
Metformin Treatment or PRODH/POX-Knock out Similarly Induces Apoptosis by Reprograming of Amino Acid Metabolism, TCA, Urea Cycle and Pentose Phosphate Pathway in MCF-7 Breast Cancer Cells

 , , , , , and
, , , , , and
Abstract
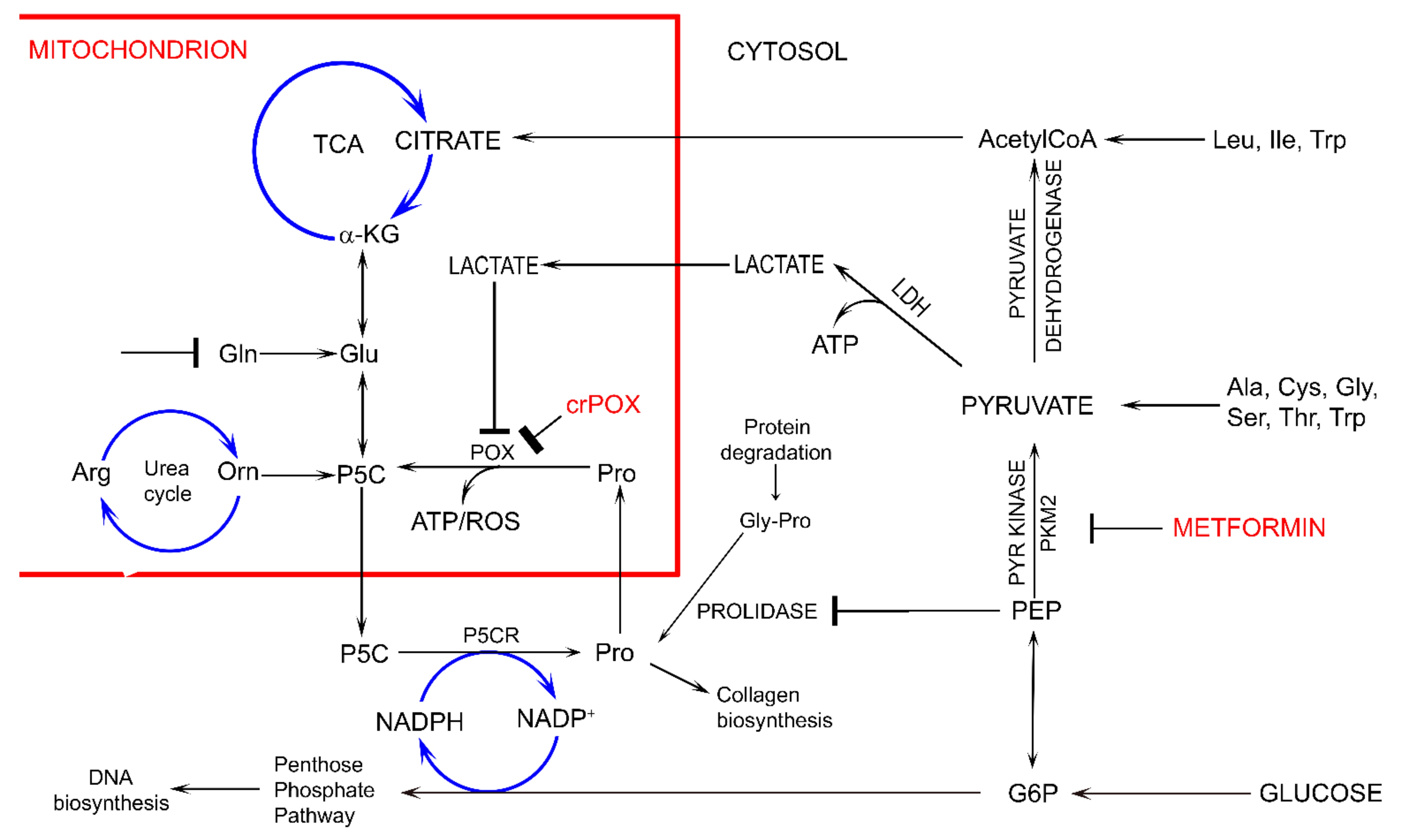
:1. Introduction
2. Materials and Methods
2.1. PRODH/POX Knock out CRISPR-cas9 DNA Plasmid Purification
2.2. Transfection into MCF-7 Breast Cancer Cell Line
2.3. Cell Culture
2.4. Metabolite Extraction
2.5. Targeted Metabolomics Quantitative Analysis (LC-MS/MS(QqQ))
2.6. Data Pre-Treatment
2.7. Cell Lysate Preparation
2.8. Cell Proliferation Assay
2.9. Cell Cycle Analysis
2.10. Western Immunoblotting
2.11. Statistical Analyses
2.11.1. Targeted Analysis
2.11.2. Biological Analysis
3. Results
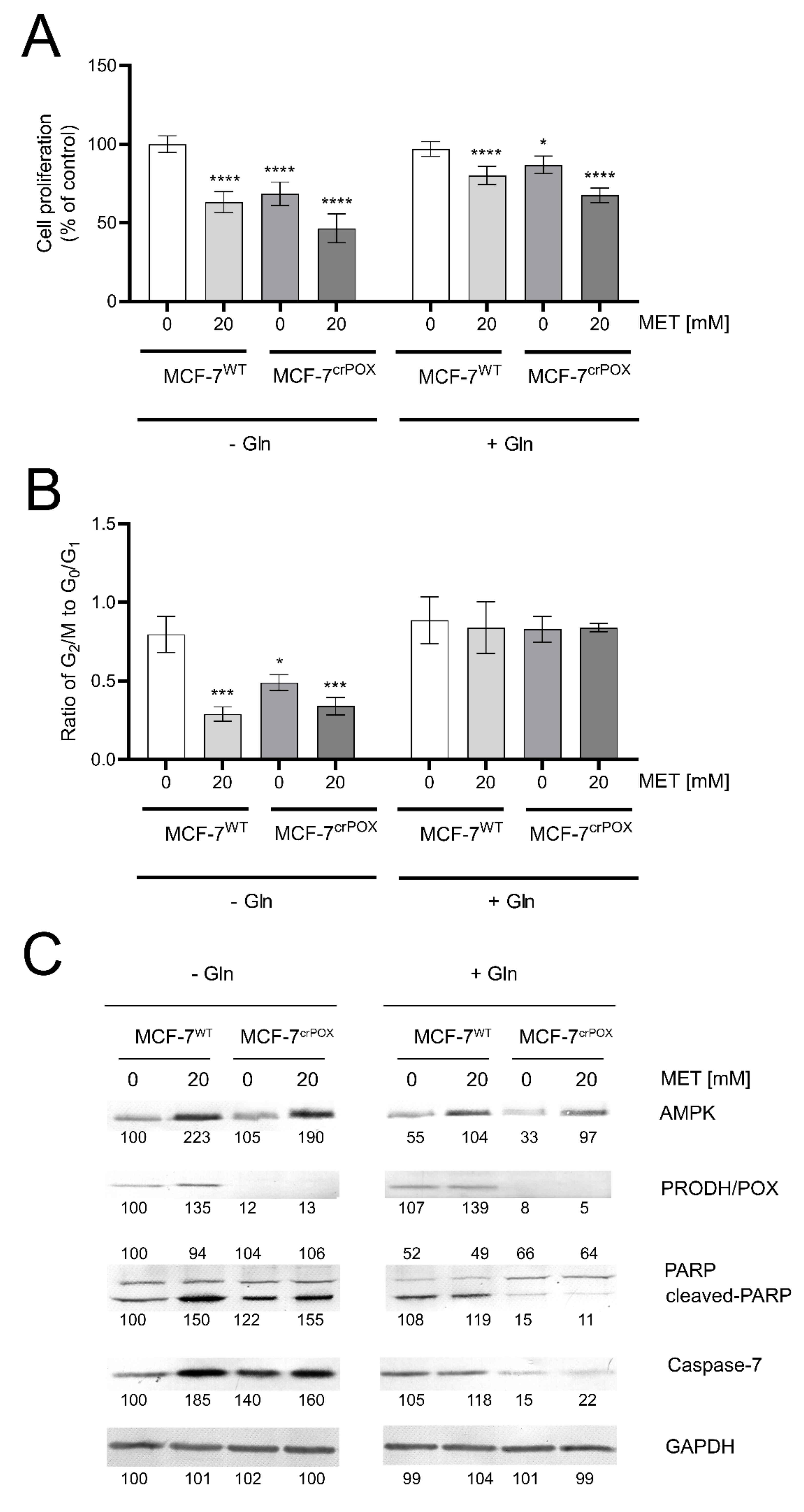
