
Myocardial and Arrhythmic Spectrum of Neuromuscular Disorders in Children


 , ,
, ,
Abstract
:1. Introduction
2. Methods
3. Results
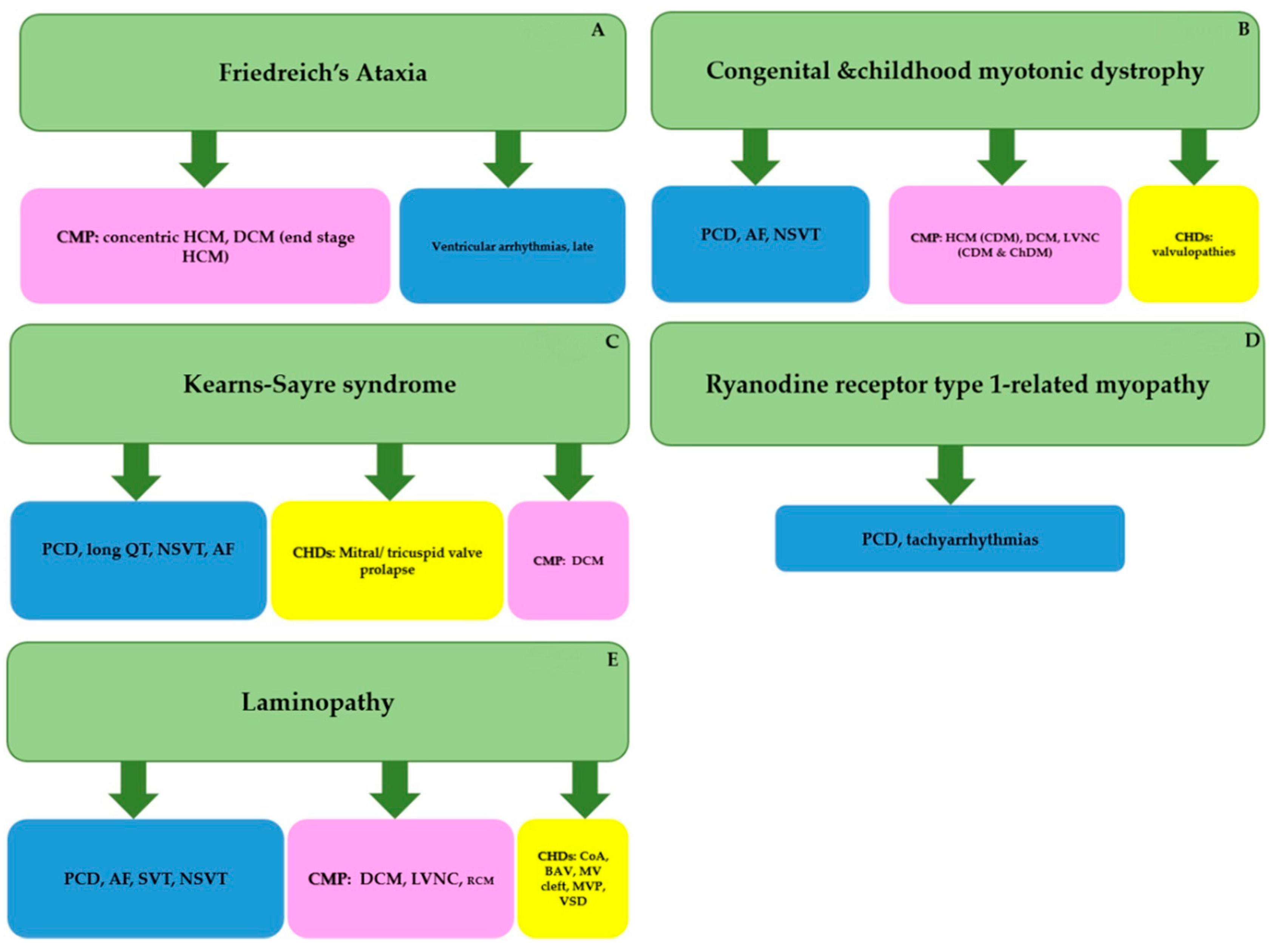
3.1. Friedreich’s Ataxia (FRDA) (Prevalence 1-9/100000, OMIM # 229300,601992)
3.2. Myotonic Dystrophy Type 1 (DM1) in Children (Congenital: Prevalence Very Rare; OMIM 160900)
3.3. Kearns Sayre Syndrome (KSS) (Prevalence 1-9/100000 OMIM #530000)
3.4. Ryanodine Receptor Type 1-Related Myopathies (RYR1-RM) (Pediatric Prevalence US 1:90.000, OMIM #11700, 255320)
3.5. Laminopathy (Prevalence Rare, LAMIN A/C OMIM-150330)

{kind=link}
{kind=link}
| NMD (OMIM) | Prevalence (in Children) | Gene | Mechanism | Inheritance | Structural HD | CMP | Heart Rhythm Changes | SCD | Cardiac Surveillace | Therapy |
|---|---|---|---|---|---|---|---|---|---|---|
| FRDA(229300 601992) | 1–9/100000 (rare) | FXN | GAA-expansion in first intron (anticipation) | AR | - | Concentric HCM, DCM (End stage HCM) | ventricular arrhythmias | yes | At diagnosis: PE, ECG, echo, & 24 h ECG monitoring. Asymptomatic: at least annual screening. Symptomatic: consider more frequently. Symptomatic+ arrhythmic events: consider 24 h ECG monitoring and event recorder. Consider CMR. | Coenzyme Q, Idebenone, antioxidant and iron-chelating agents, frataxin increase inducers, HT in end-stage CMP. |
| CDM and ChDM (#160900) | Very rare, rare | DMPK | CTG- expansion in UTR (anticipation) | AD | valvulopathies | HCM, DCM, NCLV | PCD, AF, NSVT | yes | At diagnosis: PE, ECG, echo, & 24 h ECG monitoring, EST and SA-ECG. Asymptomatic:+ normal ECG and LVEF: annual PE, ECG; 24 h ECG monitoring, EST and SA-ECG; every 2-4 years echo. Symptomatic + ECG anomalies: at least annual screening (consider EPS for PM/ICD). Consider CMR. | Antiarrhythmic drugs, anticoagulant drugs, radiofrequency catheter ablation, HT in end-stage CMP. Several experimental trials in progress. Caution is recommended in anesthetic management. |
| KSS (#530000) | 1–9/100000 | m.8470_13446del4977 | mtDNA deletion | matrilinear | MV or TV prolapse | DCM | PCD, long QT, NSVT, AF | yes | At diagnosis: PE, ECG, echo, & 24 h ECG monitoring, EST Asymptomatic: at least annual screening. Symptomatic + ECG anomalies: consider more frequent screening (+EPS for PM/ICD). Consider CMR. | Folic acid, coenzyme Q10, antioxidants, HT. |
| RYR1-RM(-) | 1:90.000 (rare) | RYR1 | Missense/nonsense variants | AR, AD | - | - | PCD, tachyarrhythmias | - | At diagnosis: PE, ECG, echo. Asymptomatic: annual screening. | Dantrolene. Several experimental trials in progress. Caution is recommended in anesthetic management. |
| Laminopathy | rare | LMNA | Missense/nonsense variants | AR, AD | CoA, BAV, MV cleft, MVP, VSD | DCM, LVNC, RCM, | PCD, AF, SVT, NSVT | yes | At diagnosis: PE, ECG, echo, & 24 h ECG monitoring, EST. Asymptomatic: at least annual screening. Symptomatic + ECG anomalies: consider more frequent screening (+EPS for PM/ICD). Consider CMR. | Antiarrhymic drugs, synmptomatic therapy for HF, anticoagulant drugs. HT. Several experimental trials in progress. Caution is recommended in anesthetic management. |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hsu, D.T. Cardiac manifestations of neuromuscular disorders in children. Paediatr. Respir. Rev. 2010, 11, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Baban, A.; Cicenia, M.; Travaglini, L.; Calí, F.; Vasco, G.; Francalanci, P. Remember friedreich ataxia even in a toddler with apparently isolated dilated (not hypertrophic!) cardiomyopathy: Revisited. Minerva Pediatr. 