F.; Fan, S.; Zhang, J.; Ma, J.; Ai, L.; Zhang, Y. Rapid determination of 103 common veterinary drug residues in milk and dairy products by ultra performance liquid chromatography tandem mass spectrometry. Front. Nutr. 2022, 9, 879518. [Google Scholar] [CrossRef]
- Li, F.; Lv, H.; Zhu, F.; Zhang, Q.; Xu, Q.; Ji, W. High throughput detection of veterinary drug residues in chicken and eggs. Food Chem. 2025, 463, 141267. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Lee, D.J.; Jo, A.; Yun, S.H.; Eun, J.B.; Im, M.H.; Shim, J.H.; Abd El-Aty, A.M. Onsite/on-field analysis of pesticide and veterinary drug residues by a state-of-art technology: A review. J. Sep. Sci. 2021, 44, 2310–2327. [Google Scholar] [CrossRef]
- Liu, L.; Gao, Y.; Liu, J.; Li, Y.; Yin, Z.; Zhang, Y.; Pi, F.; Sun, X. Sensitive techniques for poct sensing on the residues of pesticides and veterinary drugs in food. Bull. Environ. Contam. Toxicol. 2021, 107, 206–214. [Google Scholar] [CrossRef]
- Li, M.; Zhang, X. Nanostructure-based surface-enhanced raman spectroscopy techniques for pesticide and veterinary drug residues screening. Bull. Environ. Contam. Toxicol. 2021, 107, 194–205. [Google Scholar] [CrossRef]
- Zhang, B.; Lang, Y.; Guo, B.; Cao, Z.; Cheng, J.; Cai, D.; Shentu, X.; Yu, X. Indirect competitive enzyme-linked immunosorbent assay based on broad-spectrum antibody for simultaneous determination of thirteen fluoroquinolone antibiotics in Rana catesbeianus. Foods 2023, 12, 2530. [Google Scholar] [CrossRef]
- Park, D.; Choi, Y.S.; Kim, J.Y.; Choi, J.D.; Moon, G.I. Determination of flunixin and 5-hydroxy flunixin residues in livestock and fishery products using liquid chromatography-tandem mass spectrometry (lc-ms/ms). Food Sci. Anim. Resour. 2024, 44, 873–884. [Google Scholar] [CrossRef]
- Gens, K.D.; Singer, R.S.; Dilworth, T.J.; Heil, E.L.; Beaudoin, A.L. Antimicrobials in animal agriculture in the united states: A multidisciplinary overview of regulation and utilization to foster collaboration: On behalf of the society of infectious diseases pharmacists. Open Forum Infect. Dis. 2022, 9, ofac542. [Google Scholar] [CrossRef]
- Li, R.; Wen, Y.; Wang, F.; He, P. Recent advances in immunoassays and biosensors for mycotoxins detection in feedstuffs and foods. J. Anim. Sci. Biotechnol. 2021, 12, 108. [Google Scholar] [CrossRef]
- Park, H.; Kim, E.; Lee, T.H.; Park, S.; Choi, J.D.; Moon, G. Multiclass method for the determination of anthelmintic and antiprotozoal drugs in livestock products by ultra-high-performance liquid chromatography-tandem mass spectrometry. Food Sci. Anim. Resour. 2023, 43, 914–937. [Google Scholar] [CrossRef]
- Beguiristain, I.; Alongi, A.; Rubies, A.; Granados, M. Analysis of corticosteroids in samples of animal origin using QuEChERS and ultrahigh-performance liquid chromatography coupled to high-resolution mass spectrometry. Anal. Bioanal. Chem. 2019, 411, 449–457. [Google Scholar] [CrossRef]
- Okerman, L.; Noppe, H.; Cornet, V.; De Zutter, L. Microbiological detection of 10 quinolone antibiotic residues and its application to artificially contaminated poultry samples. Food Addit. Contam. 2007, 24, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Liao, G.Q.; Chen, L.; Qian, Y.Z.; Yan, X.; Qiu, J. Pesticide residues in animal-derived food: Current state and perspectives. Food Chem. 2024, 438, 137974. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Zhao, Z.; Lv, Y.K.; Shen, S.; Liang, S.X. Recent advances in porous organic frameworks for sample pretreatment of pesticide and veterinary drug residues: A review. Analyst 2021, 146, 7394–7417. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; E, Z.; Zhai, F.; Bing, X. Rapid multi-residue detection methods for pesticides and veterinary drugs. Molecules 2020, 25, 3590. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Peng, Y.; Liu, J. Nanomaterial-based fluorescent biosensors for veterinary drug detection in foods. J. Food Drug Anal. 2020, 28, 575–594. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, B. Liquid-liquid extraction (lle). In Separation and Purification Technologies in Biorefineries; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 61–78. [Google Scholar]
- Tshepho, R.; Dube, S.; Nindi, M.M. Ionic liquid-based dispersive liquid-liquid microextraction of anthelmintic drug residues in small-stock meat followed by LC-ESI-MS/MS detection. Food Sci. Nutr. 2023, 11, 6288–6302. [Google Scholar] [CrossRef]
- Araujo, P.; Iqbal, S.; Arno, A.; Espe, M.; Holen, E. Validation of a liquid-liquid extraction method to study the temporal production of d-series resolvins by head kidney cells from atlantic salmon (Salmon salar) exposed to docosahexaenoic acid. Molecules 2023, 28, 4728. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Xie, Y.; Chen, Z.; Cao, M.; Lei, X.; Le, T. A comprehensive review on the pretreatment and detection methods of nitrofurans and their metabolites in animal-derived food and environmental samples. Food Chem. X 2024, 24, 101928. [Google Scholar] [CrossRef] [PubMed]
- Doria, S.; Yost, J.; Gagnon, Z. Free-flow biomolecular concentration and separation of proteins and nucleic acids using teíchophoresis. Talanta 2023, 255, 124198. [Google Scholar] [CrossRef]
- Kannouma, R.E.; Hammad, M.A.; Kamal, A.H.; Mansour, F.R. Miniaturization of liquid-liquid extraction; the barriers and the enablers. Microchem. J. 2022, 182, 107863. [Google Scholar] [CrossRef]
- Faraji, H. Advancements in overcoming challenges in dispersive liquid-liquid microextraction: An overview of advanced strategies. TrAC Trends Anal. Chem. 2024, 170, 117429. [Google Scholar] [CrossRef]
- Cantwell, F.F.; Losier, M. Chapter 11 Liquid—Liquid extraction. In Comprehensive Analytical Chemistry; Elsevier: Amsterdam, The Netherlands, 2002; Volume 37, pp. 297–340. [Google Scholar]
- Sun, Q.; Zhang, S.; Chen, Q.; Cao, L. Salting-out assisted liquid-liquid extraction for the simple and rapid determination of veterinary antibiotic residues in aquatic products by HPLC-MS/MS. Food Chem. 2024, 460, 140775. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, J.; Ji, X.; Wu, H.; Yang, H.; Zhang, H.; Zhang, X.; Li, Z.; Ni, X.; Qian, M. Determination of veterinary drug/pesticide residues in livestock and poultry excrement using selective accelerated solvent extraction and magnetic material purification combined with ultra-high-performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2020, 1617, 460808. [Google Scholar] [CrossRef]