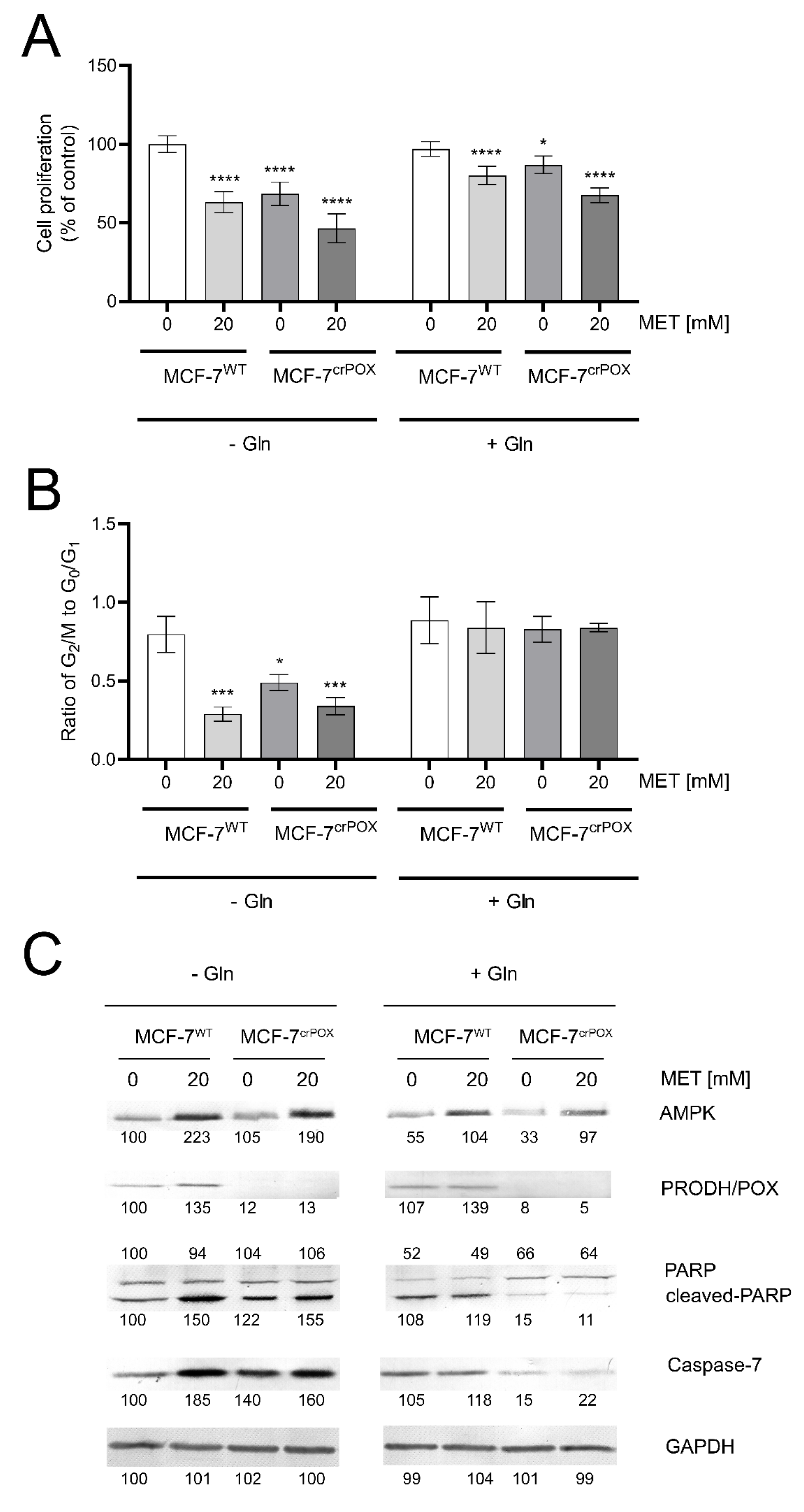
3.1. Metformin Inhibits MCF-7 Cell Proliferation and Induces Apoptosis
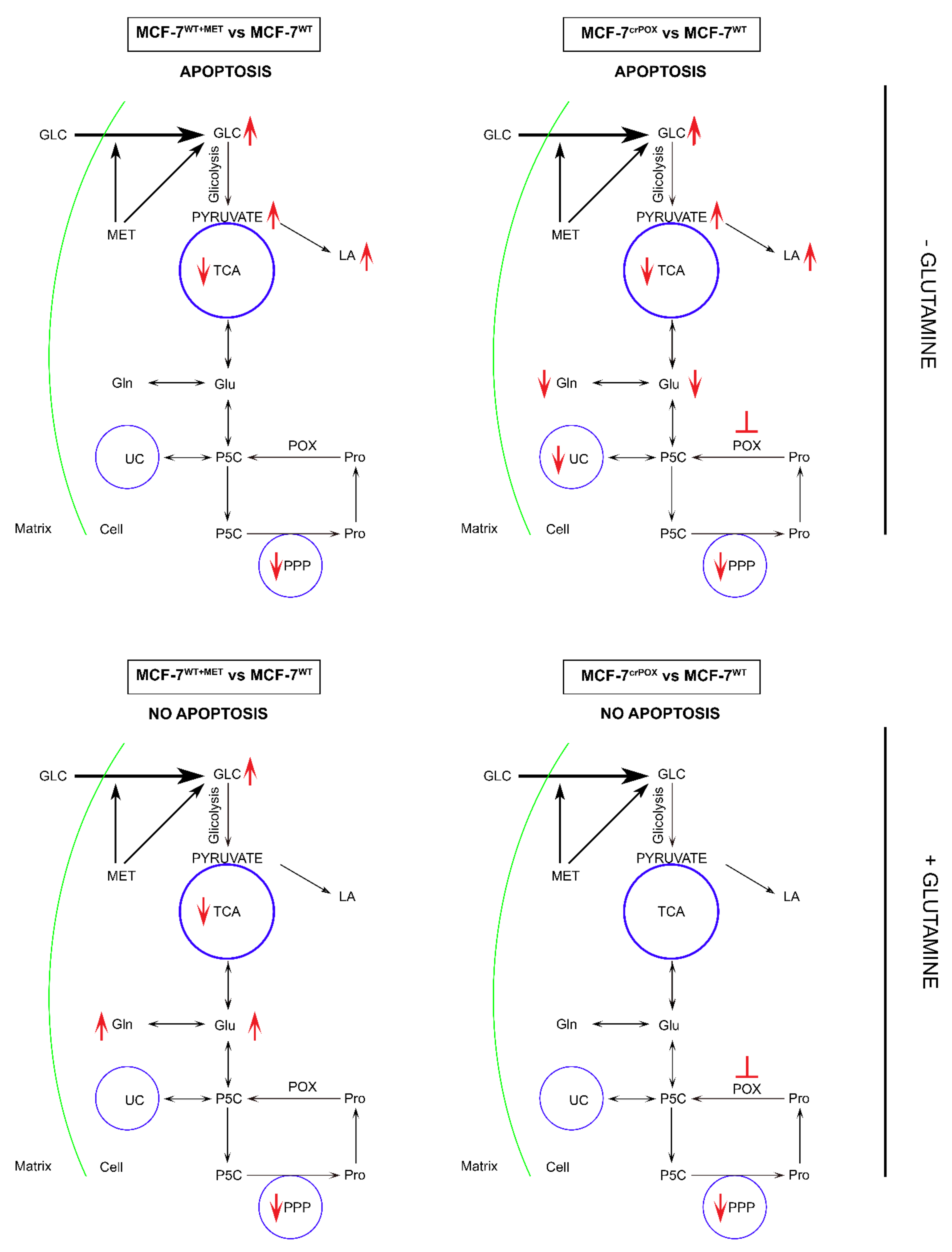
3.2. Targeted Metabolic Profiles of Some Metabolites of Glycolysis, Pentose Phosphate Pathway, TCA and Urea Cycles in PRODH/POX-Knock out of MCF-7 Cells (MCF-7crPOX) and Wild Type MCF-7 Cells (MCF-7WT) Cultured in Glutamine (Gln) Free Medium
3.3. Targeted Metabolic Profiles of Some Metabolites of Glycolysis, Pentose Phosphate Pathway, TCA and Urea Cycles in Metformin (MET) Treated Wild Type MCF-7 Cells (MCF-7WT+MET) and in MCF-7WT Cells Cultured in Gln Free Medium
3.4. Targeted Metabolic Profiles of Some Metabolites of Glycolysis, Pentose Phosphate Pathway, TCA and Urea Cycles in MCF-7crPOX Treated with MET (MCF-7crPOX+MET) and in MCF-7WT Cultured in Gln Free Medium