2021. [Google Scholar] [CrossRef]
- Baban, A.; Cicenia, M.; Magliozzi, M.; Gnazzo, M.; Cantarutti, N.; Silvetti, M.S.; Adorisio, R.; Dallapiccola, B.; Bertini, E.; Novelli, A.; et al. Cardiovascular Involvement in Pediatric Laminopathies. Report of Six Patients and Literature Revision. Front. Pediatr. 2020, 8, 374. [Google Scholar] [CrossRef] [PubMed]
- Adorisio, R.; Mencarelli, E.; Cantarutti, N.; Calvieri, C.; Amato, L.; Cicenia, M.; Silvetti, M.; D’Amico, A.; Grandinetti, M.; Drago, F.; et al. Duchenne Dilated Cardiomyopathy: Cardiac Management from Prevention to Advanced Cardiovascular Therapies. J. Clin. Med. 2020, 9, 3186. [Google Scholar] [CrossRef]
- Norrish, G.; Field, E.; Mcleod, K.; Ilina, M.; Stuart, G.; Bhole, V.; Uzun, O.; Brown, E.; Daubeney, P.E.F.; Lota, A.; et al. Clinical presentation and survival of childhood hypertrophic cardiomyopathy: A retrospective study in United Kingdom. Eur. Heart J. 2019, 40, 986–993. [Google Scholar] [CrossRef]
- Rupp, S.; Felimban, M.; Schänzer, A.; Schranz, D.; Marschall, C.; Zenker, M.; Logeswaran, T.; Neuhäuser, C.; Thul, J.; Jux, C.; et al. Genetic basis of hypertrophic cardiomyopathy in children. Clin. Res. Cardiol. 2019, 108, 282–289. [Google Scholar] [CrossRef]
- Sharma, A.; Singh, S.; Mishra, S.K. Cardiac Abnormalities in Congenital and Childhood Myotonic Muscular Dystrophy Type 1. Neuropediatrics 2017, 48, 42–44. [Google Scholar] [CrossRef]
- Forsberg, H.; Olofsson, B.O.; Eriksson, A.; Andersson, S. Cardiac involvement in congenital myotonic dystrophy. Heart 1990, 63, 119–121. [Google Scholar] [CrossRef] [Green Version]
- English, K.M.; Gibbs, J.L. Cardiac monitoring and treatment for children and adolescents with neuromuscular disorders. Dev. Med. Child Neurol. 2006, 48, 231–235. [Google Scholar] [CrossRef]
- Ho, G.; Cardamone, M.; Farrar, M. Congenital and childhood myotonic dystrophy: Current aspects of disease and future directions. World J. Clin. Pediatr. 2015, 4, 66–80. [Google Scholar] [CrossRef]
- Ho, G.; Carey, K.A.; Cardamone, M.; A Farrar, M. Myotonic dystrophy type 1: Clinical manifestations in children and adolescents. Arch. Dis. Child. 2019, 104, 48–52. [Google Scholar] [CrossRef]
- Feingold, B.; Mahle, W.T.; Auerbach, S.; Clemens, P.; Domenighetti, A.A.; Jefferies, J.L.; Judge, D.P.; Lal, A.K.; Markham, L.W.; Parks, W.J.; et al. Management of Cardiac Involvement Associated with Neuromuscular Diseases: A Scientific Statement from the American Heart Association. Circulation 2017, 136, e200–e231. [Google Scholar] [CrossRef] [Green Version]