- Pang, M.; Xu, J.; Tang, Y.; Guo, Y.; Ding, H.; Wang, R.; Zhang, T.; Zhang, G.; Guo, X.; Dai, G.; et al. Combining gc-ms/mS with a lle-spe sample pretreatment step to simultaneously analyse enrofloxacin and ofloxacin residues in chicken tissues and pork. Food Chem. 2024, 456, 139972. [Google Scholar] [CrossRef]
- Zhang, Y.; Xue, X.; Su, S.; Guo, Z.; Wang, J.; Ding, L.; Liu, Y.; Zhu, J. A multi-class, multi-residue method for detection of veterinary drugs in multiple meat using a pass-through cleanup spe technique and uplc-ms/mS analysis. Food Anal. Methods 2018, 11, 2865–2884. [Google Scholar] [CrossRef]
- Badawy, M.E.I.; El-Nouby, M.A.M.; Kimani, P.K.; Lim, L.W.; Rabea, E.I. A review of the modern principles and applications of solid-phase extraction techniques in chromatographic analysis. Anal. Sci. Int. J. Jpn. Soc. Anal. Chem. 2022, 38, 1457–1487. [Google Scholar] [CrossRef]
- Cheng, R.; Mao, X.; Yu, J.; Liu, F.; Guo, L.; Luo, D.; Wan, Y. A dispersive solid-phase extraction method for the determination of Aristolochic acids in Houttuynia cordata based on MIL-101(Fe): An analytes-oriented adsorbent selection design. Food Chem. 2023, 407, 135074. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Okoli, C.P. Development of a solid-phase extraction method based on biocompatible starch polyurethane polymers for gc-ms analysis of polybrominated diphenyl ethers in ambient water samples. Molecules 2022, 27, 3253. [Google Scholar] [CrossRef]
- Islas, G.; Ibarra, I.S.; Hernandez, P.; Miranda, J.M.; Cepeda, A. Dispersive solid phase extraction for the analysis of veterinary drugs applied to food samples: A review. Int. J. Anal. Chem. 2017, 2017, 8215271. [Google Scholar] [CrossRef] [PubMed]
- Brandi, J.; Siragusa, G.; Robotti, E.; Marengo, E.; Cecconi, D. Analysis of veterinary drugs and pesticides in food using liquid chromatography-mass spectrometry. TrAC Trends Anal. Chem. 2024, 179, 117888. [Google Scholar] [CrossRef]
- Zhu, Y.; Jiang, X.; Shen, D.; Mao, J.; Cao, Y.; Zhang, K.; Peng, J.; Dong, F.; Wang, N.; He, K. A one-step solid-phase extraction with uhplc-ms/ms for fast and accurate determination of multi-class veterinary drugs in animal muscles. Food Chem. 2023, 428, 136712. [Google Scholar] [CrossRef]
- Mehl, A.; Hudel, L.; Bucker, M.; Morlock, G.E. Validated screening method for 81 multiclass veterinary drug residues in food via online-coupling high-throughput planar solid-phase extraction to high-performance liquid chromatography-orbitrap tandem mass spectrometry. J. Agric. Food Chem. 2022, 70, 10886–10898. [Google Scholar] [CrossRef]
- Rodriguez-Mozaz, S.; Lopez de Alda, M.J.; Barceló, D. Advantages and limitations of on-line solid phase extraction coupled to liquid chromatography–mass spectrometry technologies versus biosensors for monitoring of emerging contaminants in water. J. Chromatogr. A 2007, 1152, 97–115. [Google Scholar] [CrossRef]
- Talib, N.; Mohd-Setapar, S.; Khamis, A. The benefits and limitations of methods development in solid phase extraction: Mini review. J. Teknol. (Sci. Eng.) 2014, 69, 69–72. [Google Scholar] [CrossRef]
- Zheng, M.; Tang, S.; Bao, Y.; Daniels, K.D.; How, Z.T.; El-Din, M.G.; Wang, J.; Tang, L. Fully-automated spe coupled to uhplc-ms/ms method for multiresidue analysis of 26 trace antibiotics in environmental waters: SPE optimization and method validation. Environ. Sci. Pollut. Res. Int. 2022, 29, 16973–16987. [Google Scholar] [CrossRef]
- Sharmeen, S.; Suh, K.; Kyei, I.; Jones, J.; Olupathage, H.; Campbell, A.; Hage, D.S. Immunoaffinity chromatography for protein purification and analysis. Curr. Protoc. 2023, 3, e867. [Google Scholar] [CrossRef]
- Ren, J.; Xiong, H.; Huang, C.; Ji, F.; Jia, L. An engineered peptide tag-specific nanobody for immunoaffinity chromatography application enabling efficient product recovery at mild conditions. J. Chromatogr. A 2022, 1676, 463274. [Google Scholar] [CrossRef] [PubMed]
- Poddar, S.; Sharmeen, S.; Hage, D.S. Affinity monolith chromatography: A review of general principles and recent developments. Electrophoresis 2021, 42, 2577–2598. [Google Scholar] [CrossRef]
- Fitzgerald, J.; Leonard, P.; Darcy, E.; Sharma, S.; O’Kennedy, R. Immunoaffinity chromatography: Concepts and applications. Methods Mol. Biol. 2017, 1485, 27–51. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A. Immunoaffinity chromatography. Mol. Biotechnol. 2002, 20, 41–47. [Google Scholar] [CrossRef]
- Moser, A.C.; Hage, D.S. Immunoaffinity chromatography: An introduction to applications and recent developments. Bioanalysis 2010, 2, 769–790. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Zhang, X.; Wang, Y.; Pan, Y.; Liu, Z.; Chen, D.; Sheng, F.; Yuan, Z. An immunoaffinity column for the selective purification of 3-methyl-quinoxaline-2-carboxylic acid from swine tissues and its determination by high-performance liquid chromatography with ultraviolet detection and a colloidal gold-based immunochromatographic assay. Food Chem. 2017, 237, 290–296. [Google Scholar] [CrossRef]
- Wang, Q.; Zhao, H.; Xi, C.; Wang, G.; Chen, D.; Ding, S. Determination of chloramphenicol and zeranols in pig muscle by immunoaffinity column clean-up and LC-MS/MS analysis. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2014, 31, 1177–1186. [Google Scholar] [CrossRef]
- Nelson, M.A.; Reiter, W.S.; Hage, D.S. Chromatographic competitive binding immunoassays: A comparison of the sequential and simultaneous injection methods. Biomed. Chromatogr. 2003, 17, 188–200. [Google Scholar] [CrossRef]
- Thompson, N.E.; Burgess, R.R. Immunoaffinity chromatography: Advantages and limitations. In Analytical Separation Science; Wiley: Hoboken, NJ, USA, 2015; pp. 483–502. [Google Scholar] [CrossRef]
- Kavianpour, A.; Ashjari, M.; Hosseini, S.N.; Khatami, M. Quantitative assessment of LPS-HBsAg interaction by introducing a novel application of immunoaffinity chromatography. Prep. Biochem. Biotechnol. 2023, 53, 672–682. [Google Scholar] [CrossRef]
- Gonzalez-Curbelo, M.A.; Varela-Martinez, D.A.; Riano-Herrera, D.A. Pesticide-residue analysis in soils by the QuEChERS method: A Review. Molecules 2022, 27, 4323. [Google Scholar] [CrossRef]