3.5. Targeted Metabolic Profiles of Some Metabolites of Glycolysis, Pentose Phosphate Pathway, TCA and Urea Cycles in PRODH/POX-Knock out of MCF-7 Cells (MCF-7crPOX) and Wild Type MCF-7 Cells (MCF-7WT) Cultured in Medium Containing Gln
3.6. Targeted Metabolic Profiles of Some Metabolites of Glycolysis, Pentose Phosphate Pathway, TCA and Urea Cycles in MET Treated Wild Type MCF-7 Cells (MCF-7WT+MET) and in MCF-7WT Cells Cultured in Medium Containing Gln
3.7. Targeted Metabolic Profiles of Some Metabolites of Glycolysis, Pentose Phosphate Pathway, TCA and Urea Cycles in MCF-7crPOX Treated with MET (MCF-7crPOX+MET) and in MCF-7WT Cultured in the Medium Containing Gln
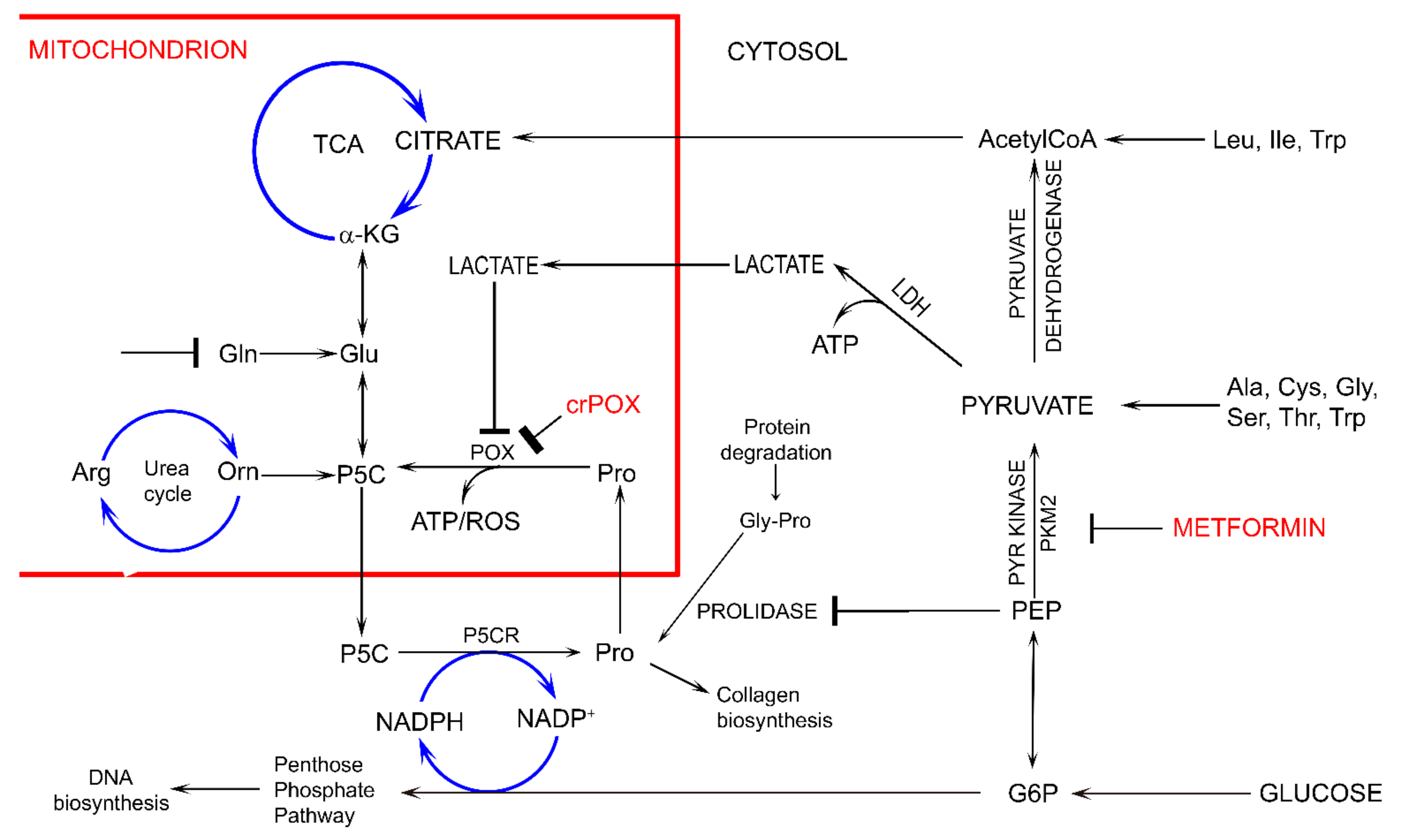
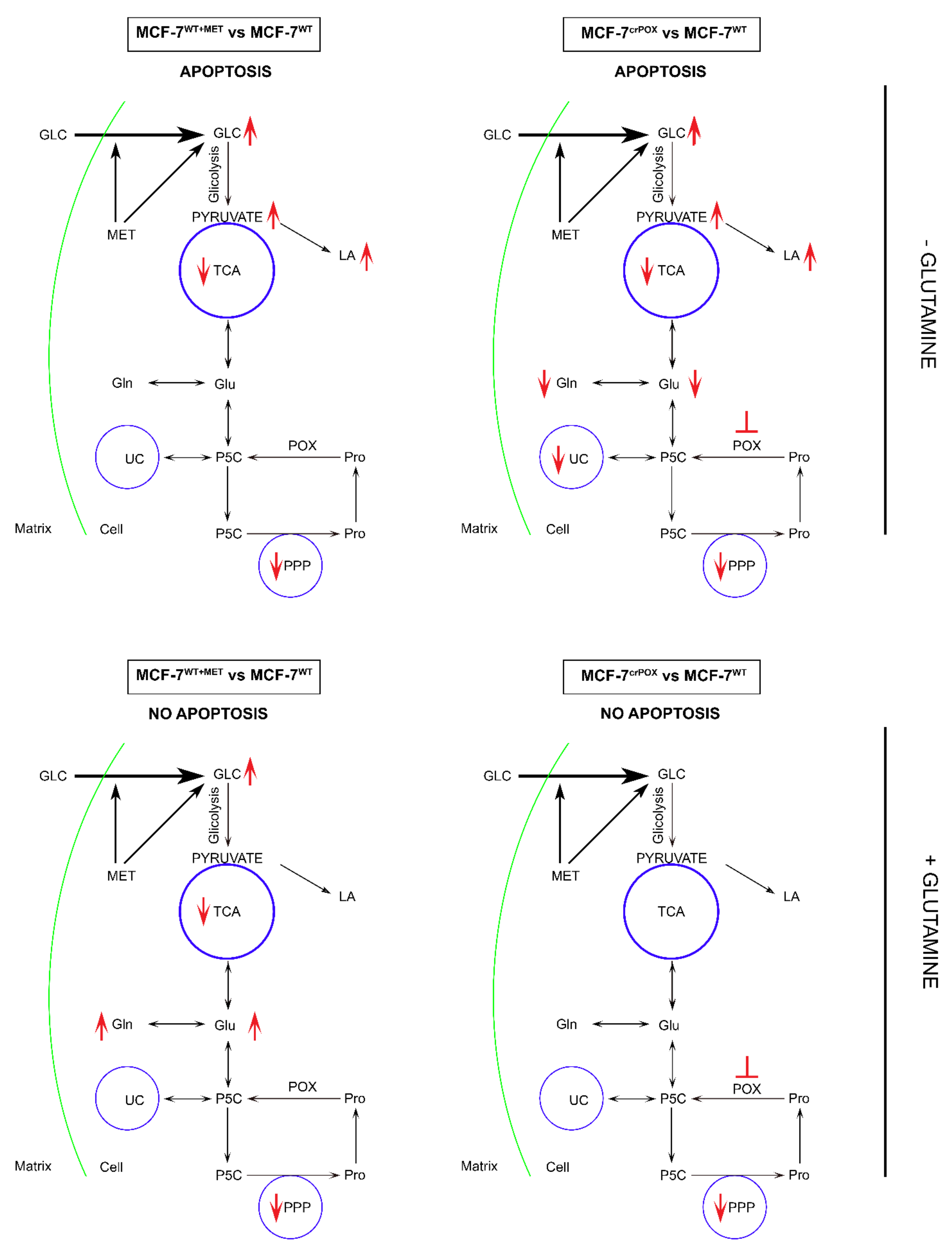
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef] [PubMed]
- De Flora, S.; Ganchev, G.; Iltcheva, M.; La Maestra, S.; Micale, R.T.; Steele, V.E.; Balansky, R. Pharmacological Modulation of Lung Carcinogenesis in Smokers: Preclinical and Clinical Evidence. Trends Pharm. Sci. 2016, 37, 120–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.K.; Arya, A.; Malecek, M.K.; Shin, D.S.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Altman, J.K.; Platanias, L.; et al. Repurposing metformin for cancer treatment: Current clinical studies. Oncotarget 2016, 7, 40767–40780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Effect of Metformin on Breast Cancer Metabolism. Available online: http://www.clinicaltrials.gov/ct2/show/NCT01266486 (accessed on 30 April 2017).