- Adorisio, R.; Calvieri, C.; Cantarutti, N.; D’Amico, A.; Catteruccia, M.; Bertini, E.; Baban, A.; Filippelli, S.; Perri, G.; Amodeo, A.; et al. Heart rate reduction strategy using ivabradine in end-stage Duchenne cardiomyopathy. Int. J. Cardiol. 2019, 280, 99–103. [Google Scholar] [CrossRef]
- Maffei, R.T.L.N.; Fortuna, G.D.L.S.; Rosso, L.C.; Pires, P.D.; Rondelli, I. Cardiomyopathy as the first manifestation of Friedreich s ataxia. Autops. Case Rep. 2020, 10, e2020204. [Google Scholar] [CrossRef]
- Amini, O.; Lakziyan, R.; Abavisani, M.; Sarchahi, Z. The cardiomyopathy of Friedreich’s ataxia common in a family: A case report. Ann. Med. Surg. 2021, 66, 102408. [Google Scholar] [CrossRef]
- Yoon, G.; Soman, T.; Wilson, J.; George, K.; Mital, S.; Dipchand, A.I.; McCabe, J.; Logan, W.; Kantor, P. Cardiac Transplantation in Friedreich Ataxia. J. Child Neurol. 2012, 27, 1193–1196. [Google Scholar] [CrossRef] [Green Version]
- Plehn, J.F.; Hasbani, K.; Ernst, I.; Horton, K.D.; Drinkard, B.E.; Di Prospero, N.A. The Subclinical Cardiomyopathy of Friedreich’s Ataxia in a Pediatric Population. J. Card. Fail. 2018, 24, 672–679. [Google Scholar] [CrossRef]
- Peverill, R.E.; Romanelli, G.; Donelan, L.; Hassam, R.; Corben, L.A.; Delatycki, M.B. Left ventricular structural and functional changes in Friedreich ataxia—Relationship with body size, sex, age and genetic severity. PLoS ONE 2019, 14, e0225147. [Google Scholar] [CrossRef]
- Child, J.S.; Perloff, J.K.; Bach, P.M.; Wolfe, A.D.; Perlman, S.; Kark, R.P. Cardiac involvement in Friedreich’s ataxia: A clinical study of 75 patients. J. Am. Coll. Cardiol. 1986, 7, 1370–1378. [Google Scholar] [CrossRef] [Green Version]
- Kipps, A.; Alexander, M.; Colan, S.D.; Gauvreau, K.; Smoot, L.; Crawford, L.; Darras, B.; Blume, E.D. The Longitudinal Course of Cardiomyopathy in Friedreich’s Ataxia During Childhood. Pediatr. Cardiol. 2009, 30, 306–310. [Google Scholar] [CrossRef]
- Zesiewicz, T.A.; Hancock, J.; Ghanekar, S.D.; Kuo, S.-H.; Dohse, C.A.; Vega, J. Emerging therapies in Friedreich’s Ataxia. Expert Rev. Neurother. 2020, 20, 1215–1228. [Google Scholar] [CrossRef]
- Johnson, N.E.; Aldana, E.Z.; Angeard, N.; Ashizawa, T.; Berggren, K.N.; Marini-Bettolo, C.; Duong, T.; Ekström, A.-B.; Sansone, V.; Tian, C.; et al. Consensus-based care recommendations for congenital and childhood-onset myotonic dystrophy type 1. Neurol. Clin. Pract. 2019, 9, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Bird, T.D. Myotonic Dystrophy Type 1. In GeneReviews®; Adam, M.P., Ed.; University of Washington: Seattle, WA, USA, 1999. [Google Scholar]
- Jain, A.; Al Khalili, Y. Congenital Myotonic Dystrophy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Harper, P.S. Major Problems in Neurology: Myotonic Dystrophy; WB Saunders: London, UK, 2001. [Google Scholar]
- Groh, W.J.; Groh, M.R.; Saha, C.; Kincaid, J.C.; Simmons, Z.; Ciafaloni, E.; Pourmand, R.; Otten, R.F.; Bhakta, D.; Nair, G.V.; et al. Electrocardiographic Abnormalities and Sudden Death in Myotonic Dystrophy Type 1. N. Engl. J. Med. 2008, 358, 2688–2697. [Google Scholar] [CrossRef]
- Schon, E.A. Rearrangements of mitonchondrial DNA. In Genetics of Mitochondrial Diseases; Holt, I.J., Ed.; Oxford University Press: Oxford, UK, 2003; pp. 111–124. [Google Scholar]