- Kim, K.; Choi, Y.; Mok, S.; Moon, H.B.; Jeon, J. Optimization of the QuEChERS method for multi-residue analysis of pharmaceuticals and pesticides in aquaculture products. Food Chem. 2023, 399, 133958. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhang, J.; Tang, Y.; Kong, C.; Li, S.; Wang, S.; Ding, S.; Gu, L.; Shen, X.; Martin, A.A.; et al. Development and validation of rapid screening of 192 veterinary drug residues in aquatic products using HPLC-HRMS coupled with QuEChERS. Food Chem. X 2024, 22, 101504. [Google Scholar] [CrossRef]
- Wang, H.; Tian, H.; Ai, L.F.; Liang, S.X. Screening and quantification of 146 veterinary drug residues in beef and chicken using QuEChERS combined with high performance liquid chromatography-quadrupole orbitrap mass spectrometry. Food Chem. 2023, 408, 135207. [Google Scholar] [CrossRef]
- Mu, P.; Xu, N.; Chai, T.; Jia, Q.; Yin, Z.; Yang, S.; Qian, Y.; Qiu, J. Simultaneous determination of 14 antiviral drugs and relevant metabolites in chicken muscle by uplc-ms/ms after QuEChERS preparation. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1023–1024, 17–23. [Google Scholar] [CrossRef]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Assessment and validation of the p-QuEChERS sample preparation methodology for the analysis of >200 veterinary drugs in various animal-based food matrices. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2023, 40, 356–372. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Sun, J.; Yu, H.; Feng, R.; Zhang, J.; Zhou, H.; Ji, S. Simultaneous Screening of 172 Veterinary drugs by modified QuEChERS-LC-MS/MS in TCM galli gigerii endothelium corneum. J. Chromatogr. Sci. 2024, 62, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Liu, J.; Gou, Y.; Chen, T.; Shen, X.; Wang, T.; Li, Y.; He, H.; Deng, H.; Hua, Y. Determination of veterinary drugs in foods of animal origin by QuEChERS coupled with ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). J. Chromatogr. A 2025, 1744, 465726. [Google Scholar] [CrossRef]
- Chen, L.; Wang, X.; Lu, W.; Wu, X.; Li, J. Molecular imprinting: Perspectives and applications. Chem. Soc. Rev. 2016, 45, 2137–2211. [Google Scholar] [CrossRef]
- Ye, K.; Xu, S.; Zhou, Q.; Wang, S.; Xu, Z.; Liu, Z. Advances in molecular imprinting technology for the extraction and detection of quercetin in plants. Polymers 2023, 15, 2107. [Google Scholar] [CrossRef]
- Xu, S.; Xu, Z.; Liu, Z. Paper-based molecular-imprinting technology and its application. Biosensors 2022, 12, 595. [Google Scholar] [CrossRef]
- Song, X.; Zhou, T.; Li, J.; Su, Y.; Xie, J.; He, L. Determination of macrolide antibiotics residues in pork using molecularly imprinted dispersive solid-phase extraction coupled with LC-MS/MS. J. Sep. Sci. 2018, 41, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Wang, J.; Shi, S.; Hu, S.; Yuan, L. Determination of beta-agonist residues in animal-derived food by a liquid chromatography-tandem mass spectrometric method combined with molecularly imprinted stir bar sorptive extraction. J. Anal. Methods Chem. 2018, 2018, 9053561. [Google Scholar] [CrossRef]
- Elfadil, D.; Lamaoui, A.; Della Pelle, F.; Amine, A.; Compagnone, D. Molecularly Imprinted polymers combined with electrochemical sensors for food contaminants analysis. Molecules 2021, 26, 4607. [Google Scholar] [CrossRef] [PubMed]
- Farooq, S.; Xu, L.; Ullah, S.; Li, J.; Nie, J.; Ping, J.; Ying, Y. Advancements and greenification potential of magnetic molecularly imprinted polymers for chromatographic analysis of veterinary drug residues in milk. Compr. Rev. Food Sci. Food Saf. 2024, 23, e13399. [Google Scholar] [CrossRef]
- Tang, Y.; Meng, H.; Wang, W.; Song, Y.; Wang, S.; Li, Z.; Wang, X.; Hu, X. Off-line magnetic Fe3O4@SiO2@MIPs-based solid phase dispersion extraction coupling with HPLC for the simultaneous determination of olaquindox and its metabolite in fish muscle and milk samples. Food Chem. X 2023, 17, 100611. [Google Scholar] [CrossRef]
- Norman, R.L.; Singh, R.; Muskett, F.W.; Parrott, E.L.; Rufini, A.; Langridge, J.I.; Runau, F.; Dennison, A.; Shaw, J.A.; Piletska, E.; et al. Mass spectrometric detection of KRAS protein mutations using molecular imprinting. Nanoscale 2021, 13, 20401–20411. [Google Scholar] [CrossRef]
- Li, D.; Luo, K.; Zhang, L.; Gao, J.; Liang, J.; Li, J.; Pan, H. Research and application of highly selective molecular imprinting technology in chiral separation analysis. Crit. Rev. Anal. Chem. 2023, 53, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Khatibi, S.A.; Hamidi, S.; Siahi-Shadbad, M.R. Application of liquid-liquid extraction for the determination of antibiotics in the foodstuff: Recent trends and developments. Crit. Rev. Anal. Chem. 2022, 52, 327–342. [Google Scholar] [CrossRef]
- Wang, J.; Yang, H.Y.; Wang, X.D.; Lv, Y.F.; Wei, N. Application of QuEChERS for analysis of contaminants in dairy products: A review. J. Food Prot. 2025, 88, 100453. [Google Scholar] [CrossRef]
- Xiao, Z.; Wang, J.; Cao, Y.; Yao, T.; Wang, S.; Liu, J.; Suo, D.; Tian, J.; Jia, Z.; Li, Y.; et al. Quick and high-throughput quantification of 22 beta-agonists residues in animal-derived foods using enzymatic probe sonication. Food Chem. 2023, 408, 135262. [Google Scholar] [CrossRef]
- Zhang, Q.; Du, H.; Zhang, Y. Recent progress on the detection of animal-derived food stimulants using mass spectrometry-based techniques. Front. Nutr. 2023, 10, 1226530. [Google Scholar] [CrossRef] [PubMed]
- Hajrulai-Musliu, Z.; Uzunov, R.; Jovanov, S.; Musliu, D.; Dimitrieska-Stojkovikj, E.; Stojanovska-Dimzoska, B.; Angeleska, A.; Stojkovski, V.; Sasanya, J.J. Multi-class/residue method for determination of veterinary drug residues, mycotoxins and pesticide in urine using LC-MS/MS technique. BMC Vet. Res. 2023, 19, 156. [Google Scholar] [CrossRef]
- Li, G.; Li, H.; Zhai, J.; Guo, J.; Li, Q.; Wang, C.F.; Chen, S. Microfluidic fluorescent platform for rapid and visual detection of veterinary drugs. RSC Adv. 2022, 12, 8485–8491. [Google Scholar] [CrossRef]
- Mondello, L.; Cordero, C.; Janssen, H.-G.; Synovec, R.E.; Zoccali, M.; Tranchida, P.Q. Comprehensive two-dimensional gas chromatography–mass spectrometry. Nat. Rev. Methods Primers 2025, 5, 7. [Google Scholar] [CrossRef]
- Brettell, T.A.; Lum, B.J. Analysis of drugs of abuse by gas chromatography-mass spectrometry (gc-ms). Methods Mol. Biol. 2018, 1810, 29–42. [Google Scholar] [CrossRef]
- Gruber, B.; David, F.; Sandra, P. Capillary gas chromatography-mass spectrometry: Current trends and perspectives. TrAC Trends Anal. Chem. 2020, 124, 115475. [Google Scholar] [CrossRef]