- Metformin Clinical Trial. Available online: https://clinicaltrials.gov/ct2/show/NCT01101438 (accessed on 30 April 2017).
- Han, D.; Li, S.J.; Zhu, Y.T.; Liu, L.; Li, M.X. LKB1/AMPK/mTOR signaling pathway in non-small-cell lung cancer. Asian Pac. J. Cancer Prev. 2013, 14, 4033–4039. [Google Scholar] [CrossRef] [Green Version]
- Salani, B.; Maffioli, S.; Hamoudane, M.; Parodi, A.; Ravera, S.; Passalacqua, M.; Alama, A.; Nhiri, M.; Cordera, R.; Maggi, D. Caveolin-1 is essential for metformin inhibitory effect on IGF1 action in non-small-cell lung cancer cells. FASEB J. 2012, 26, 788–798. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Q.; Wang, D.; Wang, Z.; Hu, C. Metformin inhibits growth of lung adenocarcinoma cells by inducing apoptosis via the mitochondria-mediated pathway. Oncol. Lett. 2015, 10, 1343–1349. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Liu, Z.; Jiang, L.; Liu, M.; Ma, J.; Yang, C.; Han, L.; Nan, K.; Liang, X. Metformin inhibits growth of human non-small cell lung cancer cells via liver kinase B-1-independent activation of adenosine monophosphate-activated protein kinase. Mol. Med. Rep. 2016, 13, 2590–2596. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. Minireview: The AMP-activated protein kinase cascade: The key sensor of cellular energy status. Endocrinology 2003, 144, 5179–5183. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Palma, F.R.; Ratti, B.A.; Paviani, V.; Coelho, D.R.; Miguel, R.; Danes, J.M.; Zaichik, S.V.; de Abreu, A.L.; Silva, S.O.; Chen, Y.; et al. AMPK-deficiency forces metformin-challenged cancer cells to switch from carbohydrate metabolism to ketogenesis to support energy metabolism. Oncogene 2021, 40, 5455–5467. [Google Scholar] [CrossRef]
- Pandhare, J.; Donald, S.P.; Cooper, S.K.; Phang, J.M. Regulation and function of proline oxidase under nutrient stress. J. Cell Biochem. 2009, 107, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Kononczuk, J.; Czyzewska, U.; Moczydlowska, J.; Surażyński, A.; Palka, J.; Miltyk, W. Proline Oxidase (POX) as A Target for Cancer Therapy. Curr. Drug Targets 2015, 16, 1464–1469. [Google Scholar] [CrossRef]
- Liu, W.; Phang, J.M. Proline dehydrogenase (oxidase), a mitochondrial tumor suppressor, and autophagy under the hypoxia microenvironment. Autophagy 2012, 8, 1407–1409. [Google Scholar] [CrossRef] [Green Version]
- Phang, J.M.; Liu, W.; Hancock, C.; Christian, K.J. The proline regulatory axis and cancer. Front. Oncol. 2012, 2, 60. [Google Scholar] [CrossRef] [Green Version]
- Phang, J.M.; Donald, S.P.; Pandhare, J.; Liu, Y. The metabolism of proline, a stress substrate, modulates carcinogenic pathways. Amino Acids 2008, 35, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Donald, S.P.; Sun, X.Y.; Hu, C.A.; Yu, J.; Mei, J.M.; Valle, D.; Phang, J.M. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res. 2001, 61, 1810–1815. [Google Scholar]
- Hu, C.A.; Donald, S.P.; Yu, J.; Lin, W.W.; Liu, Z.; Steel, G.; Obie, C.; Valle, D.; Phang, J.M. Overexpression of proline oxidase induces proline-dependent and mitochondria-mediated apoptosis. Mol. Cell Biochem. 2007, 295, 85–92. [Google Scholar] [CrossRef]
- Liu, Y.; Borchert, G.L.; Surazynski, A.; Hu, C.A.; Phang, J.M. Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: The role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene 2006, 25, 5640–5647. [Google Scholar] [CrossRef] [Green Version]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, and apoptosis. Am. J. Med. Genet. 2001, 106, 62–70. [Google Scholar] [CrossRef]
- Rippe, R.A.; Schrum, L.W.; Stefanovic, B.; Solís-Herruzo, J.A.; Brenner, D.A. NF-kappaB inhibits expression of the alpha1(I) collagen gene. DNA Cell Biol. 1999, 18, 751–761. [Google Scholar] [CrossRef]
- Maxwell, S.A.; Rivera, A. Proline oxidase induces apoptosis in tumor cells, and its expression is frequently absent or reduced in renal carcinomas. J. Biol. Chem. 2003, 278, 9784–9789. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Borchert, G.L.; Donald, S.P.; Surazynski, A.; Hu, C.A.; Weydert, C.J.; Oberley, L.W.; Phang, J.M. MnSOD inhibits proline oxidase-induced apoptosis in colorectal cancer cells. Carcinogenesis 2005, 26, 1335–1342. [Google Scholar] [CrossRef]
- Karna, E.; Miltyk, W.; Wołczyński, S.; Pałka, J.A. The potential mechanism for glutamine-induced collagen biosynthesis in cultured human skin fibroblasts. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2001, 130, 23–32. [Google Scholar] [CrossRef]
- Miltyk, W.; Palka, J.A. Potential role of pyrroline 5-carboxylate in regulation of collagen biosynthesis in cultured human skin fibroblasts. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2000, 125, 265–271. [Google Scholar] [CrossRef]