- Berardo, A.; DiMauro, S.; Hirano, M. A Diagnostic Algorithm for Metabolic Myopathies. Curr. Neurol. Neurosci. Rep. 2010, 10, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Ashrafzadeh, F.; Ghaemi, N.; Akhondian, J.; BeiraghiToosi, M.; Elmi, S. Hypoparathyroidism as the first manifesta-tion of kearns-sayre syndrome: A case report. Iran. J. Child Neurol. 2013, 7, 53–57. [Google Scholar]
- Ergül, Y.; Nişli, K.; Saygılı, A.; Dindar, A. Kearns-Sayre syndrome presenting as somatomedin C deficiency and complete heart block. Turk. Kardiyol. Dern. Ars. 2010, 38, 568–571. [Google Scholar]
- Kane, J.M.; Rossi, J.; Tsao, S.; Burton, B.K. Metabolic Cardiomyopathy and Mitochondrial Disorders in the Pediatric Intensive Care Unit. J. Pediatr. 2007, 151, 538–541. [Google Scholar] [CrossRef]
- Chertkof, J.; Hufnagel, R.B.; Blain, D.; Gropman, A.L.; Brooks, B. Retinoschisis associated with Kearns-Sayre syndrome. Ophthalmic Genet. 2020, 41, 497–500. [Google Scholar] [CrossRef]
- Auré, K.; De Baulny, H.O.; Laforêt, P.; Jardel, C.; Eymard, B.; Lombès, A. Chronic progressive ophthalmoplegia with large-scale mtDNA rearrangement: Can we predict progression? Brain 2007, 130, 1516–1524. [Google Scholar] [CrossRef] [Green Version]
- Di Nora, C.; Paldino, A.; Miani, D.; Finato, N.; Pizzolitto, S.; De Maglio, G.; Vendramin, I.; Sponga, S.; Nalli, C.; Sinagra, G.; et al. Heart Transplantation in Kearns-Sayre Syndrome. Transplantation 2019, 103, e393–e394. [Google Scholar] [CrossRef]
- Berenberg, R.A.; Pellock, J.M.; DiMauro, S.; Schotlan, D.L.; Bonilla, E.; Eastwood, A.; Hays, A.; Vicale, C.T.; Behrens, M.; Chutorian, A.; et al. Lumping or splitting? “ophthalmoplegia-plus” or kearns-sayre syndrome? Ann. Neurol. 1977, 1, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.; Çoku, J.; Forbes, T.; Kannan, S. Kearns-Sayre Syndrome Presenting as Complete Heart Block. Pediatr. Cardiol. 2008, 29, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Di Mambro, C.; Tamborrino, P.P.; Silvetti, M.S.; Yammine, M.L.; Marcolin, C.; Righi, D.; Baban, A.; Martinelli, D.; Vici, C.D.; Drago, F. Progressive involvement of cardiac conduction system in paediatric patients with Kearns–Sayre syndrome: How to predict occurrence of complete heart block and sudden cardiac death? Europace 2021, 23, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Wittich, C.; Khambatta, S.; Nguyen, D.; Beckman, T. Kearns–Sayre syndrome: A case series of 35 adults and children. Int. J. Gen. Med. 2014, 7, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Larsson, N.-G.; Holme, E.; Kristiansson, B.; Oldfors, A.; Tulinius, M. Progressive Increase of the Mutated Mitochondrial DNA Fraction in Kearns-Sayre Syndrome. Pediatr. Res. 1990, 28, 131–136. [Google Scholar] [CrossRef]
- Polak, P.E.; Zulstra, F.; Roelandt, J.R.T.C. Indications for pacemaker implantation in the Kearns-Sayre syndrome. Eur. Heart J. 1989, 10, 281–282. [Google Scholar] [CrossRef]