- Welke, J.E.; Hernandes, K.C.; Lago, L.O.; Silveira, R.D.; Marques, A.T.B.; Zini, C.A. Flavoromic analysis of wines using gas chromatography, mass spectrometry and sensory techniques. J. Chromatogr. A 2024, 1734, 465264. [Google Scholar] [CrossRef]
- Liu, S.; Tang, Y.; Lu, Y.; Guo, Y.; Xie, K.; Guan, F.; Gao, P.; Zhu, Y.; Dong, Y.; Zhang, T.; et al. Qualitative and quantitative determination of decoquinate in chicken tissues by gas chromatography tandem mass spectrometry. Molecules 2023, 28, 3875. [Google Scholar] [CrossRef]
- Wang, B.; Wang, Y.; Xie, X.; Diao, Z.; Xie, K.; Zhang, G.; Zhang, T.; Dai, G. Quantitative analysis of spectinomycin and lincomycin in poultry eggs by accelerated solvent extraction coupled with gas chromatography tandem mass spectrometry. Foods 2020, 9, 651. [Google Scholar] [CrossRef]
- Guo, Y.; Xie, X.; Diao, Z.; Wang, Y.; Wang, B.; Xie, K.; Wang, X.; Zhang, P. Detection and determination of spectinomycin and lincomycin in poultry muscles and pork by ASE-SPE-GC–MS/MS. J. Food Compos. Anal. 2021, 101, 103979. [Google Scholar] [CrossRef]
- Yu, H.; Tao, Y.; Le, T.; Chen, D.; Ishsan, A.; Liu, Y.; Wang, Y.; Yuan, Z. Simultaneous determination of amitraz and its metabolite residue in food animal tissues by gas chromatography-electron capture detector and gas chromatography-mass spectrometry with accelerated solvent extraction. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2010, 878, 1746–1752. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.-J.; Schultz, A.W.; Wang, J.; Johnson, C.H.; Yannone, S.M.; Patti, G.J.; Siuzdak, G. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the metlin database. Nat. Protoc. 2013, 8, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Glish, G.L.; Burinsky, D.J. Hybrid mass spectrometers for tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2008, 19, 161–172. [Google Scholar] [CrossRef]
- Chernushevich, I.V.; Loboda, A.V.; Thomson, B.A. An introduction to quadrupole-time-of-flight mass spectrometry. J. Mass Spectrom. JMS 2001, 36, 849–865. [Google Scholar] [CrossRef]
- Allen, D.R.; McWhinney, B.C. Quadrupole Time-of-Flight Mass Spectrometry: A paradigm shift in toxicology screening applications. Clin. Biochem. Rev. 2019, 40, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Du, M.; Wei, K.; Dai, C.; Yang, R.Y.H.; Zhou, B.; Luo, Z.; Yang, X.; Yu, Y.; Lin, W.; et al. Study of Xuanhuang Pill in protecting against alcohol liver disease using ultra-performance liquid chromatography/time-of-flight mass spectrometry and network pharmacology. Front. Endocrinol. 2023, 14, 1175985. [Google Scholar] [CrossRef]
- Zhang, H.; Chang, Q.; Li, J.; Qiu, G.; Wu, F.; Zhu, R.; Wang, X.; Su, M. A liquid chromatography-time-of-flight/mass spectrometry method for analysis of pesticides and transfer behavior in Radix Codonopsis and Angelica sinensis decoctions. Anal. Methods 2023, 15, 2121–2131. [Google Scholar] [CrossRef]
- Li, X.; Chi, Q.; Xia, S.; Pan, Y.; Chen, Y.; Wang, K. Untargeted multi-residue method for the simultaneous determination of 141 veterinary drugs and their metabolites in pork by high-performance liquid chromatography time-of-flight mass spectrometry. J. Chromatogr. A 2020, 1634, 461671. [Google Scholar] [CrossRef] [PubMed]
- Vardali, S.C.; Samanidou, V.F.; Kotzamanis, Y.P. Development and validation of an ultra performance liquid chromatography-quadrupole time of flight-mass spectrometry (in MS(E) mode) method for the quantitative determination of 20 antimicrobial residues in edible muscle tissue of European sea bass. J. Chromatogr. A 2018, 1575, 40–48. [Google Scholar] [CrossRef]
- Choi, B.S.; Lee, D.U.; Kim, W.S.; Park, C.W.; Choe, W.J.; Moon, M.J. Simultaneous screening of 322 residual pesticides in fruits and vegetables using gc-ms/ms and deterministic health risk assessments. Foods 2023, 12, 3001. [Google Scholar] [CrossRef]
- Petrarca, M.H.; Braga, P.A.C.; Reyes, F.G.R.; Bragotto, A.P.A. Exploring miniaturized sample preparation approaches combined with LC-QToF-MS for the analysis of sulfonamide antibiotic residues in meat- and/or egg-based baby foods. Food Chem. 2022, 366, 130587. [Google Scholar] [CrossRef] [PubMed]
- Stachniuk, A.; Kozub, A.; Czeczko, R.; Montowska, M.; Fornal, E. LC-QTOF-MS evaluation of rabbit-specific peptide markers for meat quantitation. J. Food Drug Anal. 2022, 30, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Yaseen, S.B.; Akram, A.; Musharraf, S.G.; Wajidi, M.; Tabassum, N.; Nazir, N.; Shah, S.M.Z. The lc-qtof-ms/ms analysis of acid degradation products of rifaximin, an antibiotic. MethodsX 2022, 9, 101735. [Google Scholar] [CrossRef]
- Zhang, M.; Li, E.; Su, Y.; Zhang, Y.; Xie, J.; He, L. Quick Multi-Class Determination of Residues of Antimicrobial Veterinary Drugs in Animal Muscle by LC-MS/MS. Molecules 2018, 23, 1736. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Kanda, M.; Yoshikawa, S.; Nakajima, T.; Hayashi, H.; Matsushima, Y.; Ohba, Y.; Koike, H.; Nagano, C.; Otsuka, K.; et al. Single-laboratory Validation Study and Surveillance Using an Improved Multiresidue Analytical Method for Veterinary Drugs in Livestock Products by lc-ms/ms. Shokuhin Eiseigaku Zasshi 2023, 64, 53–60. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, Z.; Yu, H.; Chen, Y.; Yuan, F.; Zhang, X.; Fang, S. Furazolidone and nitrofurazone metabolic studies in crucian carp by ultra-performance liquid chromatography Tandem Mass Spectrometry. J. Chromatogr. Sci. 2022, 60, 963–969. [Google Scholar] [CrossRef]
- Cheng, Y.H.; Lee, C.H.; Wang, S.Y.; Chou, C.Y.; Yang, Y.J.; Kao, C.C.; Wu, H.Y.; Dong, Y.; Hung, W.Y.; Su, C.Y.; et al. Multiplexed antibody glycosylation profiling using dual enzyme digestion and liquid chromatography-triple quadrupole mass spectrometry method. Mol. Cell Proteom. 2024, 23, 100710. [Google Scholar] [CrossRef]
- Jung, Y.S.; Kim, D.B.; Nam, T.G.; Seo, D.; Yoo, M. Identification and quantification of multi-class veterinary drugs and their metabolites in beef using LC-MS/MS. Food Chem. 2022, 382, 132313. [Google Scholar] [CrossRef]
- Lehotay, S.J. Comparison of analyte identification criteria and other aspects in triple quadrupole tandem mass spectrometry: Case study using UHPLC-MS/MS for regulatory analysis of veterinary drug residues in liquid and powdered eggs. Anal. Bioanal. Chem. 2022, 414, 287–302. [Google Scholar] [CrossRef]
- Na, T.W.; Seo, H.J.; Jang, S.N.; Kim, H.; Yun, H.; Kim, H.; Ahn, J.; Cho, H.; Hong, S.H.; Kim, H.J.; et al. Multi-residue analytical method for detecting pesticides, veterinary drugs, and mycotoxins in feed using liquid- and gas chromatography coupled with mass spectrometry. J. Chromatogr. A 2022, 1676, 463257. [Google Scholar] [CrossRef]
