
Omics Technologies to Decipher Regulatory Networks in Granulocytic Cell Differentiation

 , ,
, ,  and
and
Abstract
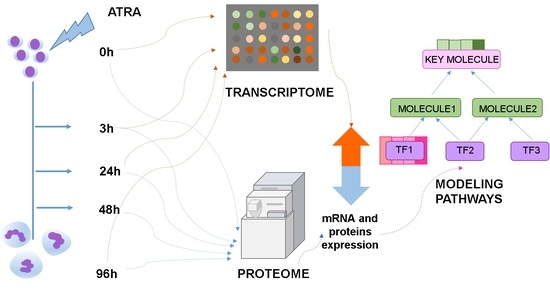
:
1. Introduction
2. Materials and Methods
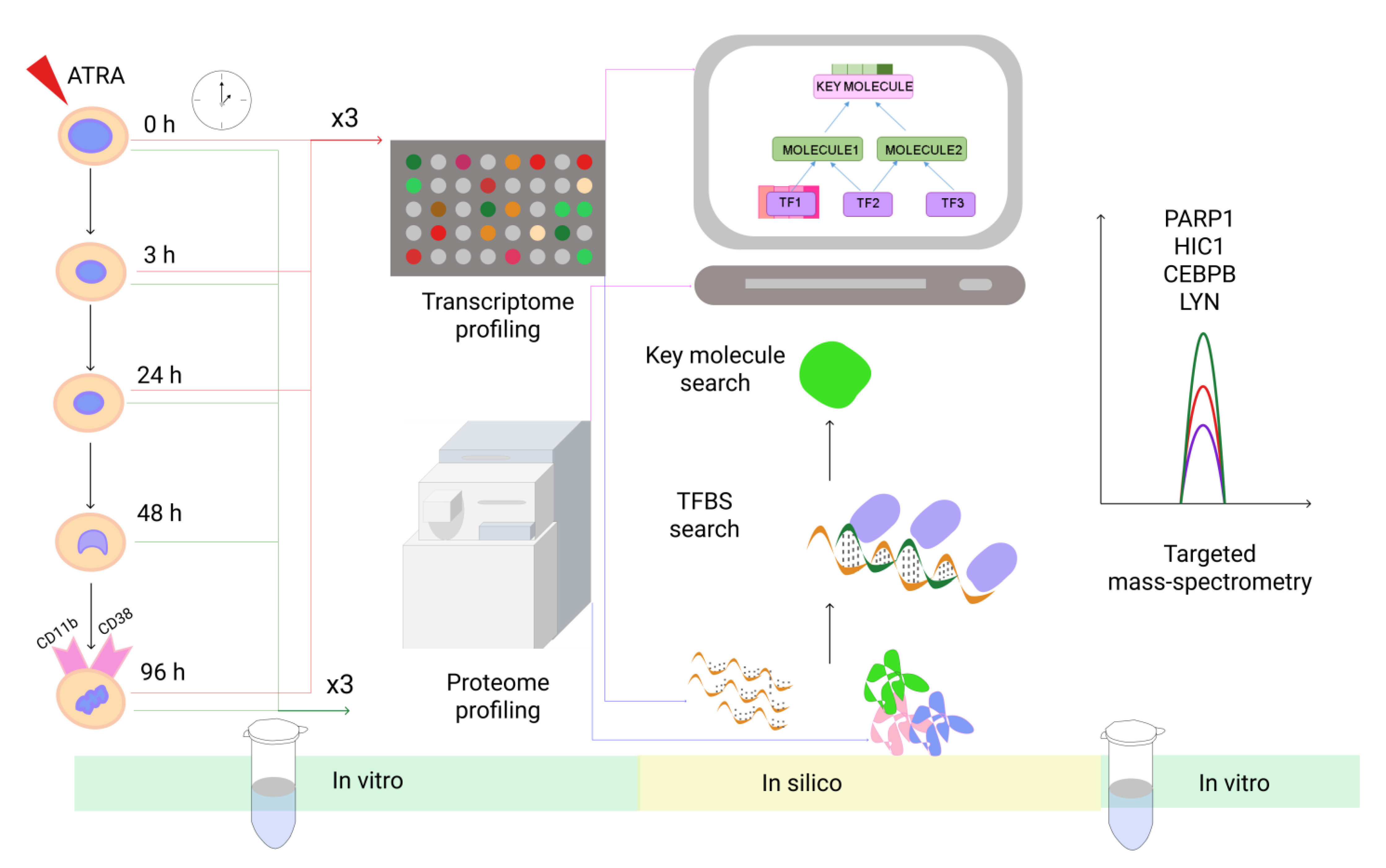
2.1. Experimental Design
2.2. HL60 Cells Cultures
2.3. Transcriptome Analysis
2.4. Preparation of HL60 Cells Lysates and In-Solution Digestion with Trypsin
2.5. Shotgun Mass Spectrometry
2.6. Data Analysis
2.7. Functional Classification of Differentially Expressed Genes and Proteins
2.8. Search for Transcription Factors, Putatively Regulating Gene and Protein Expression during ATRA-Induced Differentiation of HL-60 Cells
2.9. Generation of Regulatory Networks
2.10. Selected Reaction Monitoring (SRM)
3. Results
3.1. Transcriptome Analysis and Functional Annotation of Differentially Expressed Genes during ATRA-Induced Differentiation of HL-60 Cells