- Imamura, T.; Sumitomo, N.; Muraji, S.; Mori, H.; Osada, Y.; Oyanagi, T.; Kojima, T.; Yoshiba, S.; Kobayashi, T.; Ono, K. The necessity of implantable cardioverter defibrillators in patients with Kearns-Sayre syndrome—Systematic review of the articles. Int. J. Cardiol. 2019, 279, 105–111. [Google Scholar] [CrossRef]
- Goldstein, A.; Falk, M.J. Mitochondrial DNA Deletion Syndromes. In GeneReviews®; Adam, M.P., Ed.; University of Washington: Seattle, WA, USA, 2003. [Google Scholar]
- Ms, K.A.; McNamara, N.; Bennett, L.R.; DO, M.E.M.; Acsadi, G.; Dowling, J.J. Prevalence of congenital myopathies in a representative pediatric united states population. Ann. Neurol. 2011, 70, 662–665. [Google Scholar] [CrossRef] [Green Version]
- Hermans, M.; Pinto, Y.; Merkies, I.; de Die-Smulders, C.; Crijns, H.; Faber, C.G. Hereditary muscular dystrophies and the heart. Neuromuscul. Disord. 2010, 20, 479–492. [Google Scholar] [CrossRef]
- Zahradnikova, A.; Zahradník, I.; Györke, I.; Györke, S. Rapid Activation of the Cardiac Ryanodine Receptor by Submillisecond Calcium Stimuli. J. Gen. Physiol. 1999, 114, 787–798. [Google Scholar] [CrossRef]
- Lanner, J.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine Receptors: Structure, Expression, Molecular Details, and Function in Calcium Release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef] [Green Version]
- Dowling, J.J.; Arbogast, S.; Hur, J.; Nelson, D.D.; McEvoy, A.; Waugh, T.; Marty, I.; Lunardi, J.; Brooks, S.; Kuwada, J.Y.; et al. Oxidative stress and successful antioxidant treatment in models of RYR1-related myopathy. Brain 2012, 135, 1115–1127. [Google Scholar] [CrossRef]
- Lawal, T.A.; Todd, J.; Meilleur, K.G. Ryanodine Receptor 1-Related Myopathies: Diagnostic and Therapeutic Approaches. Neurotherapeutics 2018, 15, 885–899. [Google Scholar] [CrossRef] [Green Version]
- Magee, K.R.; Shy, G.M. A new congenital non-progressive myopathy. Brain 1956, 79, 610–621. [Google Scholar] [CrossRef]
- Quane, K.A.; Healy, J.; Keating, K.E.; Manning, B.M.; Couch, F.J.; Palmucci, L.M.; Doriguzzi, C.; Fagerlund, T.H.; Berg, K.; Ording, H.; et al. Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat. Genet. 1993, 5, 51–55. [Google Scholar] [CrossRef]
- Ferreiro-Girani, A.; Monnier, N.; Romero, N.B.; Leroy, J.-P.; Bönnemann, C.; Haenggeli, C.-A.; Straub, V.; Voss, W.D.; Nivoche, Y.; Jungbluth, H.; et al. A recessive form of central core disease, transiently presenting as multi-minicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann. Neurol. 2002, 51, 750–759. [Google Scholar] [CrossRef]
- Fattori, F.; Maggi, L.; Bruno, C.; Cassandrini, D.; Codemo, V.; Catteruccia, M.; Tasca, G.; Berardinelli, A.; Magri, F.; Pane, M.; et al. Centronuclear myopathies: Genotype–phenotype correlation and frequency of defined genetic forms in an Italian cohort. J. Neurol. 2015, 262, 1728–1740. [Google Scholar] [CrossRef]
- Scacheri, P.C.; Hoffman, E.P.; Fratkin, J.D.; Semino-Mora, C.; Senchak, A.; Davis, M.R.; Laing, N.G.; Vedanarayanan, V.; Subramony, S.H. A novel ryanodine receptor gene mutation causing both cores and rods in congenital myopathy. Neurology 2000, 55, 1689–1696. [Google Scholar] [CrossRef]