- Wittenberg, J.B.; Simon, K.A.; Wong, J.W. Targeted multiresidue analysis of veterinary drugs in milk-based powders using liquid chromatography-tandem mass spectrometry (lc-ms/ms). J. Agric. Food Chem. 2017, 65, 7288–7293. [Google Scholar] [CrossRef] [PubMed]
- Desmarchelier, A.; Bessaire, T.; Savoy, M.C.; Tarres, A.; Mujahid, C.; Beck, A.; Mottier, P.; Delatour, T. Screening of 154 veterinary drug residues in foods of animal origin using lc-ms/ms: First action 2020.04. J. AOAC Int. 2021, 104, 650–681. [Google Scholar] [CrossRef]
- Jang, S.; Seo, H.; Kim, H.; Kim, H.; Ahn, J.; Cho, H.; Hong, S.; Lee, S.; Na, T. Development of a quantitative method for detection of multiclass veterinary drugs in feed using modified quppe extraction and lc-ms/ms. Molecules 2022, 27, 4483. [Google Scholar] [CrossRef] [PubMed]
- Khadim, A.; Yaseen Jeelani, S.U.; Khan, M.N.; Kumari, S.; Raza, A.; Ali, A.; Zareena, B.; Zaki Shah, S.M.; Musharraf, S.G. Targeted analysis of veterinary drugs in food samples by developing a high-resolution tandem mass spectral library. J. Agric. Food Chem. 2023, 71, 12839–12848. [Google Scholar] [CrossRef] [PubMed]
- Al-Shakliah, N.S.; Kadi, A.A.; Aljohar, H.I.; AlRabiah, H.; Attwa, M.W. Profiling of in vivo, in vitro and reactive zorifertinib metabolites using liquid chromatography ion trap mass spectrometry. RSC Adv. 2022, 12, 20991–21003. [Google Scholar] [CrossRef]
- Berendsen, B.J.A.; Meijer, T.; Mol, H.G.J.; van Ginkel, L.; Nielen, M.W.F. A global inter-laboratory study to assess acquisition modes for multi-compound confirmatory analysis of veterinary drugs using liquid chromatography coupled to triple quadrupole, time of flight and orbitrap mass spectrometry. Anal. Chim. Acta 2017, 962, 60–72. [Google Scholar] [CrossRef]
- Guo, Y.; Mao, R.; Zhang, Y.; Li, R.; Oduro, P.K.; Si, D.; Han, L.; Huang, Y.; Pan, G. An integrated strategy for the systematic chemical characterization of Salvianolate lyophilized injection using four scan modes based on the ultra-high performance liquid chromatography-triple quadrupole-linear ion trap mass spectrometry. J. Pharm. Biomed. Anal. 2022, 215, 114769. [Google Scholar] [CrossRef]
- Chen, G.Y.; Zhang, Q. Quantification of plasma oxylipins using solid-phase extraction and reversed-phase liquid chromatography-triple quadrupole mass spectrometry. Methods Mol. Biol. 2021, 2306, 171–186. [Google Scholar] [CrossRef]
- Kang, J.; Park, S.J.; Park, H.C.; Hossain, M.A.; Kim, M.A.; Son, S.W.; Lim, C.M.; Kim, T.W.; Cho, B.H. Multiresidue screening of veterinary drugs in meat, milk, egg, and fish using liquid chromatography coupled with ion trap time-of-flight mass spectrometry. Appl. Biochem. Biotechnol. 2017, 182, 635–652. [Google Scholar] [CrossRef]
- Casado, N.; Morante-Zarcero, S.; Perez-Quintanilla, D.; Sierra, I. Application of a hybrid ordered mesoporous silica as sorbent for solid-phase multi-residue extraction of veterinary drugs in meat by ultra-high-performance liquid chromatography coupled to ion-trap tandem mass spectrometry. J. Chromatogr. A 2016, 1459, 24–37. [Google Scholar] [CrossRef]
- Sabei, F.Y.; Khardali, I.; Al-Kasim, M.A.; Shaheen, E.S.; Oraiby, M.; Alamir, A.; David, B.; Alshahrani, S.; Jali, A.M.; Attafi, M.; et al. Disposition kinetics of cathinone and its metabolites after oral administration in rats. Curr. Drug Metab. 2024, 25, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, B.; Xu, L.; Xiong, L.; Guo, D.; Cui, W.; Xu, X.; Lin, Y. Quality consistency evaluation on three species of North Patrininae herba by high-performance liquid chromatography coupled with electrospray ion trap time-of-flight mass spectrometry and network pharmacology approaches. J. Sep. Sci. 2022, 45, 3852–3865. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, X.; Geng, Y.; Dai, J.; Tang, S.; Adams, E.; Chen, D.; Yuan, Y. Analysis of impurity profiling of arbekacin sulfate by ion-pair liquid chromatography coupled with pulsed electrochemical detection and online ion suppressor-ion trap-time off light mass spectrometry. J. Pharm. Biomed. Anal. 2022, 221, 115061. [Google Scholar] [CrossRef]
- Saade, J.; Biacchi, M.; Giorgetti, J.; Lechner, A.; Beck, A.; Leize-Wagner, E.; Francois, Y.N. Analysis of monoclonal antibody glycopeptides by capillary electrophoresis-mass spectrometry coupling (ce-ms). Methods Mol. Biol. 2021, 2271, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Ferre, S.; Boccard, J.; Rudaz, S.; Gonzalez-Ruiz, V. Evaluation of prototype ce-ms interfaces. Methods Mol. Biol. 2022, 2531, 1–13. [Google Scholar] [CrossRef]
- Latosinska, A.; Siwy, J.; Mischak, H.; Frantzi, M. Peptidomics and proteomics based on ce-ms as a robust tool in clinical application: The past, the present, and the future. Electrophoresis 2019, 40, 2294–2308. [Google Scholar] [CrossRef]
- Fang, P.; Pan, J.Z.; Fang, Q. A robust and extendable sheath flow interface with minimal dead volume for coupling ce with esi-ms. Talanta 2018, 180, 376–382. [Google Scholar] [CrossRef]
- Sanchez-Lopez, E.; Marina, M.L. Applications of capillary electrophoresis-mass spectrometry to chiral analysis. Methods Mol. Biol. 2022, 2531, 211–225. [Google Scholar] [CrossRef]
- Blasco, C.; Pico, Y.; Andreu, V. Analytical method for simultaneous determination of pesticide and veterinary drug residues in milk by CE-MS. Electrophoresis 2009, 30, 1698–1707. [Google Scholar] [CrossRef]
- Seyfinejad, B.; Jouyban, A. Capillary electrophoresis-mass spectrometry in pharmaceutical and biomedical analyses. J. Pharm. Biomed. Anal. 2022, 221, 115059. [Google Scholar] [CrossRef]
- Shamsi, S.A.; Akter, F. Capillary electrophoresis mass spectrometry: Developments and applications for enantioselective analysis from 2011–2020. Molecules 2022, 27, 4126. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lu, Y.; Zhang, L.; Qin, L.; Wen, H.; Fan, X.; Peng, D. Highly sensitive magnetic-nanoparticle-based immunochromatography assay for rapid detection of amantadine in chicken and eggs. Biosensors 2023, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yin, X.; Yang, H.; Wen, H.; Han, S.; Pan, X.; Li, H.; Peng, D. A sensitive and specific monoclonal antibody based enzyme-linked immunosorbent assay for the rapid detection of pretilachlor in grains and the environment. Foods 2023, 13, 12. [Google Scholar] [CrossRef]
