
Molecular Mechanism of Vitamin K2 Protection against Amyloid-β-Induced Cytotoxicity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of β–CTF/C6 Cell Line
2.2. Real-Time PCR (qPCR) Analyses
2.3. Cell Viability under Vitamin K2 Treatment
2.4. Warfarin Assay
2.5. Reactive Oxygen Species (ROS) Assay
2.6. Caspase Assay
2.7. Inhibitor Assay
2.8. Western Blot Assay
2.9. siRNA Silencing Gas6
2.10. Statistical Analysis
3. Results
3.1. Construction of β-CTF/C6 Cell Lines
3.2. Vitamin K2 Protects Cells against Aβ-Induced Cytotoxicity
3.3. The Protective Effect of Vitamin K2 on Aβ Toxicity Is Abolished by Adding Warfarin
3.4. Vitamin K2 Reduces the Free Radical Level
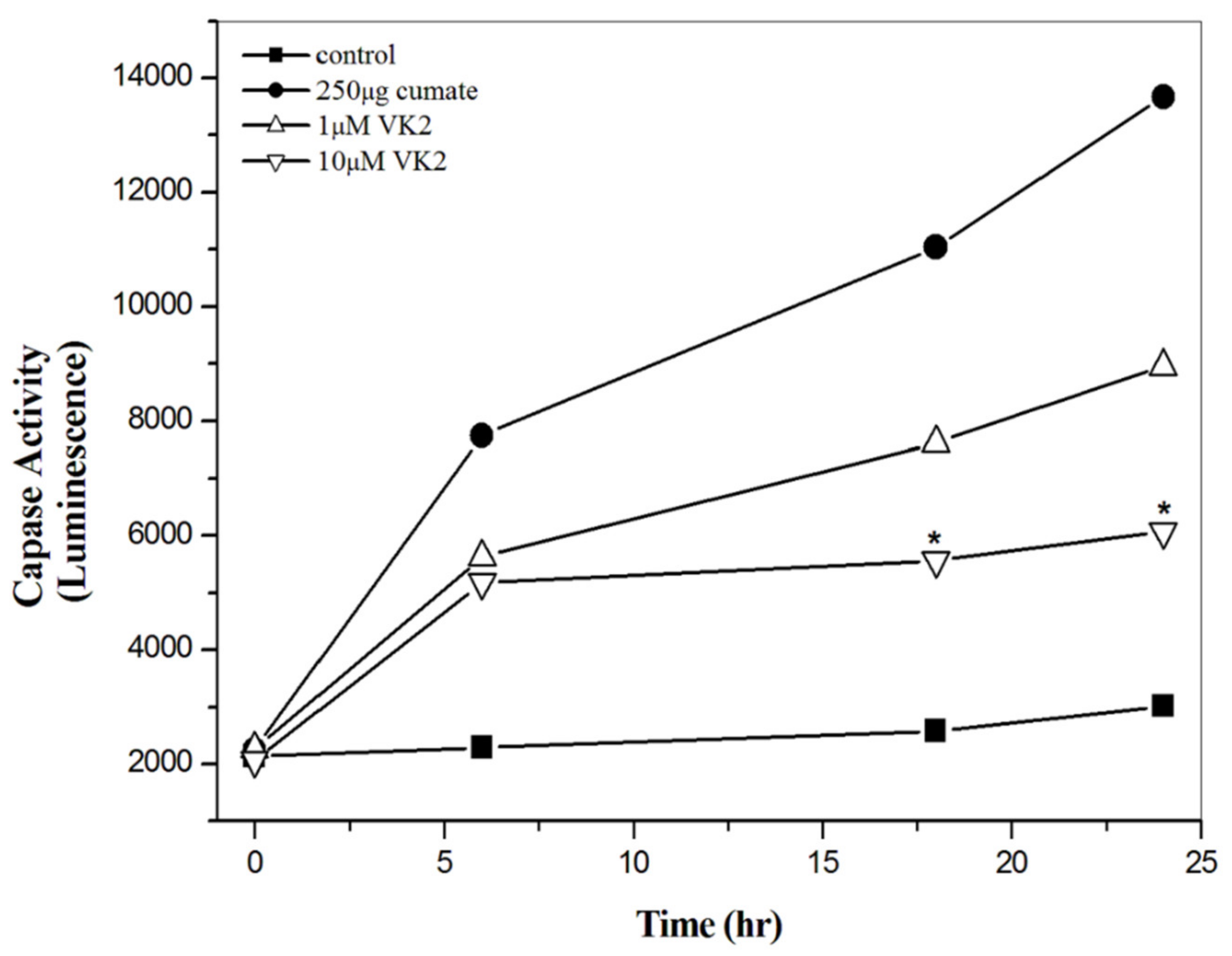
3.5. Vitamin K2 Effectively Inhibit Aβ-Mediated Apoptosis
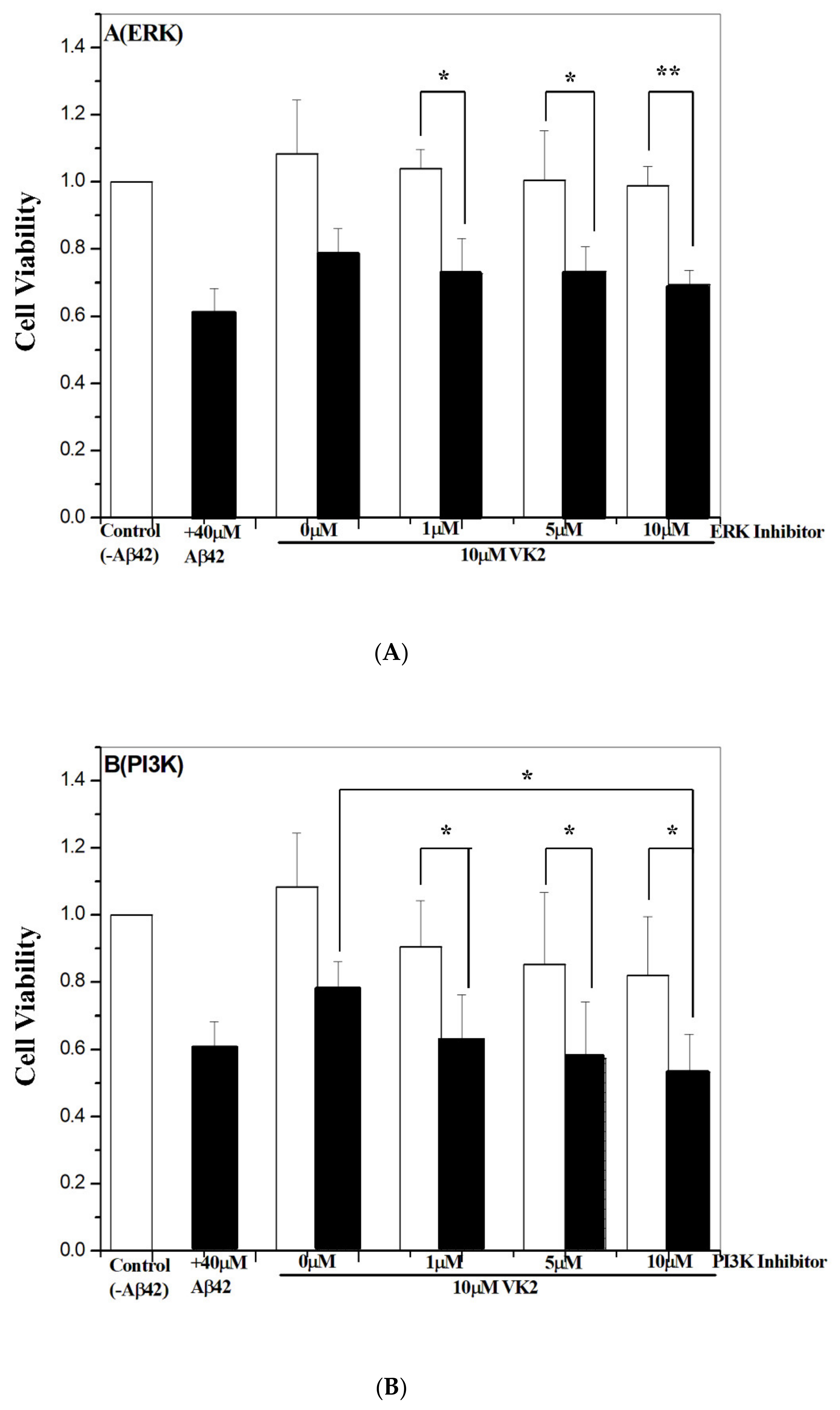
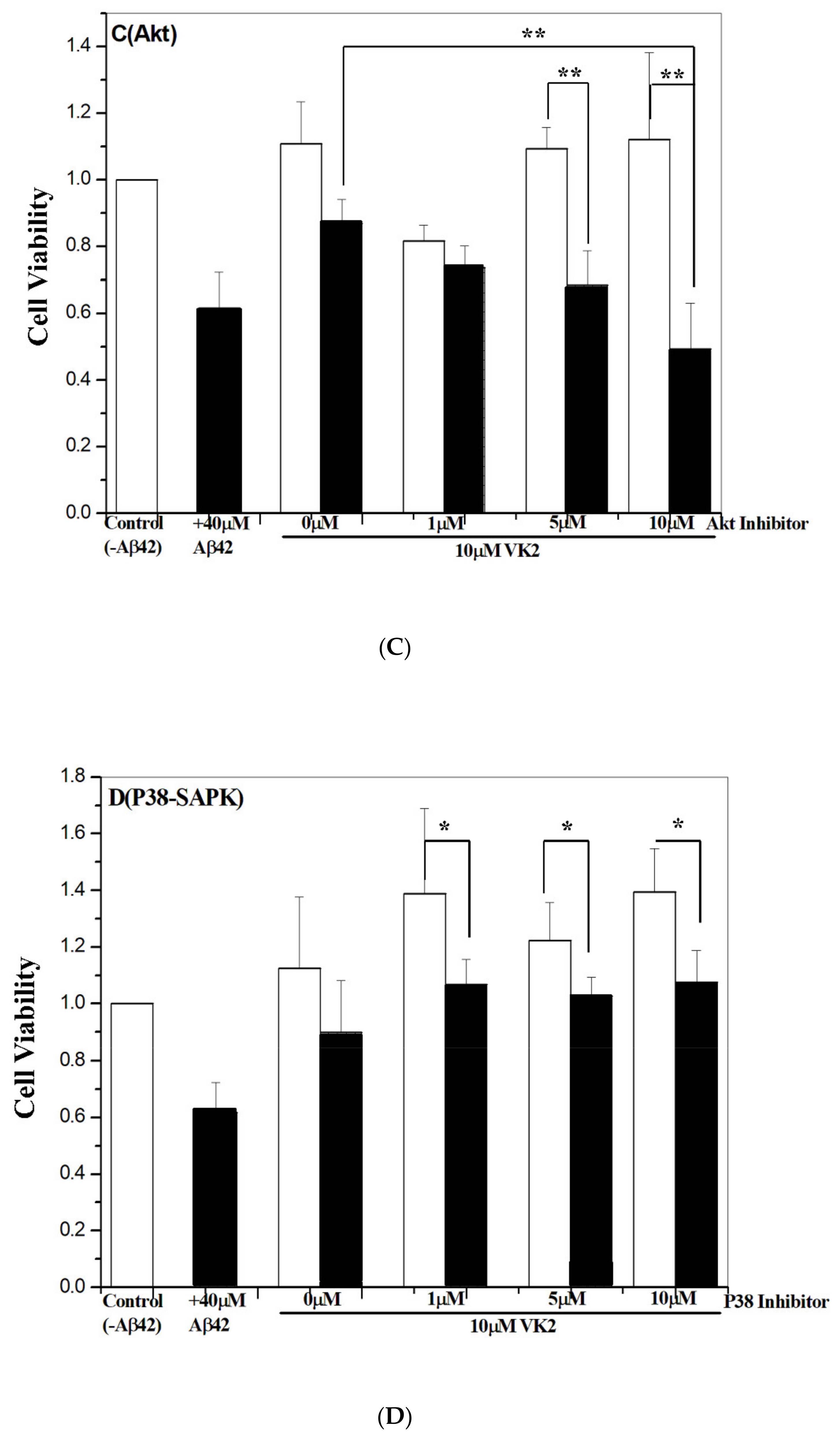
3.6. Vitamin K2-Mediated Protection against Aβ Toxicity Is via Activation of the PI3K/Akt Pathway
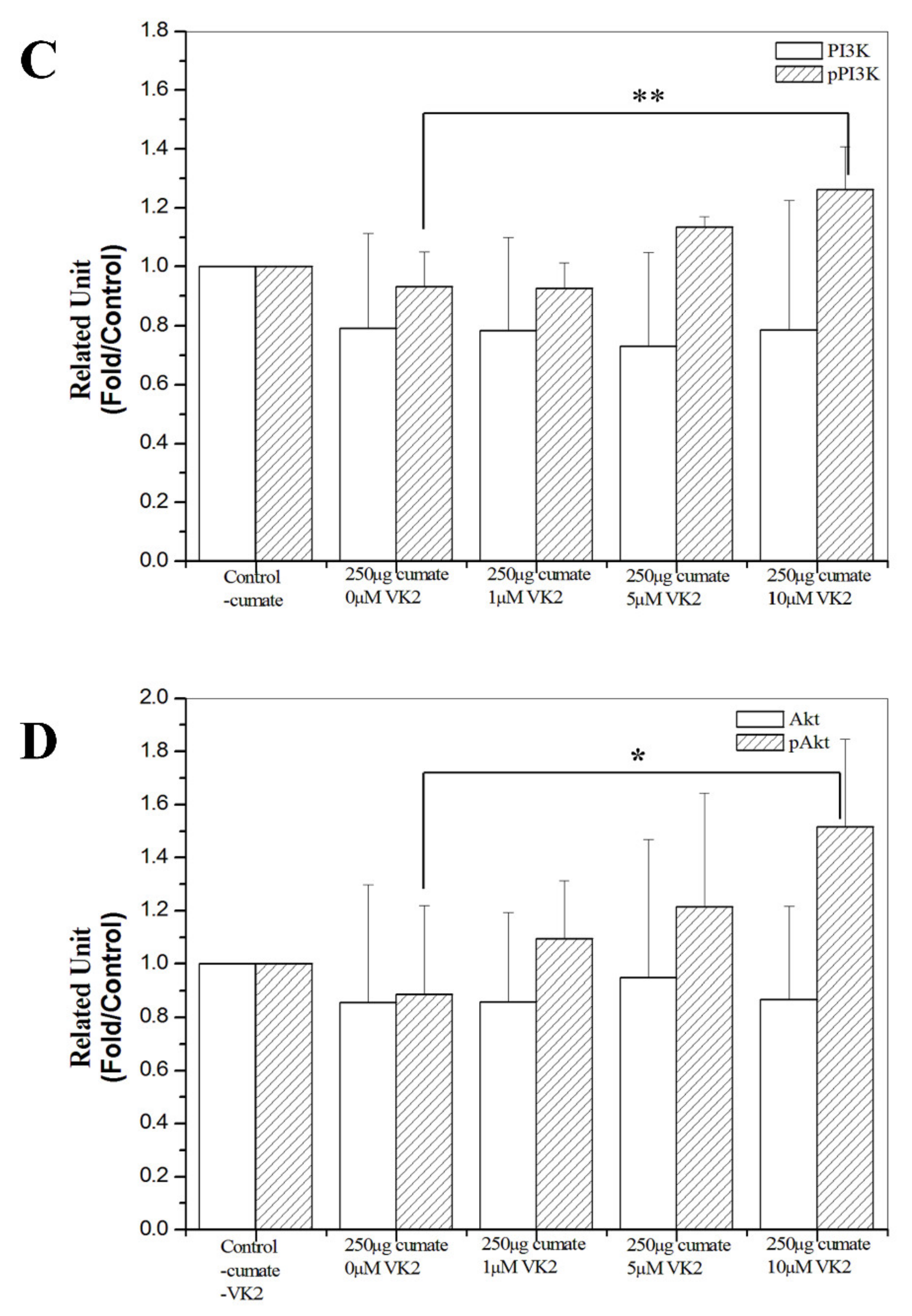
3.7. Western Blot Analysis of Phosphorylated PI3K and Akt Proteins
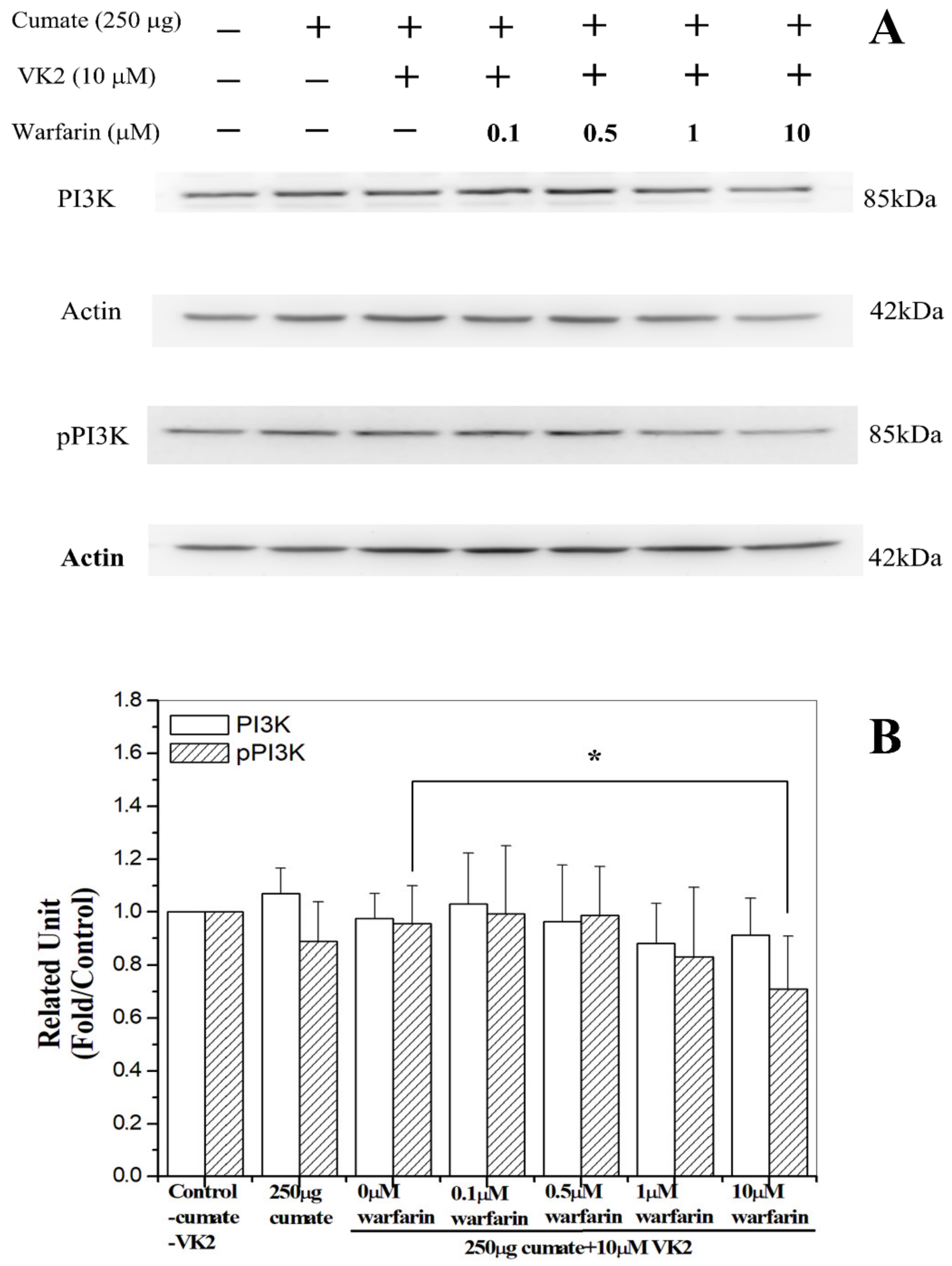
3.8. Warfarin Abolished Vitamin K2-Mediated Phosphorylation of PI3K
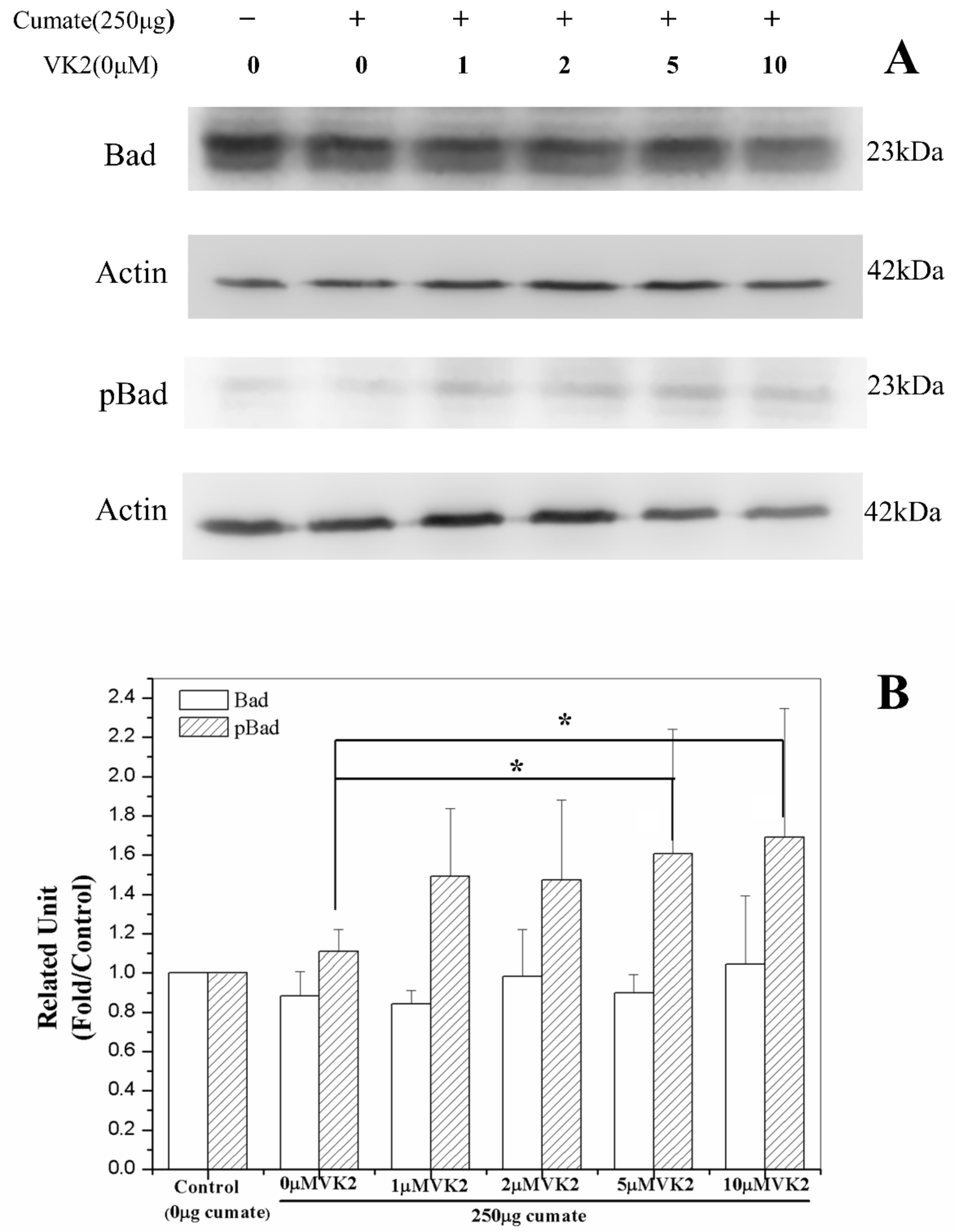
3.9. Vitamin K2 Activates Antiapoptosis via Phosphorylation of Bad
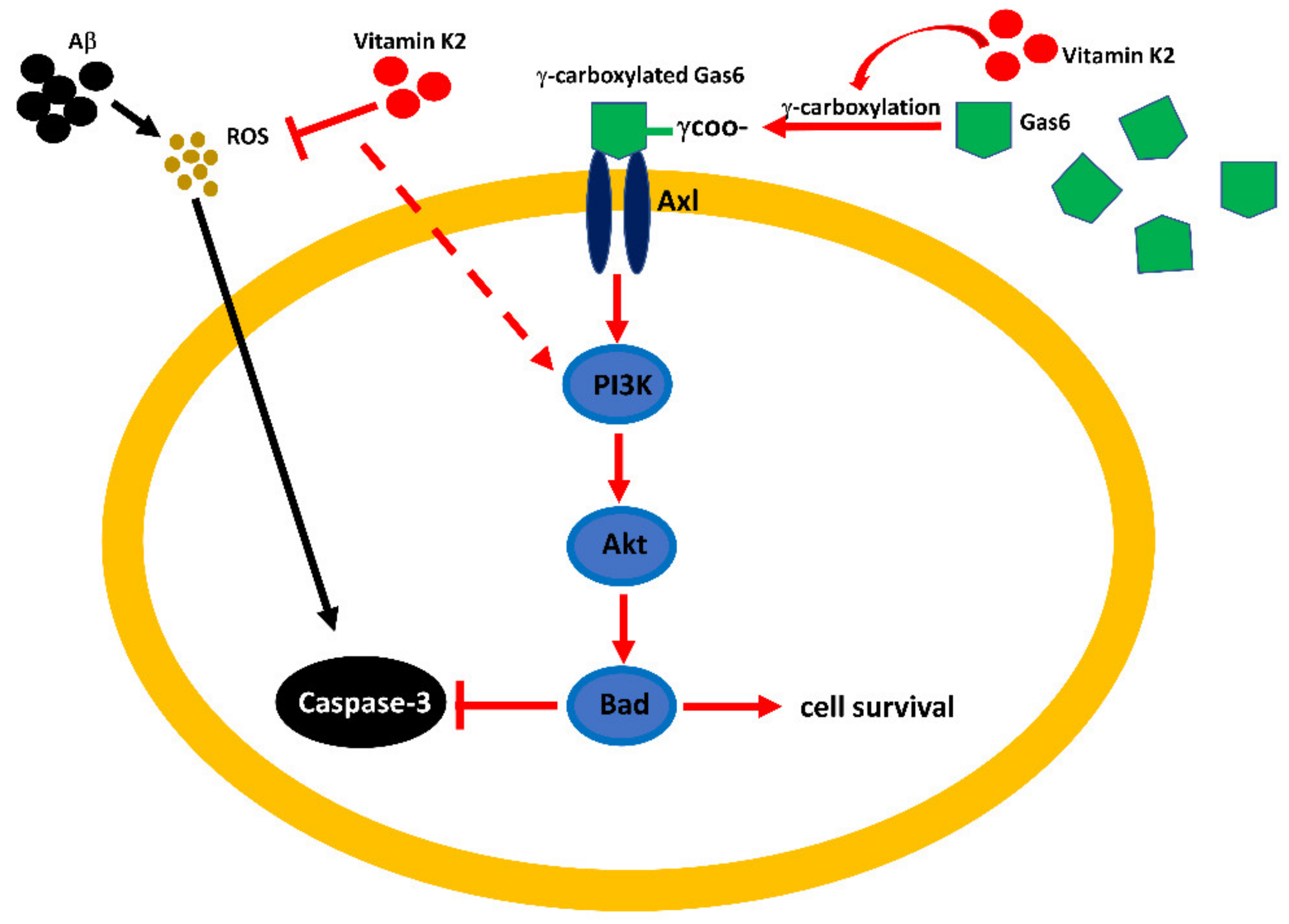
3.10. Vitamin K2-Mediated PI3K/Akt Pathway Upregulated via Gas6/Axl Receptor
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A6730 | Akt 1/2 inhibitor |
| AD | Alzheimer’s disease |
| Aβ | Amyloid-β |
| Akt | Protein kinase B |
| ApoE4 | Apolipoprotein E |
| APP | Amyloid precursor protein |
| Axl | Axl receptor tyrosine kinase |
| Bad | Bcl-2-associated death promoter protein, tyros3, Axl and Mer (TAM) family |
| C99 | C-terminal 99 residues of APP |
| β-CTF | C-terminal 99-residue fragment of APP |
| β-CTF/C6 | C6 cell line-containing β-CTF gene |
| ERK1/2 | Extracellular signal-regulated protein kinases 1 and 2 |
| Gas6 | Growth arrest-specific protein 6 |
| HEK293 | Human embryonic kidney 293 |
| c-JNK | C-Jun N-terminal kinase |
| LY294002 | PI3K inhibitor |
| MAPK | Mitogen-activated protein kinase |