- Clarke, N.F.; Waddell, L.B.; Cooper, S.T.; Perry, M.; Smith, R.L.; Kornberg, A.J.; Muntoni, F.; Lillis, S.; Straub, V.; Bushby, K.; et al. Recessive mutations in RYR1 are a common cause of congenital fiber type disproportion. Hum. Mutat. 2010, 31, E1544–E1550. [Google Scholar] [CrossRef]
- Witting, N.; Werlauff, U.; Duno, M.; Vissing, J. Phenotypes, genotypes, and prevalence of congenital myopathies older than 5 years in Denmark. Neurol. Genet. 2017, 3, e140. [Google Scholar] [CrossRef] [Green Version]
- Dowling, J.J.; Lillis, S.; Amburgey, K.; Zhou, H.; Al-Sarraj, S.; Buk, S.J.; Wraige, E.; Chow, G.; Abbs, S.; Leber, S.; et al. King–Denborough syndrome with and without mutations in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul. Disord. 2011, 21, 420–427. [Google Scholar] [CrossRef]
- Matthews, E.; Neuwirth, C.; Jaffer, F.; Scalco, R.; Fialho, D.; Parton, M.; Rayan, D.R.; Suetterlin, K.; Sud, R.; Spiegel, R.; et al. Atypical periodic paralysis and myalgia. Neurology 2018, 90, e412–e418. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, H.; Davis, M.; James, D.; Pollock, N.; Stowell, K. Malignant hyperthermia. Orphanet J. Rare Dis. 2007, 2, 21. [Google Scholar] [CrossRef] [Green Version]
- Jungbluth, H.; Treves, S.; Zorzato, F.; Sarkozy, A.; Ochala, J.; Sewry, C.; Phadke, R.; Gautel, M.; Muntoni, F. Congenital myopathies: Disorders of excitation—contraction coupling and muscle contraction. Nat. Rev. Neurol. 2018, 14, 151–167. [Google Scholar] [CrossRef]
- Rosenberg, H.; Pollock, N.; Schiemann, A.H.; Bulger, T.; Stowell, K.M. Malignant hyperthermia: A review. Orphanet J. Rare Dis. 2015, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Greiser, M.; Kerfant, B.-G.; Williams, G.S.; Voigt, N.; Harks, E.; Dibb, K.; Giese, A.; Meszaros, J.; Verheule, S.; Ravens, U.; et al. Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. J. Clin. Investig. 2014, 124, 4759–4772. [Google Scholar] [CrossRef] [Green Version]
- Van Petegem, F. Ryanodine Receptors: Structure and Function. J. Biol. Chem. 2012, 287, 31624–31632. [Google Scholar] [CrossRef] [Green Version]
- North, K.N.; Wang, C.H.; Clarke, N.; Jungbluth, H.; Vainzof, M.; Dowling, J.J.; Amburgey, K.; Quijano-Roy, S.; Beggs, A.; Sewry, C.; et al. Approach to the diagnosis of congenital myopathies. Neuromuscul. Disord. 2014, 24, 97–116. [Google Scholar] [CrossRef] [Green Version]
- Petri, H.; Wahbi, K.; Witting, N.; Køber, L.; Bundgaard, H.; Kamoun, E.; Vellieux, G.; Stojkovic, T.; Béhin, A.; Laforet, P.; et al. Congenital myopathies are mainly associated with a mild cardiac phenotype. J. Neurol. 2019, 266, 1367–1375. [Google Scholar] [CrossRef]
- Steele, H.E.; Harris, E.; Barresi, R.; Marsh, J.; Beattie, A.; Bourke, J.P.; Straub, V.; Chinnery, P.F. Cardiac involvement in hereditary myopathy with early respiratory failure. Neurology 2016, 87, 1031–1035. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, I.; Abe, Y.; Ono, H.; Kubota, M. Severe congenital RYR1-associated myopathy complicated with atrial tachycardia and sinus node dysfunction: A case report. Ital. J. Pediatr. 2019, 45, 165–173. [Google Scholar] [CrossRef]