- Lee, M.J.; Aliya, S.; Lee, E.S.; Jeon, T.J.; Oh, M.H.; Huh, Y.S. Simple and rapid detection of ractopamine in pork with comparison of LSPR and LFIA sensors. J. Food Drug Anal. 2022, 30, 590–602. [Google Scholar] [CrossRef]
- Ahmed, S.; Ning, J.; Peng, D.; Chen, T.; Ahmad, I.; Ali, A.; Lei, Z.; Abu bakr Shabbir, M.; Cheng, G.; Yuan, Z. Current advances in immunoassays for the detection of antibiotics residues: A review. Food Agric. Immunol. 2020, 31, 268–290. [Google Scholar] [CrossRef]
- Calidonio, J.M.; Hamad-Schifferli, K. Biophysical and biochemical insights in the design of immunoassays. Biochim. Biophys. Acta Gen. Subj. 2023, 1867, 130266. [Google Scholar] [CrossRef]
- Sun, B.; Chen, Z.; Feng, B.; Chen, S.; Feng, S.; Wang, Q.; Niu, X.; Zhang, Z.; Zheng, P.; Lin, M.; et al. Development of a colloidal gold-based immunochromatographic assay for rapid detection of nasal mucosal secretory IgA against SARS-CoV-2. Front. Microbiol. 2024, 15, 1386891. [Google Scholar] [CrossRef]
- Ziyang, H.; Shaoen, Z.; Qingzhou, C.; Hong, L.; Jianxin, S.; Kaiqiang, W.; Huiying, W.; Limin, C. Novel solid-phase extraction promotes simultaneous colloidal gold immunochromatographic assay of malachite green, leuco-malachite green, chloramphenicol, and semi-carbazone metabolites in aquatic products. Food Chem. 2025, 465, 142118. [Google Scholar] [CrossRef] [PubMed]
- Na, G.; Hu, X.; Yang, J.; Sun, Y.; Kwee, S.; Tang, L.; Xing, G.; Xing, Y.; Zhang, G. A rapid colloidal gold-based immunochromatographic strip assay for monitoring nitroxynil in milk. J. Sci. Food Agric. 2020, 100, 1860–1866. [Google Scholar] [CrossRef]
- Chen, L.; Sun, Y.; Hu, X.; Xing, Y.; Kwee, S.; Na, G.; Zhang, G. Colloidal gold-based immunochromatographic strip assay for the rapid detection of diminazene in milk. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2020, 37, 1667–1677. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, S.; Cai, X.; Cao, M.; Lu, Q.; Hu, D.; Chen, Q.; Xiong, X. A rapid colloidal gold immunochromatographic assay based on polyclonal antibodies against HtpsC protein for the detection of Streptococcus suis. Front. Microbiol. 2023, 14, 1294368. [Google Scholar] [CrossRef] [PubMed]
- Na, G.; Hu, X.; Yang, J.; Sun, Y.; Kwee, S.; Tang, L.; Xing, G.; Xing, Y.; Zhang, G. Colloidal gold-based immunochromatographic strip assay for the rapid detection of bacitracin zinc in milk. Food Chem. 2020, 327, 126879. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Cao, Y.; Yan, M.; Sun, M.; Zhang, Q.; Wang, J.; Fu, G.; Liu, R.; Huang, Y.; Su, J. Development of a colloidal gold immunochromatographic assay for duck enteritis virus detection using monoclonal antibodies. Pathogens 2021, 10, 365. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Qian, C.; Jiang, H.; Yang, H. Development of an enzyme-linked immunosorbent assay for the determination of maduramicin in broiler chicken tissues. J. Agric. Food Chem. 2001, 49, 2697–2701. [Google Scholar] [CrossRef]
- Chen, R.; Shang, H.; Niu, X.; Huang, J.; Miao, Y.; Sha, Z.; Qin, L.; Huang, H.; Peng, D.; Zhu, R. Establishment and evaluation of an indirect ELISA for detection of antibodies to goat Klebsiella pneumonia. BMC Vet. Res. 2021, 17, 107. [Google Scholar] [CrossRef]
- Qin, L.; Xiao, J.; Yang, H.; Liang, J.; Li, L.; Wu, S.; Peng, D. Rapid immunoassays for the detection of quinoxalines and their metabolites residues in animal-derived foods: A review. Food Chem. 2024, 443, 138539. [Google Scholar] [CrossRef]
- Wernike, K.; Aebischer, A.; Michelitsch, A.; Hoffmann, D.; Freuling, C.; Balkema-Buschmann, A.; Graaf, A.; Muller, T.; Osterrieder, N.; Rissmann, M.; et al. Multi-species ELISA for the detection of antibodies against SARS-CoV-2 in animals. Transbound. Emerg. Dis. 2021, 68, 1779–1785. [Google Scholar] [CrossRef]
- Fu, B.; Gao, H.; Fang, C.; Cheng, G.; Wang, H.; Wang, Y.; Hao, H.; Wang, X.; Huang, L.; Peng, D. Development of a monoclonal antibody-based indirect competitive enzyme-linked immunosorbent assay for screening of diethylstilbestrol in animal-derived foods. Heliyon 2024, 10, e39769. [Google Scholar] [CrossRef]
- Yin, X.; Li, H.; Wu, S.; Lu, Y.; Yang, Y.; Qin, L.; Li, L.; Xiao, J.; Liang, J.; Si, Y.; et al. A sensitive and specific enzyme-linked immunosorbent assay for the detection of pymetrozine in vegetable, cereal, and meat. Food Chem. 2023, 418, 135949. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Yang, H. Development of a nucleocapsid protein-based blocking elisa for the detection of porcine deltacoronavirus antibodies. Viruses 2022, 14, 1815. [Google Scholar] [CrossRef]
- He, D.; Sun, M.; Jiang, X.; Zhang, S.; Wei, F.; Wu, B.; Diao, Y.; Tang, Y. Development of an indirect competitive ELISA method based on ORF2 detecting the antibodies of novel goose astrovirus. J. Virol. Methods 2023, 311, 114643. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, J.; Li, Z.; He, X.; Zhao, S.; Ma, Q.; Li, X.; Liu, J.; Liu, A.; Li, Y.; et al. A universal ELISA assay for detecting six strains of ovine Babesia species in China. Vet. Parasitol. 2021, 300, 109616. [Google Scholar] [CrossRef]
- Guo, L.; Liu, M.; Li, Q.; Dong, B.; Li, H.; Mari, G.M.; Liu, R.; Yu, W.; Yu, X.; Wang, Z.; et al. Synthesis and characterization of tracers and development of a fluorescence polarization immunoassay for amantadine with high sensitivity in chicken. J. Food Sci. 2021, 86, 4754–4767. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liang, D.; Xiong, J.; Wang, Z.; Zheng, P.; Zhang, H.; Ren, Z.; Jiang, H. Development of a sensitive and rapid fluorescence polarization immunoassay for high throughput screening eight glucocorticoids in beef. J. Pharm. Biomed. Anal. 2022, 214, 114719. [Google Scholar] [CrossRef]
- Mukhametova, L.I.; Eremin, S.A.; Arutyunyan, D.A.; Goryainova, O.S.; Ivanova, T.I.; Tillib, S.V. Fluorescence Polarization Immunoassay of Human Lactoferrin in Milk Using Small Single-Domain Antibodies. Biochemistry 2022, 87, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, O.D.; Mukhametova, L.I.; Zvereva, E.A.; Zherdev, A.V.; Eremin, S.A. A sensitive fluorescence polarization immunoassay for the rapid detection of okadaic acid in environmental waters. Biosensors 2023, 13, 477. [Google Scholar] [CrossRef]
- Smith, D.S.; Eremin, S.A. Fluorescence polarization immunoassays and related methods for simple, high-throughput screening of small molecules. Anal. Bioanal. Chem. 2008, 391, 1499–1507. [Google Scholar] [CrossRef]