| MK4 | Menaquinone-4 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| p38-SAPK | Stress-activated protein kinase p38 |
| PD98059 | Extracellular signal-regulated protein kinases 1 and 2 (ERK) inhibitor |
| PI3K | Phosphatidylinositol 3-kinase |
| R428 | Axl inhibitor |
| ROS | Reactive oxygen species |
| SB202190 | Stress-activated protein kinase p38 (p38-SAPK) inhibitor |
| SN50 | NF-κB inhibitor |
| SP600125 | JNK inhibitor |
| VKD | Vitamin K-dependent |
References
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s Disease: Past, Present, and Future. J. Int. Neuropsychol. Soc. 2017, 23, 818–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garad, M.; Edelmann, E.; Leßmann, V. Impairment of Spike-Timing-Dependent Plasticity at Schaffer Collateral-CA1 Synap-ses in Adult APP/PS1 Mice Depends on Proximity of Aβ Plaques. Int. J. Mol. Sci. 2021, 22, 1378. [Google Scholar] [CrossRef]
- Brito, L.M.; Ribeiro-dos-Santos, Â.; Vidal, A.F.; de Araújo, G.S. Differential Expression and miRNA–Gene Interactions in Early and Late Mild Cognitive Impairment. Biology 2020, 9, 251. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [Green Version]
- Viola, K.L.; Klein, W.L. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Multhaup, G.; Bayer, T.A. A modified β-amyloid hypothesis: Intraneuronal accumulation of the β-amyloid pep-tide —The first step of a fatal cascade. J. Neurochem. 2004, 91, 513–520. [Google Scholar] [CrossRef]
- Guillozet, A.L.; Weintraub, S.; Mash, D.C.; Mesulam, M.M. Neurofibrillary Tangles, Amyloid, and Memory in Aging and Mild Cognitive Impairment. Arch. Neurol. 2003, 60, 729–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanz, R.E. The synaptic A β hypothesis of Alzherimer’s disease. Nat. Neurosic. 2005, 8, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.Q.; Tzeng, Y.J.; Chang, Y.X.; Huang, H.B.; Lin, T.H.; Chyan, C.L.; Chen, Y.C. The correlation between neurotox-icity, aggregative ability and secondary structure studied by sequence truncated Aβ peptides. FEBS Lett. 2007, 581, 1161–1165. [Google Scholar] [CrossRef] [Green Version]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dys-function and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoweri, A.O.; Gagolewicz, P.; Frazier, H.N.; Gant, J.C.; Andrew, R.D.; Bennett, B.M.; Thibault, O. Neuronal