- Rendu, J.; Brocard, J.; Denarier, E.; Monnier, N.; Pietri-Rouxel, F.; Beley, C.; Roux-Buisson, N.; Gilbert-Dussardier, B.; Perez, M.J.; Romero, N.; et al. Exon Skipping as a Therapeutic Strategy Applied to an RYR1 Mutation with Pseudo-Exon Inclusion Causing a Severe Core Myopathy. Hum. Gene Ther. 2013, 24, 702–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worman, H.J. Nuclear lamins and laminopathies. J. Pathol. 2012, 226, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Wydner, K.L.; McNeil, J.A.; Lin, F.; Worman, H.J.; Lawrence, J.B. Chromosomal Assignment of Human Nuclear Envelope Protein Genes LMNA, LMNB1, and LBR by Fluorescencein SituHybridization. Genomics 1996, 32, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar] [CrossRef]
- Rodriguez, S.; Coppedè, F.; Sagelius, H.; Eriksson, M. Increased expression of the Hutchinson–Gilford progeria syndrome truncated lamin A transcript during cell aging. Eur. J. Hum. Genet. 2009, 17, 928–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moir, R.D.; Spann, T.P.; Goldman, R.D. The Dynamic Properties and Possible Functions of Nuclear Lamins. Int. Rev. Cytol. 1995, 162, 141–182. [Google Scholar] [CrossRef]
- Worman, H.J.; Bonne, G. “Laminopathies”: A wide spectrum of human diseases. Exp. Cell Res. 2007, 313, 2121–2133. [Google Scholar] [CrossRef] [Green Version]
- Arbustini, E.; Pilotto, A.; Repetto, A.; Grasso, M.; Negri, A.; Diegoli, M.; Campana, C.; Scelsi, L.; Baldini, E.; Gavazzi, A.; et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: A lamin A/C defect-related disease. J. Am. Coll. Cardiol. 2002, 39, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Parks, S.B.; Kushner, J.D.; Nauman, D.; Burgess, D.; Ludwigsen, S.; Peterson, A.; Li, D.; Jakobs, P.; Litt, M.; Porter, C.B.; et al. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am. Heart J. 2008, 156, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Perrot, A.; Hussein, S.; Ruppert, V.; Schmidt, H.H.J.; Wehnert, M.S.; Duong, N.T.; Posch, M.G.; Panek, A.; Dietz, R.; Kindermann, I.; et al. Identification of mutational hot spots in LMNA encoding lamin A/C in patients with familial dilated cardiomyopathy. Basic Res. Cardiol. 2009, 104, 90–99. [Google Scholar] [CrossRef]
- Karkkainen, S.; Reissell, E.; Heliö, T.; Kaartinen, M.; Tuomainen, P.; Toivonen, L.; Kuusisto, J.; Kupari, M.; Nieminen, M.S.; Laakso, M.; et al. Novel mutations in the lamin A/C gene in heart transplant recipients with end stage dilated cardiomyopathy. Heart 2006, 92, 524–526. [Google Scholar] [CrossRef] [Green Version]
- Meune, C.; van Berlo, J.; Anselme, F.; Bonne, G.; Pinto, Y.M.; Duboc, D. Primary Prevention of Sudden Death in Patients with Lamin A/C Gene Mutations. N. Engl. J. Med. 2006, 354, 209–210. [Google Scholar] [CrossRef]
- van Rijsingen, I.A.; Arbustini, E.; Elliott, P.; Mogensen, J.; Ast, J.F.H.-V.; van der Kooi, A.J.; van Tintelen, J.P.; Berg, M.V.D.; Pilotto, A.; Pasotti, M.; et al. Risk Factors for Malignant Ventricular Arrhythmias in Lamin A/C Mutation Carriers. J. Am. Coll. Cardiol. 2012, 59, 493–500. [Google Scholar] [CrossRef]