- Mukhametova, L.I.; Eremin, S.A. Fluorescence polarization assays for organic compounds in food safety. Front. Biosci. (Elite Ed.) 2024, 16, 4. [Google Scholar] [CrossRef]
- Duan, C.; Zhang, H.; Zhang, Y.; Li, Q.; Li, P.; Mari, G.M.; Eremin, S.A.; Shen, J.; Wang, Z. A robust homogeneous fluorescence polarization immunoassay for rapid determination of erythromycin in milk. Foods 2023, 12, 1581. [Google Scholar] [CrossRef]
- Duan, C.; Zhang, Y.; Li, P.; Li, Q.; Yu, W.; Wen, K.; Eremin, S.A.; Shen, J.; Yu, X.; Wang, Z. Dual-wavelength fluorescence polarization immunoassay for simultaneous detection of sulfonamides and antibacterial synergists in milk. Biosensors 2022, 12, 1053. [Google Scholar] [CrossRef]
- Zhang, Q.; Zou, M.; Wang, W.; Li, J.; Liang, X. Design, synthesis, and characterization of tracers and development of a fluorescence polarization immunoassay for rapid screening of 4,4′-dinitrocarbanilide in chicken muscle. Foods 2021, 10, 1822. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yang, J.; Tao, Z.; Eremin, S.A.; Hua, X.; Wang, M. Development of fluorescence polarization immunoassay for imidacloprid in environmental and agricultural samples. Front. Chem. 2020, 8, 615594. [Google Scholar] [CrossRef]
- Chowdhury, J.; Ranc, V.; Sarkar, S. Editorial: Surface enhanced raman scattering: Theory and applications. Front. Chem. 2023, 11, 1176495. [Google Scholar] [CrossRef]
- Liu, H.L.; Zhan, K.; Wang, K.; Xia, X.H. Nanopore-based surface-enhanced Raman scattering technologies. Sci. Bull. 2022, 67, 1539–1541. [Google Scholar] [CrossRef]
- Tanwar, S.; Kim, J.H.; Bulte, J.W.M.; Barman, I. Surface-enhanced Raman scattering: An emerging tool for sensing cellular function. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2022, 14, e1802. [Google Scholar] [CrossRef]
- Dhakal, S.; Chao, K.; Huang, Q.; Kim, M.; Schmidt, W.; Qin, J.; Broadhurst, C.L. A simple surface-enhanced raman spectroscopic method for on-site screening of tetracycline residue in whole milk. Sensors 2018, 18, 424. [Google Scholar] [CrossRef]
- Fan, R.; Tang, S.; Luo, S.; Liu, H.; Zhang, W.; Yang, C.; He, L.; Chen, Y. Duplex surface enhanced raman scattering-based lateral flow immunosensor for the low-level detection of antibiotic residues in milk. Molecules 2020, 25, 5249. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, Y.; Jing, X.; Ma, W. SERS-active plasmonic metal NP-CsPbX(3) films for multiple veterinary drug residues detection. Food Chem. 2023, 412, 135420. [Google Scholar] [CrossRef]
- Xu, M.L.; Gao, Y.; Han, X.X.; Zhao, B. Innovative Application of sers in food quality and safety: A brief review of recent trends. Foods 2022, 11, 5249. [Google Scholar] [CrossRef]
- Wang, L.; Ma, P.; Chen, H.; Chang, M.; Lu, P.; Chen, N.; Yuan, Y.; Chen, N.; Zhang, X. Rapid determination of mixed pesticide residues on apple surfaces by surface-enhanced raman spectroscopy. Foods 2022, 11, 1089. [Google Scholar] [CrossRef]
- Lee, K.M.; Yarbrough, D.; Kozman, M.M.; Herrman, T.J.; Park, J.; Wang, R.; Kurouski, D. A rapid and convenient screening method for detection of restricted monensin, decoquinate, and lasalocid in animal feed by applying SERS and chemometrics. Food Chem. Toxicol. 2020, 144, 111633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Lv, H.; Wang, J.; Yang, Z.; Ding, Y.; Zhao, B.; Tian, Y. An aptamer-based colorimetric/SERS dual-mode sensing strategy for the detection of sulfadimethoxine residues in animal-derived foods. Anal. Methods 2023, 15, 1047–1053. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, S.; Xie, Y.; Zhang, L.; Shao, Y.; Lin, J.; Wu, A. Advances in SERS detection method combined with microfluidic technology for bio-analytical applications. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2025, 332, 125797. [Google Scholar] [CrossRef] [PubMed]
- Vanaja, P.; Moorthy, N.; Rajpoot, V.S.; Rao, H.; Goswami, R.K.; Subash, P.; Khute, S.; Rao, K.S. Metabolite profiling, antimalarial potentials of Schleichera oleosa using LC-MS and GC-MS: In vitro, molecular docking and molecular dynamics. Front. Mol. Biosci. 2025, 12, 1543939. [Google Scholar] [CrossRef]
- Johnson, K.R.; Gao, Y.; Gregus, M.; Ivanov, A.R. On-capillary cell lysis enables top-down proteomic analysis of single mammalian cells by ce-ms/ms. Anal. Chem. 2022, 94, 14358–14367. [Google Scholar] [CrossRef]
- Tiseo, K.; Huber, L.; Gilbert, M.; Robinson, T.P.; Van Boeckel, T.P. Global Trends in Antimicrobial Use in Food Animals from 2017 to 2030. Antibiotics 2020, 9, 918. [Google Scholar] [CrossRef]
- Rapacz, D.; Smolinska-Kempisty, K.; Wolska, J. Preparation and characterization of SPE column with smart green molecularly imprinted polymers materials for selective determination of S-metolachlor herbicide. Sci. Rep. 2025, 15, 3153. [Google Scholar] [CrossRef]
- Zad, N.; Tell, L.A.; Ampadi Ramachandran, R.; Xu, X.; Riviere, J.E.; Baynes, R.; Lin, Z.; Maunsell, F.; Davis, J.; Jaberi-Douraki, M. Development of machine learning algorithms to estimate maximum residue limits for veterinary medicines. Food Chem. Toxicol. 2023, 179, 113920. [Google Scholar] [CrossRef]
- Wu, P.Y.; Chou, W.C.; Wu, X.; Kamineni, V.N.; Kuchimanchi, Y.; Tell, L.A.; Maunsell, F.P.; Lin, Z. Development of machine learning-based quantitative structure-activity relationship models for predicting plasma half-lives of drugs in six common food animal species. Toxicol. Sci. 2025, 203, 52–66. [Google Scholar] [CrossRef]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Improving the QuEChERS liquid/liquid extraction of analytes with widely varying physicochemical properties: Example of 201 veterinary drugs in milk. J. AOAC Int. 2022, 105, 1030–1042. [Google Scholar] [CrossRef]
- Fodor, A.; Cseppento, D.C.N.; Badea, G.E.; Petrehele, A.I.; Groze, A.; Tit, D.M.; Bungau, S.G. Colloidal gold immunochromatography and elisa traceability of tetracycline residues from raw milk to its dairy products. In Vivo 2023, 37, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Ge, L.; Li, J.; Qiu, G.; Wu, F.; Zhang, H.; Xu, F.; Zhu, R.; Qi, P.; Bai, R.; et al. Automated QuEChERS for the determination of 482 pesticide residues in Radix codonopsis by GC-Q-TOF/MS and LC-Q-TOF/MS. Anal. Methods 2021, 13, 5660–5669. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhang, S.; Chen, J.; Luo, W.; Kang, F.; Ren, Y.; Zhou, W. Development and application of a new QuEChERS method coupled with uplc-qtof-ms/ms for analysis of tiafenacil and its photolysis products in water. J. Agric. Food Chem. 2024, 72, 27007–27018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, H.; Chen, W.; Zhang, J. Nanomaterials used in fluorescence polarization based biosensors. Int. J. Mol. Sci. 2022, 23, 8625. [Google Scholar] [CrossRef]