Calcium Imaging, Excitability, and Plasticity Changes in the Aldh2−/− Mouse Model of Sporadic Alzheimer’s Disease. J. Alzheimers Dis. 2020, 77, 1623–1637. [Google Scholar] [CrossRef]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catanesi, M.; D’Angelo, M.; Tupone, M.G.; Benedetti, E.; Giordano, A.; Castelli, V.; Cimini, A. MicroRNAs Dysregulation and Mitochondrial Dysfunction in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 5986. [Google Scholar] [CrossRef] [PubMed]
- Rehker, J.; Rodhe, J.; Nesbitt, R.R.; Boyle, E.A.; Martin, B.K.; Lord, J.; Karaca, I.; Naj, A.; Jessen, F.; Helisalmi, S.; et al. Caspase-8, association with Alzheimer’s Disease and functional analysis of rare variants. PLoS ONE 2017, 12, e0185777. [Google Scholar] [CrossRef] [Green Version]
- Calissano, P.; Matrone, C.; Amadoro, G. Apoptosis and in vitro Alzheimer’s disease neuronal models. Commun. Integr. Biol. 2009, 2, 163–169. [Google Scholar] [CrossRef]
- Chen, T.J.; Wang, D.C.; Chen, S.S. Amyloid-beta interrupts the PI3K-Akt-mTOR signaling pathway that could be involved in brain-derived neurotrophic factor-induced Arc expression in rat cortical neurons. J. Neurosci. Res. 2009, 87, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.-G.; Lv, J.; Yang, W.-N.; Chang, K.-W.; Hu, X.-D.; Shi, L.-L.; Zhai, W.-Y.; Zong, H.-F.; Qian, Y.-H. The p38 mitogen activated protein kinase regulates β-amyloid protein internalization through the α7 nicotinic acetylcholine receptor in mouse brain. Brain Res. Bull. 2018, 137, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.-H.; So, S.-P.; Kim, N.-Y.; Kim, H.-J.; Yoon, S.-Y.; Kim, D.-H. c-Jun N-terminal Kinase (JNK) induces phosphorylation of amyloid precursor protein (APP) at Thr668, in okadaic acid-induced neurodegeneration. BMB Rep. 2016, 49, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.T.; Xu, J.M.; Ku, G.; Han, X.; Yang, D.I.; Chen, S.; Hsu, C.Y. Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase -ceramide pathway. J. Cell Biol. 2004, 164, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Lee, J.W.; Lee, H.; Yoo, H.S.; Yun, Y.P.; Oh, K.W.; Ha, T.Y.; Hong, J.T. Inhibitory effect of green tea extract on beta-amyloid-induced PC12 cell death by inhibition of the activation of NF-kappaB and ERK/p38 MAP kinase pathway through antioxidant mechanism. Mol. Brain Res. 2005, 140, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Singh, D.; Kh, R. Sinapic Acid Alleviates Oxidative Stress and Neuro-Inflammatory Changes in Sporadic Model of Alzheimer’s Disease in Rats. Brain Sci. 2020, 10, 923. [Google Scholar] [CrossRef]
- Song, H.-C.; Chen, Y.; Chen, Y.; Park, J.; Zheng, M.; Surh, Y.-J.; Kim, U.-H.; Park, J.W.; Yu, R.; Chung, H.T.; et al. GSK-3β inhibition by curcumin mitigates amyloidogenesis via TFEB activation and anti-oxidative activity in human neuroblastoma cells. Free. Radic. Res. 2020, 1–13. [Google Scholar] [CrossRef]
- Ju, T.C.; Chen, S.D.; Liu, C.C.; Yang, D.I. Protective effects of S-nitrosoglutathione against smyloid β-peptide neurotoxicity. Free Radic. Biol. Med. 2005, 38, 938–949. [Google Scholar] [CrossRef]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Vitamin A exhibits potent antiamyloidogenic and fibril-destabilizing effects in vitro. Exp. Neurol. 2004, 189, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.-H.; Hyon-Lee; Lee, K.-M. The Possible Role of Antioxidant Vitamin C in Alzheimer’s Disease Treatment and Prevention. Am. J. Alzheimer’s Dis. Other Dement. 2013, 28, 120–125. [Google Scholar] [CrossRef]
- Marinelli, R.; Torquato, P.; Bartolini, D.; Mas-Bargues, C.; Bellezza, G.; Gioiello, A.; Borras, C.; De Luca, A.; Fallarino, F.; Se-bastiani, B.; et al. Garcinoic acid pre-vents β-amyloid (Aβ) deposition in the mouse brain. J. Biol. Chem. 2020, 295, 11866–11876. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.W.; Mett, J.; Hartmann, T. The Impact of Vitamin E and Other Fat-Soluble Vitamins on Alzheimer´s Disease. Int. J. Mol. Sci. 2016, 17, 1785. [Google Scholar] [CrossRef] [Green Version]
- Ferland, G. Vitamin K and the Nervous system: An overview of its action. Adv. Nutr. 2012, 3, 204–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shearer, M.J.; Fu, X.; Booth, S.L. Vitamin K Nutrition, Metabolism, and Requirements: Current Concepts and Future Research. Adv. Nutr. 2012, 3, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Capozzi, A.; Scambia, G.; Lello, S. Calcium, vitamin D, vitamin K2, and magnesium supplementation and skeletal health. Maturitas 2020, 140, 55–63. [Google Scholar] [CrossRef]