- Dai, Y. Discovery MetabolomicsLC/MS Methods Optimized for Polar Metabolites; Hsiao, J.J., Ed.; Agilent Technologies Application Note: Santa Clara, CA, USA, 2019; Volume 5994-1492EN. [Google Scholar]
- Misiura, M.; Oscilowska, I.; Bielawska, K.; Palka, J.; Miltyk, W. PRODH/POX-Dependent Celecoxib-Induced Apoptosis in MCF-7 Breast Cancer. Pharmaceuticals 2021, 14, 874. [Google Scholar] [CrossRef]
- Gandini, S.; Puntoni, M.; Heckman-Stoddard, B.M.; Dunn, B.K.; Ford, L.; DeCensi, A.; Szabo, E. Metformin and cancer risk and mortality: A systematic review and meta-analysis taking into account biases and confounders. Cancer Prev. Res. 2014, 7, 867–885. [Google Scholar] [CrossRef] [Green Version]
- Landman, G.W.; Kleefstra, N.; van Hateren, K.J.; Groenier, K.H.; Gans, R.O.; Bilo, H.J. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care 2010, 33, 322–326. [Google Scholar] [CrossRef] [Green Version]
- Libby, G.; Donnelly, L.A.; Donnan, P.T.; Alessi, D.R.; Morris, A.D.; Evans, J.M. New users of metformin are at low risk of incident cancer: A cohort study among people with type 2 diabetes. Diabetes Care 2009, 32, 1620–1625. [Google Scholar] [CrossRef] [Green Version]
- Bo, S.; Ciccone, G.; Rosato, R.; Villois, P.; Appendino, G.; Ghigo, E.; Grassi, G. Cancer mortality reduction and metformin: A retrospective cohort study in type 2 diabetic patients. Diabetes Obes Metab. 2012, 14, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.J.; Zheng, Y.X.; Zhou, P.C.; Xiao, Y.N.; Tan, H.Z. Metformin use improves survival of diabetic liver cancer patients: Systematic review and meta-analysis. Oncotarget 2016, 7, 66202–66211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998, 352, 854–865. [Google Scholar] [CrossRef]
- Ahn, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Frattaruolo, L.; Brindisi, M.; Curcio, R.; Marra, F.; Dolce, V.; Cappello, A.R. Targeting the Mitochondrial Metabolic Network: A Promising Strategy in Cancer Treatment. Int. J. Mol. Sci. 2020, 21, 14. [Google Scholar] [CrossRef]
- Filipp, F.V.; Ratnikov, B.; De Ingeniis, J.; Smith, J.W.; Osterman, A.L.; Scott, D.A. Glutamine-fueled mitochondrial metabolism is decoupled from glycolysis in melanoma. Pigment. Cell Melanoma Res. 2012, 25, 732–739. [Google Scholar] [CrossRef] [Green Version]
- Catchpole, G.; Platzer, A.; Weikert, C.; Kempkensteffen, C.; Johannsen, M.; Krause, H.; Jung, K.; Miller, K.; Willmitzer, L.; Selbig, J.; et al. Metabolic profiling reveals key metabolic features of renal cell carcinoma. J. Cell Mol. Med. 2011, 15, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, A.; Kami, K.; Sugimoto, M.; Sugawara, M.; Toki, N.; Onozuka, H.; Kinoshita, T.; Saito, N.; Ochiai, A.; Tomita, M.; et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009, 69, 4918–4925. [Google Scholar] [CrossRef] [Green Version]
- Kakkad, S.M.; Solaiyappan, M.; O’Rourke, B.; Stasinopoulos, I.; Ackerstaff, E.; Raman, V.; Bhujwalla, Z.M.; Glunde, K. Hypoxic tumor microenvironments reduce collagen I fiber density. Neoplasia 2010, 12, 608–617. [Google Scholar] [CrossRef] [Green Version]
- Zareba, I.; Huynh, T.Y.L.; Kazberuk, A.; Teul, J.; Klupczynska, A.; Matysiak, J.; Surazynski, A.; Palka, J. Overexpression of Prolidase Induces Autophagic Death in MCF-7 Breast Cancer Cells. Cell Physiol. Biochem. 2020, 54, 875–887. [Google Scholar] [CrossRef]
- Kazberuk, A.; Zareba, I.; Palka, J.; Surazynski, A. A novel plausible mechanism of NSAIDs-induced apoptosis in cancer cells: The implication of proline oxidase and peroxisome proliferator-activated receptor. Pharmacol. Rep. 2020, 72, 1152–1160. [Google Scholar] [CrossRef]
- Kowaloff, E.M.; Phang, J.M.; Granger, A.S.; Downing, S.J. Regulation of proline oxidase activity by lactate. Proc. Natl. Acad. Sci. USA 1977, 74, 5368–5371. [Google Scholar] [CrossRef] [Green Version]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Meena, A.S.; Bhat, M.K. Targeting metabolic flexibility by simultaneously inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget 2015, 6, 37281–37299. [Google Scholar] [CrossRef] [Green Version]
- Granchi, C.; Roy, S.; Giacomelli, C.; Macchia, M.; Tuccinardi, T.; Martinelli, A.; Lanza, M.; Betti, L.; Giannaccini, G.; Lucacchini, A.; et al. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J. Med. Chem. 2011, 54, 1599–1612. [Google Scholar] [CrossRef]
- Maftouh, M.; Avan, A.; Sciarrillo, R.; Granchi, C.; Leon, L.G.; Rani, R.; Funel, N.; Smid, K.; Honeywell, R.; Boggi, U.; et al. Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br. J. Cancer 2014, 110, 172–182. [Google Scholar] [CrossRef]
- Chen, Y.; Wei, L.; Zhang, X.; Liu, X.; Chen, Y.; Zhang, S.; Zhou, L.; Li, Q.; Pan, Q.; Zhao, S.; et al. 3Bromopyruvate sensitizes human breast cancer cells to TRAILinduced apoptosis via the phosphorylated AMPKmediated upregulation of DR5. Oncol. Rep. 2018, 40, 2435–2444. [Google Scholar] [CrossRef] [Green Version]