- Park, J.; Kim, Y.W.; Jeon, H.J. Machine learning-driven innovations in microfluidics. Biosensors 2024, 14, 613. [Google Scholar] [CrossRef]
- Zhang, D.; He, S.; Wang, Z.; Yan, P.; Li, H.; Xu, X.; Zeng, Q.; Wang, N.; Chen, X. Recent advances in CRISPR/Cas systems-associated surface enhanced Raman spectroscopy biosensor towards nucleic acid detection. Med. Nov. Technol. Devices 2024, 24, 100336. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | LOD (μg/kg) | Accuracy (Recovery Rate) | Applicability | Advantages | Disadvantages | Feasibility | Cost-Effectiveness |
|---|---|---|---|---|---|---|---|
| LLE | 0.5–1.0 | 71.4–120% | Suitable for lipophilic drugs in aquatic products, milk, and tissues. | Excellent selectivity for lipophilic drugs. Simple operation with minimal equipment. Low cost. | High solvent consumption. Prone to emulsification. Limited extraction efficiency for polar analytes. | Simple but requires solvent management. | Low |
| SPE | 0.2–3.0 | 60–120% | Broad applicability for diverse matrices (honey, muscle, milk, and eggs). | Effective enrichment of trace residues. High selectivity with tailored adsorbents. Reduced solvent use. | High adsorbent costs. Complex optimization for matrix effects. Susceptible to column clogging. | Requires skilled optimization. | Moderate to high |
| IAC | 0.04–0.10 | 74.5–105% | Ideal for specific targets (e.g., chloramphenicol and β-agonists) in muscle/liver. | Exceptional specificity via antibody–antigen binding. High sensitivity for trace residues. High-throughput potential. | Antibody development is costly/time-consuming. Limited column lifespan. Cross-reactivity risks. | Antibody-dependent and storage-sensitive. | High |
| QuEChERS | 0.15–3.03 | 52.1–138.2% | Effective for high-fat matrices (beef and chicken) and multi-residue analysis. | Rapid and simple. Cost-effective with minimal solvents. Effective impurity removal. | Sorbent selectivity limitations. Residual matrix interference. Optimization challenges for diverse analytes. | Easy to implement with standard lab tools. | Low to moderate |
| MIT | 0.05–0.5 | 68.6–95.5% | Customizable for antibiotics (e.g., tetracyclines and β-agonists) in complex matrices. | Tailored specificity via imprinting. Reusable and stable. Adaptable to diverse targets. | Labor-intensive synthesis. Cross-reactivity with structural analogs. Requires confirmatory methods. | Specialized expertise needed for polymer design. | Moderate |
| Method | LOD (μg/kg) | Accuracy (Recovery Rate) | Applicability | Advantages | Disadvantages | Feasibility | Costing |
|---|---|---|---|---|---|---|---|
| GC-MS | 2.3–4.3 | 77.38–95.7% | Volatile/semi-volatile compounds. | High specificity for volatile analytes. Robust qualitative capabilities. Wide applicability for small molecules. | Requires derivatization for non-volatile compounds. Limited to thermally stable analytes. | Requires derivatization expertise. | Moderate to high |
| LC-QTOF-MS | 0.5 | More than 70% | High-resolution multi-residue screening. | Ultra-high resolution for accurate mass identification. Broad-spectrum detection. Rich structural data. | High equipment/maintenance costs. Demands advanced data analysis skills. | Requires high-end infrastructure. | Very high |
| LC-MS/MS | 0.02–82 | 70–120% | Gold standard for trace-level quantification. | High sensitivity and selectivity. Reliable for multi-residue analysis. Robust quantitative accuracy. | Expensive instrumentation. Complex sample preparation. Matrix effects require mitigation. | Skilled operation and maintenance needed. | High |
| LC-IT-MS | 0.01–18.75 | 63–122% | Multi-stage fragmentation for structural elucidation. | Mul-ti-stage mass for structural insights. Compact and cost-effective. | Slower scanning speeds. Moderate resolution limits complex mixture analysis. | Suitable for targeted analysis. | Moderate |
| CE-MS | 1–9 | More than 78% | Ionizable metabolites. | High separation efficiency. Minimal sample/reagent consumption. Fast analysis. | Poor reproducibility due to buffer/temperature sensitivity. | Technically demanding for calibration. | Moderate |
| GICA | 0.01–0.5 | 84.2–112.9% | Rapid on-site screening. | Equipment-free, rapid results. Low cost and user-friendly. | Qualitative/semi-quantitative only. Limited sensitivity for trace residues. Matrix interference risks. | Ideal for field testing. | Low |
| ELISA | 1.56–2.72 | 70.1–103.1% | High-throughput screening. | High throughput and specificity. Cost-effective for batch analysis. Minimal instrumentation. | Cross-reactivity with analogs. Enzyme activity affected by environmental factors. | Requires antibody development. | Low to moderate |
| FPIA | 0.01 | 78.6–107.77% | Homogeneous assays. | Rapid and homogeneous. Minimal sample pretreatment. Moderate sensitivity. | Limited by antibody/tracer availability. Matrix interference in complex samples. | Suitable for simple matrices. | Moderate |
| SERS | 0.01–0.015 | 88.8–111.3% | Ultra-trace detection. | Ultra-high sensitivity. Rapid and minimal pretreatment. Multiplexing potential. | Poor reproducibility due to nanoparticle variability. | Requires nanoparticle optimization. | High |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, Q.; Tang, S.; Dai, C. Recent Advances in Pretreatment Methods and Detection Techniques for Veterinary Drug Residues in Animal-Derived Foods. Metabolites 2025, 15, 233. https://doi.org/10.3390/metabo15040233
Dai Q, Tang S, Dai C. Recent Advances in Pretreatment Methods and Detection Techniques for Veterinary Drug Residues in Animal-Derived Foods. Metabolites. 2025; 15(4):233. https://doi.org/10.3390/metabo15040233
Chicago/Turabian StyleDai, Qing, Shusheng Tang, and Chongshan Dai. 2025. "Recent Advances in Pretreatment Methods and Detection Techniques for Veterinary Drug Residues in Animal-Derived Foods" Metabolites 15, no. 4: 233. https://doi.org/10.3390/metabo15040233
APA StyleDai, Q., Tang, S., & Dai, C. (2025). Recent Advances in Pretreatment Methods and Detection Techniques for Veterinary Drug Residues in Animal-Derived Foods. Metabolites, 15(4), 233. https://doi.org/10.3390/metabo15040233