- Takeda, T.; Sato, Y. Effects of Vitamin K2 on Osteoporosis. Curr. Pharm. Des. 2004, 10, 2557–2576. [Google Scholar] [CrossRef]
- Berkner, K.L. Vitamin K-dependent carboxylation. Vitam. Horm. 2008, 78, 131–156. [Google Scholar] [PubMed]
- Danziger, J. Vitamin K-dependent Proteins, Warfarin, and Vascular Calcification. Clin. J. Am. Soc. Nephrol. 2008, 3, 1504–1510. [Google Scholar] [CrossRef]
- Lev, M.; Milford, A.E. Effect of vitamin K depletion and restoration on sphingolipid metabolism in Bacteroides melani-nogenicus. J. Lipid Res. 1972, 13, 364–370. [Google Scholar] [CrossRef]
- Sundaram, K.S.; Lev, M. Warfarin administration reduces synthesis of sulfatides and other sphingolipids in mouse brain. J. Lipid Res. 1988, 29, 1475–1479. [Google Scholar] [CrossRef]
- Qiu, C.; Zheng, H.; Tao, H.; Yu, W.; Jiang, X.; Li, A.; Jin, H.; Lv, A.; Li, H. Vitamin K2 inhibits rat vascular smooth muscle cell calcification by restoring the Gas6/Axl/Akt anti-apoptotic pathway. Mol. Cell. Biochem. 2017, 433, 149–159. [Google Scholar] [CrossRef]
- Shafit-Zagardo, B.; Gruber, R.C.; DuBois, J.C. The role of TAM family receptors and ligands in the nervous system: From development to pathobiology. Pharmacol. Ther. 2018, 188, 97–117. [Google Scholar] [CrossRef]
- Simes, D.C.; Viegas, C.S.B.; Araújo, N.; Marreiros, C. Vitamin K as a Diet Supplement with Impact in Human Health: Current Ev-idence in Age-Related Diseases. Nutrients 2020, 12, 138. [Google Scholar] [CrossRef] [Green Version]
- Allison, A.C. The possible role of vitamin K deficiency in the pathogenesis of Alzheimer’s disease and in augmenting brain damage associated with cardiovascular disease. Med. Hypotheses 2001, 57, 151–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presse, N.; Shatenstein, B.; Kergoat, M.-J.; Ferland, G. Low Vitamin K Intakes in Community-Dwelling Elders at an Early Stage of Alzheimer’s Disease. J. Am. Diet. Assoc. 2008, 108, 2095–2099. [Google Scholar] [CrossRef]