- Lis, P.; Jurkiewicz, P.; Cal-Bakowska, M.; Ko, Y.H.; Pedersen, P.L.; Goffeau, A.; Ulaszewski, S. Screening the yeast genome for energetic metabolism pathways involved in a phenotypic response to the anti-cancer agent 3-bromopyruvate. Oncotarget 2016, 7, 10153–10173. [Google Scholar] [CrossRef] [Green Version]
- Grant, C.M. Metabolic reconfiguration is a regulated response to oxidative stress. J. Biol. 2008, 7, 1. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Relevant Metabolic Pathways | Metabolites | Conc. Average (ppb) | % Change (MCF-7crPOX vs. MCF-7WT) | P Value (Mann–Whitney) | MCF-7crPOX | |
|---|---|---|---|---|---|---|
| MCF-7WT | MCF-7crPOX | |||||
| Glycolysis | Pyruvic acid | 6.5 | 117.2 | 1712.0 | 0.010 | ↑ |
| Glucose | 11.2 | 150.8 | 1252.0 | 0.010 | ↑ | |
| Phospho-enolpyruvic acid | 6449.0 | 0.0 | −100.0 | 0.010 | ↓ | |
| PPP | Glucose 6-phosphate | 328.6 | 0.0 | −100.0 | 0.010 | ↓ |
| 6-Phospho-gluconic acid | 269.9 | 10.9 | −96.0 | 0.010 | ↓ | |
| TCA | Malic acid | 1126.1 | 77.4 | −93.1 | 0.010 | ↓ |
| Succinic acid | 250.5 | 131.5 | −47.5 | 0.159 | ||
| Fumaric acid | 336.2 | 27.5 | −91.8 | 0.010 | ↓ | |
| cis-Aconitic acid | 43.7 | 4.2 | −90.3 | 0.010 | ↓ | |
| Citric acid | 6425.6 | 0.0 | −100.0 | 0.010 | ↓ | |
| alpha-Ketoglutaric acid | 222.3 | 97.1 | −56.3 | 0.019 | ↓ | |
| Urea Cycle | Citrulline | 10.3 | 4.6 | −55.1 | 0.035 | ↓ |
| Arginine | 14,526.1 | 437.6 | −97.0 | 0.010 | ↓ | |
| Ornithine | 2626.1 | 0.0 | −100.0 | 0.010 | ↓ | |
| AA | Proline | 2642.0 | 2694.3 | 2.0 | 0.841 | |
| Glutamine | 31.0 | 20.3 | −34.5 | 0.010 | ↓ | |
| Glutamic acid | 410.2 | 155.9 | −62.0 | 0.010 | ↓ | |
| Additional | Lactic acid | 4613.3 | 13,347.5 | 189.3 | 0.010 | ↑ |
| Fructose | 2.3 | 34.6 | 1384.5 | 0.010 | ↑ | |
| Relevant Metabolic Pathways | Metabolites | Conc. Average (ppb) | % Change (MCF-7WT+MET vs. MCF-7WT) | P Value (Mann–Whitney) | MCF-7WT+MET | |
|---|---|---|---|---|---|---|
| MCF-7WT | MCF-7WT+MET | |||||
| Glycolysis | Pyruvic acid | 6.5 | 227.9 | 3423.5 | 0.038 | ↑ |
| Glucose | 11.2 | 115.8 | 938.0 | 0.038 | ↑ | |
| Phospho-enolpyruvic acid | 6449.0 | 417.1 | −93.5 | 0.057 | ||
| PPP | Glucose 6-phosphate | 328.6 | 45.5 | −86.1 | 0.038 | ↓ |
| 6-Phospho-gluconic acid | 269.9 | 607.7 | 125.1 | 0.727 | ||
| TCA | Malic acid | 1126.1 | 514.0 | −54.4 | 0.260 | |
| Succinic acid | 250.5 | 168.8 | −32.6 | 0.420 | ||
| Fumaric acid | 336.2 | 179.2 | −46.7 | 0.260 | ||
| cis-Aconitic acid | 43.7 | 5.9 | −86.4 | 0.050 | ↓ | |
| Citric acid | 6425.6 | 600.3 | −90.7 | 0.050 | ↓ | |
| alpha-Ketoglutaric acid | 222.3 | 1818.6 | 718.3 | 0.483 | ||
| Urea Cycle | Citrulline | 10.3 | 14.1 | 36.8 | 0.500 | |
| Arginine | 14,526.1 | 4528.0 | −68.8 | 0.420 | ||
| Ornithine | 2626.1 | 1223.7 | −53.4 | 0.327 | ||
| AA | Proline | 2642.0 | 2335.2 | −11.6 | 0.841 | |
| Glutamine | 31.0 | 25.8 | −16.6 | 0.168 | ||
| L-Glutamic acid | 410.2 | 466.0 | 13.6 | 0.841 | ||
| Additional | Lactic acid | 4613.3 | 17,831.6 | 286.5 | 0.038 | ↑ |
| Fructose | 2.3 | 11.5 | 392.3 | 0.057 | ||
| Relevant Metabolic Pathways | Metabolites | Conc. Average (ppb) | % Change (MCF-7crPOX+MET vs. MCF-7WT) | P Value (Mann–Whitney) | MCF-7crPOX+MET | |
|---|---|---|---|---|---|---|
| MCF-7WT | MCF-7crPOX+MET | |||||
| Glycolysis | Pyruvic acid | 6.5 | 289.6 | 4378.6 | 0.022 | ↑ |
| Glucose | 11.2 | 303.1 | 2618.3 | 0.022 | ↑ | |
| Phospho-enolpyruvic acid | 6449.0 | 0.0 | −100.0 | 0.022 | ↓ | |
| PPP | Glucose 6-phosphate | 328.6 | 18.8 | −94.3 | 0.025 | ↓ |
| 6-Phospho-gluconic acid | 269.9 | 71.9 | −73.4 | 0.104 | ||
| TCA | Malic acid | 1126.1 | 133.0 | −88.2 | 0.025 | ↓ |
| Succinic acid | 250.5 | 134.1 | −46.5 | 0.169 | ||
| Fumaric acid | 336.2 | 27.6 | −91.8 | 0.025 | ↓ | |
| cis-Aconitic acid | 43.7 | 1.1 | −97.6 | 0.022 | ↓ | |
| Citric acid | 6425.6 | 0.0 | −100.0 | 0.022 | ↓ | |
| alpha-Ketoglutaric acid | 222.3 | 78.2 | −64.8 | 0.132 | ||
| Urea Cycle | Citrulline | 10.3 | 10.3 | 0.4 | 0.802 | |
| Arginine | 14,526.1 | 2098.1 | −85.6 | 0.118 | ||
| Ornithine | 2626.1 | 59.4 | −97.7 | 0.025 | ↓ | |
| AA | Proline | 2642.0 | 1244.8 | −52.9 | 0.121 | |
| Glutamine | 31.0 | 29.1 | −6.1 | 0.578 | ||
| Glutamic acid | 410.2 | 78.6 | −80.8 | 0.025 | ↓ | |
| Additional | Lactic acid | 4613.3 | 21,161.8 | 358.7 | 0.022 | ↑ |
| Fructose | 2.3 | 18.8 | 706.0 | 0.022 | ↑ | |
| Relevant Metabolic Pathways | Metabolites | Conc. Average (ppb) | % Change (MCF-7crPOX vs. MCF-7WT) | P Value (Mann–Whitney) | MCF-7crPOX | |
|---|---|---|---|---|---|---|
| MCF-7WT | MCF-7crPOX | |||||
| Glycolysis | Pyruvic acid | 94.3 | 130.9 | 38.9 | 0.653 | |