- Pasqualin, L.M.; Reed, U.C.; Costa, T.V.; Quedas, E.; Albuquerque, M.A.; Resende, M.B.; Rutkowski, A.; Chadi, G.; Zanoteli, E. Congenital Muscular Dystrophy with Dropped Head Linked to the LMNA Gene in a Brazilian Cohort. Pediatr. Neurol. 2014, 50, 400–406. [Google Scholar] [CrossRef]
- Komaki, H.; Hayashi, Y.K.; Tsuburaya, R.; Sugie, K.; Kato, M.; Nagai, T.; Imataka, G.; Suzuki, S.; Saitoh, S.; Asahina, N.; et al. Inflammatory changes in infantile-onset LMNA-associated myopathy. Neuromuscul. Disord. 2011, 21, 563–568. [Google Scholar] [CrossRef]
- Petillo, R.; D’Ambrosio, P.; Torella, A.; Taglia, A.; Picillo, E.; Testori, A.; Ergoli, M.; Nigro, G.; Piluso, G.; Nigro, V.; et al. Novel mutations in LMNA A/C gene and associated phenotypes. Acta Myol. 2015, 34, 116–119. [Google Scholar]
- Heller, F.; Dabaj, I.; Mah, J.K.; Bergounioux, J.; Essid, A.; Bönnemann, C.G.; Rutkowski, A.; Bonne, G.; Quijano-Roy, S.; Wahbi, K. Cardiac manifestations of congenital LMNA-related muscular dystrophy in children: Three case reports and recommendations for care. Cardiol. Young 2017, 27, 1076–1082. [Google Scholar] [CrossRef]
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtizberea, A.; Bécane, H.M.; Recan, D.; Merlini, L.; Wehnert, M.; Boor, R.; Reuner, U.; et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 2000, 48, 170–180. [Google Scholar] [CrossRef]
- Tan, D.; Yang, H.; Yuan, Y.; Bönnemann, C.; Chang, X.; Wang, S.; Wu, Y.; Wu, X.; Xiong, H. Phenotype–Genotype Analysis of Chinese Patients with Early-Onset LMNA-Related Muscular Dystrophy. PLoS ONE 2015, 10, e0129699. [Google Scholar] [CrossRef]
- Parent, J.J.; Towbin, J.A.; Jefferies, J.L. Left Ventricular Noncompaction in a Family with Lamin A/C Gene Mutation. Tex. Heart Inst. J. 2015, 42, 73–76. [Google Scholar] [CrossRef] [Green Version]
- Piekarowicz, K.; Machowska, M.; Dzianisava, V.; Rzepecki, R. Hutchinson-Gilford Progeria Syndrome-Current Status and Prospects for Gene Therapy Treatment. Cells 2019, 8, 88. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baban, A.; Lodato, V.; Parlapiano, G.; di Mambro, C.; Adorisio, R.; Bertini, E.S.; Dionisi-Vici, C.; Drago, F.; Martinelli, D. Myocardial and Arrhythmic Spectrum of Neuromuscular Disorders in Children. Biomolecules 2021, 11, 1578. https://doi.org/10.3390/biom11111578
Baban A, Lodato V, Parlapiano G, di Mambro C, Adorisio R, Bertini ES, Dionisi-Vici C, Drago F, Martinelli D. Myocardial and Arrhythmic Spectrum of Neuromuscular Disorders in Children. Biomolecules. 2021; 11(11):1578. https://doi.org/10.3390/biom11111578
Chicago/Turabian StyleBaban, Anwar, Valentina Lodato, Giovanni Parlapiano, Corrado di Mambro, Rachele Adorisio, Enrico Silvio Bertini, Carlo Dionisi-Vici, Fabrizio Drago, and Diego Martinelli. 2021. "Myocardial and Arrhythmic Spectrum of Neuromuscular Disorders in Children" Biomolecules 11, no. 11: 1578. https://doi.org/10.3390/biom11111578
APA StyleBaban, A., Lodato, V., Parlapiano, G., di Mambro, C., Adorisio, R., Bertini, E. S., Dionisi-Vici, C., Drago, F., & Martinelli, D. (2021). Myocardial and Arrhythmic Spectrum of Neuromuscular Disorders in Children. Biomolecules, 11(11), 1578. https://doi.org/10.3390/biom11111578