- Huy, P.D.Q.; Yu, Y.-C.; Ngo, S.T.; Van Thao, T.; Chen, C.-P.; Li, M.S.; Chen, Y.-C. In silico and in vitro characterization of anti-amyloidogenic activity of vitamin K3 analogues for Alzheimer’s disease. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 2960–2969. [Google Scholar] [CrossRef] [PubMed]
- Kaesler, N.; Immendorf, S.; Ouyang, C.; Herfs, M.; Drummen, N.; Carmeliet, P.; Vermeer, C.; Floege, J.; Krüger, T.; Schlieper, G. Gas6 protein: Its role in cardiovascular calcification. BMC Nephrol. 2016, 17, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkermann, R.; Aprico, A.; Perera, A.A.; Bujalka, H.; Cole, A.E.; Xiao, J.; Field, J.; Kilpatrick, T.J.; Binder, M.D. The TAM receptor Tyro3 regulates myelination in the central nervous system. Glia 2017, 65, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ye, J.; Zhao, G.; Hong, G.; Hu, X.; Cao, K.; Wu, Y.; Lu, Z. Gas6 attenuates lipopolysaccharide induced TNF α expression and apoptosis in H9C2 cells through NF κB and MAPK inhibition via the Axl/PI3K/Akt pathway. Int. J. Mol. Med. 2019, 44, 982–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, P.-Y.; Chen, X.-L.; Lei, Y.-H.; Liu, C.-Y.; Liu, Y.-W. Growth arrest-specific gene 6 protein promotes the proliferation and migration of endothelial progenitor cells through the PI3K/AKT signaling pathway. Int. J. Mol. Med. 2014, 34, 299–306. [Google Scholar] [CrossRef]
- Sainaghi, P.P.; Bellan, M.; Lombino, F.; Alciato, F.; Carecchio, M.; Galimberti, D.; Fenoglio, C.; Scarpini, E.; Cantello, R.; Pirisi, M.; et al. Growth Arrest Specific 6 Concentration is Increased in the Cerebrospinal Fluid of Patients with Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 55, 59–65. [Google Scholar] [CrossRef]
- Grommes, C.; Lee, C.Y.D.; Wilkinson, B.L.; Jiang, Q.; Koenigsknecht-Talboo, J.L.; Varnum, B.; Landreth, G.E. Regulation of Microglial Phagocytosis and Inflammatory Gene Expression by Gas6 Acting on the Axl/Mer Family of Tyrosine Kinases. J. Neuroimmune Pharmacol. 2008, 3, 130–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagami, T.; Ueda, K.; Asakura, K.; Sakaeda, T.; Nakazato, H.; Kuroda, T.; Hata, S.; Sakaguch, G.; Itoh, N.; Nakano, T.; et al. Gas6 rescues cortical neurons from amyloid beta protein-induced apoptosis. Neuropharmacology 2002, 43, 1289–1296. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, Q.; Xiao, B.; Lu, Q.; Wang, Y.; Wang, X. Involvement of receptor tyrosine kinase Tyro3 in amyloidogenic APP processing and β-amyloid deposition in Alzheimer’s disease models. PLoS ONE 2012, 7, e39035. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-L.; Chen, W.-Y.; Chen, Y.-P.; Kuo, C.-Y.; Chen, S.-D. Activation of GLP-1 Receptor Enhances Neuronal Base Excision Repair via PI3K-AKT-Induced Expression of Apurinic/Apyrimidinic Endonuclease 1. Theranostics 2016, 6, 2015–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.W.; Hua, D.H.; Shie, F.S.; Maezawa, I.; Sopher, B.; Martin, G.M. Novel tricyclic pyrone compounds prevent intra-cellular APP C99-induced cell death. J. Mol. Neurosci. 2002, 19, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Pagnotta, S.; Biferi, M.G.; Barkats, M.; Lacor, P.; Klein, W.; Bauer, C.; Checler, F. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016, 132, 257–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Montesinos, J. Increased localization of App-C99 in mitochondria associat-ed ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 2017, 36, 3356–3371. [Google Scholar] [CrossRef]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neuro-degenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, G.; Nhan, H.S.; Tyan, S.H.; Kawakatsu, Y.; Zhang, C.; Navarro, M.; Koo, E.H. Caspase Activation and Caspa-se-Mediated Cleavage of APP Is Associated with Amyloid β-Protein-Induced Synapse Loss in Alzheimer’s Disease. Cell Rep. 2020, 31, 107839. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Yang, J.; Wei, Y.; Li, J. Effects of piceatannol and pterostilbene against β-amyloid-induced apoptosis on the PI3K/Akt/Bad signaling pathway in PC12 cells. Food Funct. 2016, 7, 1014–1023. [Google Scholar] [CrossRef]
- Zhu, Y.; Tang, H.; Zhang, L.; Gong, L.; Wu, G.; Ni, J.; Tang, X. Suppression of miR-21-3p enhances TRAIL-mediated apopto-sis in liver cancer stem cells by suppressing the PI3K/Akt/Bad cascade via regulating PTEN. Cancer Manag. Res. 2019, 11, 955–968. [Google Scholar] [CrossRef] [Green Version]