| Glucose | 10.3 | 59.5 | 474.9 | 0.075 | ||
| Phospho-enolpyruvic acid | 3605.0 | 78.2 | −97.8 | 0.075 | ||
| PPP | Glucose 6-phosphate | 184.4 | 131.9 | −28.4 | 0.660 | |
| 6-Phospho-gluconic acid | 794.5 | 814.3 | 2.5 | 1.000 | ||
| TCA | Malic acid | 1361.2 | 635.2 | −53.3 | 0.172 | |
| Succinic acid | 195.5 | 158.0 | −19.2 | 0.660 | ||
| Fumaric acid | 402.0 | 184.4 | −54.1 | 0.172 | ||
| cis-Aconitic acid | 79.6 | 48.2 | −39.5 | 0.653 | ||
| Citric acid | 7462.9 | 4477.2 | −40.0 | 0.536 | ||
| alpha-Ketoglutaric acid | 949.3 | 684.7 | −27.9 | 0.660 | ||
| Urea Cycle | Citrulline | 6.1 | 11.0 | 81.5 | 0.377 | |
| Arginine | 10,138.1 | 6694.5 | −34.0 | 0.660 | ||
| Ornithine | 3957.4 | 1510.7 | −61.8 | 0.172 | ||
| AA | Proline | 3288.6 | 3373.5 | 2.6 | 1.000 | |
| Glutamine | 296.2 | 65.9 | −77.7 | 0.075 | ||
| Glutamic acid | 369.4 | 250.6 | −32.2 | 0.543 | ||
| Additional | Lactic acid | 24,919.0 | 23,080.4 | −7.4 | 1.000 | |
| Fructose | 12.0 | 21.4 | 78.1 | 0.075 | ||
| Relevant Metabolic Pathways | Metabolites | Conc. Average (ppb) | % Change (MCF-7WT+MET vs. MCF-7WT) | P Value (Mann–Whitney) | MCF-7WT+MET | |
|---|---|---|---|---|---|---|
| MCF-7WT | MCF-7WT+MET | |||||
| Glycolysis | Pyruvic acid | 94.3 | 201.7 | 114.0 | 0.132 | |
| Glucose | 10.3 | 124.2 | 1100.6 | 0.050 | ↑ | |
| Phospho-enolpyruvic acid | 3605.0 | 1502.4 | −58.3 | 0.176 | ||
| PPP | Glucose 6-phosphate | 184.4 | 35.7 | −80.6 | 0.165 | |
| 6-Phospho-gluconic acid | 794.5 | 79.2 | −90.0 | 0.050 | ↓ | |
| TCA | Malic acid | 1361.2 | 779.1 | −42.8 | 0.248 | |
| Succinic acid | 195.5 | 114.5 | −41.4 | 0.165 | ||
| Fumaric acid | 402.0 | 239.1 | −40.5 | 0.248 | ||
| cis-Aconitic acid | 79.6 | 5.7 | −92.9 | 0.050 | ↓ | |
| Citric acid | 7462.9 | 613.7 | −91.8 | 0.050 | ↓ | |
| alpha-Ketoglutaric acid | 949.3 | 1576.9 | 66.1 | 0.248 | ||
| Urea Cycle | Citrulline | 6.1 | 6.3 | 2.9 | 0.952 | |
| Arginine | 10,138.1 | 10963.0 | 8.1 | 0.578 | ||
| Ornithine | 3957.4 | 2019.8 | −49.0 | 0.248 | ||
| AA | Proline | 3288.6 | 4193.5 | 27.5 | 0.165 | |
| Glutamine | 296.2 | 1666.2 | 462.4 | 0.050 | ↑ | |
| Glutamic acid | 369.4 | 941.7 | 154.9 | 0.050 | ↑ | |
| Additional | Lactic acid | 24,919.0 | 15,892.6 | −36.2 | 0.248 | |
| Fructose | 12.0 | 9.8 | −18.3 | 0.086 | ||
| Relevant Metabolic Pathways | Metabolites | Conc. Average (ppb) | % Change (MCF-7crPOX+MET vs. MCF-7WT) | P Value (Mann–Whitney) | MCF-7crPOX+MET | |
|---|---|---|---|---|---|---|
| MCF-7WT | MCF-7crPOX+MET | |||||
| Glycolysis | Pyruvic acid | 94.3 | 131.6 | 39.6 | 0.586 | |
| Glucose | 10.3 | 204.6 | 1878.0 | 0.025 | ↑ | |
| Phospho-enolpyruvic acid | 3605.0 | 0.0 | −100.0 | 0.025 | ↓ | |
| PPP | Glucose 6-phosphate | 184.4 | 0.0 | −100.0 | 0.025 | ↓ |
| 6-Phospho-gluconic acid | 794.5 | 0.0 | −100.0 | 0.025 | ↓ | |
| TCA | Malic acid | 1361.2 | 37.0 | −97.3 | 0.025 | ↓ |
| Succinic acid | 195.5 | 91.9 | −53.0 | 0.086 | ||
| Fumaric acid | 402.0 | 6.1 | −98.5 | 0.025 | ↓ | |
| cis-Aconitic acid | 79.6 | 1.4 | −98.3 | 0.025 | ↓ | |
| Citric acid | 7462.9 | 0.0 | −100.0 | 0.025 | ↓ | |
| alpha-Ketoglutaric acid | 949.3 | 40.9 | −95.7 | 0.025 | ↓ | |
| Urea Cycle | Citrulline | 6.1 | 1.9 | −69.5 | 0.226 | |
| Arginine | 10,138.1 | 539.3 | −94.7 | 0.025 | ↓ | |
| Ornithine | 3957.4 | 0.0 | −100.0 | 0.025 | ↓ | |
| AA | Proline | 3288.6 | 3664.0 | 11.4 | 0.905 | |
| Glutamine | 296.2 | 210.6 | −28.9 | 0.461 | ||
| Glutamic acid | 369.4 | 68.4 | −81.5 | 0.025 | ↓ | |
| Additional | Lactic acid | 24,919.0 | 17,098.2 | −31.4 | 0.226 | |
| Fructose | 12.0 | 15.8 | 31.6 | 0.086 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huynh, T.Y.L.; Oscilowska, I.; Sáiz, J.; Nizioł, M.; Baszanowska, W.; Barbas, C.; Palka, J. Metformin Treatment or PRODH/POX-Knock out Similarly Induces Apoptosis by Reprograming of Amino Acid Metabolism, TCA, Urea Cycle and Pentose Phosphate Pathway in MCF-7 Breast Cancer Cells. Biomolecules 2021, 11, 1888. https://doi.org/10.3390/biom11121888
Huynh TYL, Oscilowska I, Sáiz J, Nizioł M, Baszanowska W, Barbas C, Palka J. Metformin Treatment or PRODH/POX-Knock out Similarly Induces Apoptosis by Reprograming of Amino Acid Metabolism, TCA, Urea Cycle and Pentose Phosphate Pathway in MCF-7 Breast Cancer Cells. Biomolecules. 2021; 11(12):1888. https://doi.org/10.3390/biom11121888
Chicago/Turabian StyleHuynh, Thi Yen Ly, Ilona Oscilowska, Jorge Sáiz, Magdalena Nizioł, Weronika Baszanowska, Coral Barbas, and Jerzy Palka. 2021. "Metformin Treatment or PRODH/POX-Knock out Similarly Induces Apoptosis by Reprograming of Amino Acid Metabolism, TCA, Urea Cycle and Pentose Phosphate Pathway in MCF-7 Breast Cancer Cells" Biomolecules 11, no. 12: 1888. https://doi.org/10.3390/biom11121888