- Tamadon-Nejad, S.; Ouliass, B.; Rochford, J.; Ferland, G. Vitamin K Deficiency Induced by Warfarin Is Associated With Cog-nitive and Behavioral Perturbations, and Alterations in Brain Sphingolipids in Rats. Front. Aging Neurosci. 2018, 10, 213. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Weissmiller, A.M.; White, J.A.; Fang, F.; Wang, X.; Wu, Y.; Pearn, M.L.; Zhao, X.; Sawa, M.; Chen, S.; et al. Amyloid precursor protein–mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J. Clin. Investig. 2016, 126, 1815–1833. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Wang, X.; Li, X.; Wang, X.; Zhang, C.; Li, W.; Zhang, Y.; Gu, H.; Xie, X.; Nan, F.; et al. Targeting the γ-/β-secretase interaction reduces β-amyloid generation and ameliorates Alzheimer’s disease-related pathogenesis. Cell Discov. 2015, 1, 15021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Chen, K.; Wang, H.; Zhang, Y.; Duan, X.; Xue, Y.; He, H.; Huang, Y.; Chen, Z.; Ren, H.; et al. Gastrin Attenuates Renal Ischemia/Reperfusion Injury by a PI3K/Akt/Bad-Mediated Anti-apoptosis Signaling. Front. Pharmacol. 2020, 11, 540479. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The Role of PI3K/Akt and ERK in Neurodegenerative Disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-D.; Zhang, B.; Wang, Y.; Li, H.; Xiong, R.; Zhao, Z.; Chu, X.; Li, Q.; Sun, S. Neuroprotective effects of salidroside through PI3K/Akt pathway activation in Alzheimer’s disease models. Drug Des. Dev. Ther. 2016, 10, 1335–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.-H.; Fang, S.-T.; Chen, Y.-C. Molecular Mechanism of Vitamin K2 Protection against Amyloid-β-Induced Cytotoxicity. Biomolecules 2021, 11, 423. https://doi.org/10.3390/biom11030423
Huang S-H, Fang S-T, Chen Y-C. Molecular Mechanism of Vitamin K2 Protection against Amyloid-β-Induced Cytotoxicity. Biomolecules. 2021; 11(3):423. https://doi.org/10.3390/biom11030423
Chicago/Turabian StyleHuang, Shu-Hsiang, Sheng-Ting Fang, and Yi-Cheng Chen. 2021. "Molecular Mechanism of Vitamin K2 Protection against Amyloid-β-Induced Cytotoxicity" Biomolecules 11, no. 3: 423. https://doi.org/10.3390/biom11030423