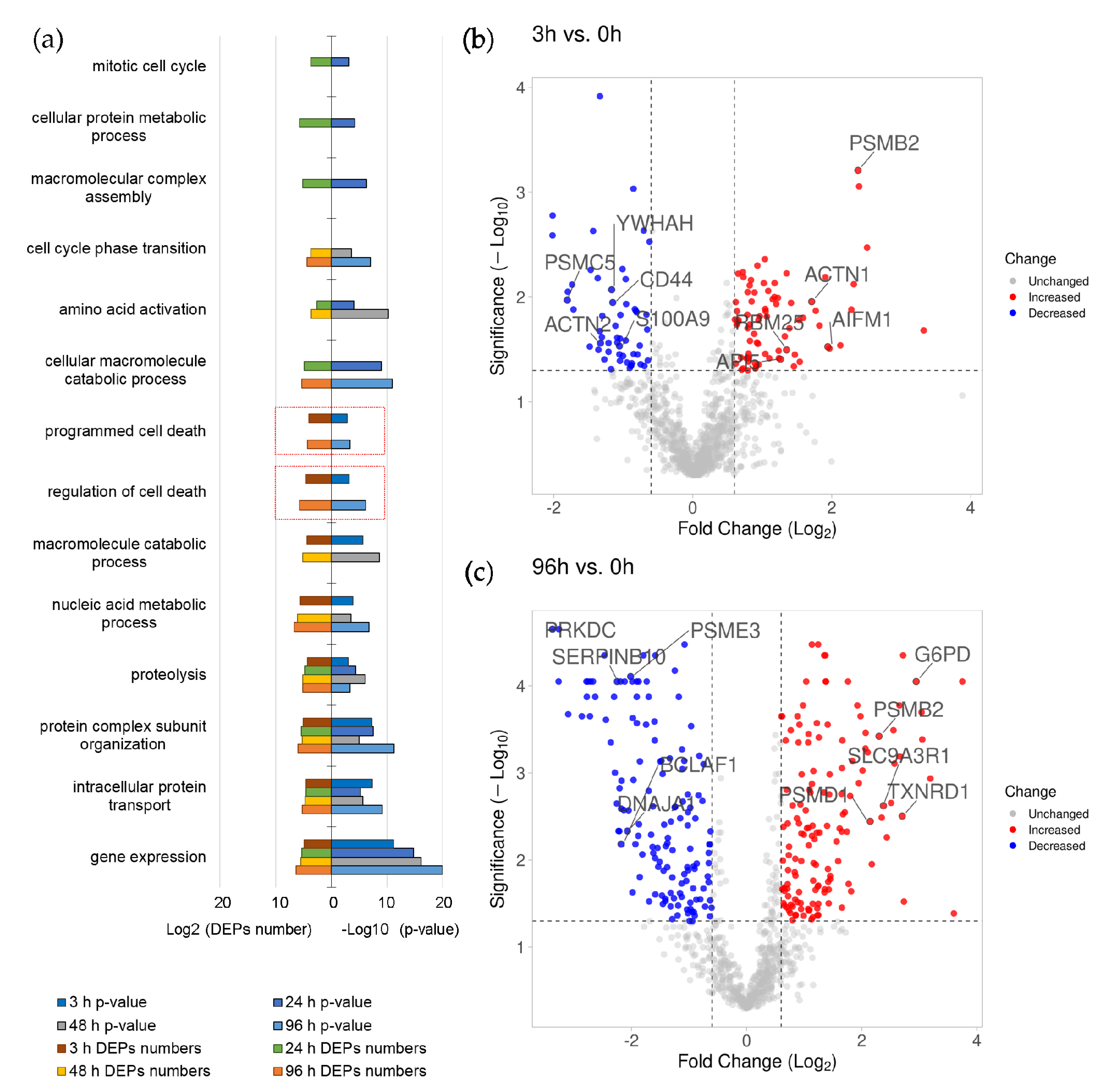
3.2. Proteomic Analysis and Functional Annotation of Differentially Expressed Proteins during ATRA-Induced Differentiation of HL-60 Cells
3.3. The Workflow of Transcriptome- and Proteome-Based Regulatory Networks Design
- Identification of TFs that can regulate the DEGs (MCD group) and DEPs at different time points after ATRA treatment using TRANSFAC@ database followed by matching putative TFs with the list of all transcripts identified (Supplemental Table S1) to cut-off the molecules that are not expressed in HL-60 cells at the mRNA level;
- The upstream prediction of key molecules that regulate the TFs determined at the previous step using TRANSFAC@ database followed by visualization of the predicted interaction as a model regulatory networks.
3.3.1. The Transcriptome-Based Modeling Pathway
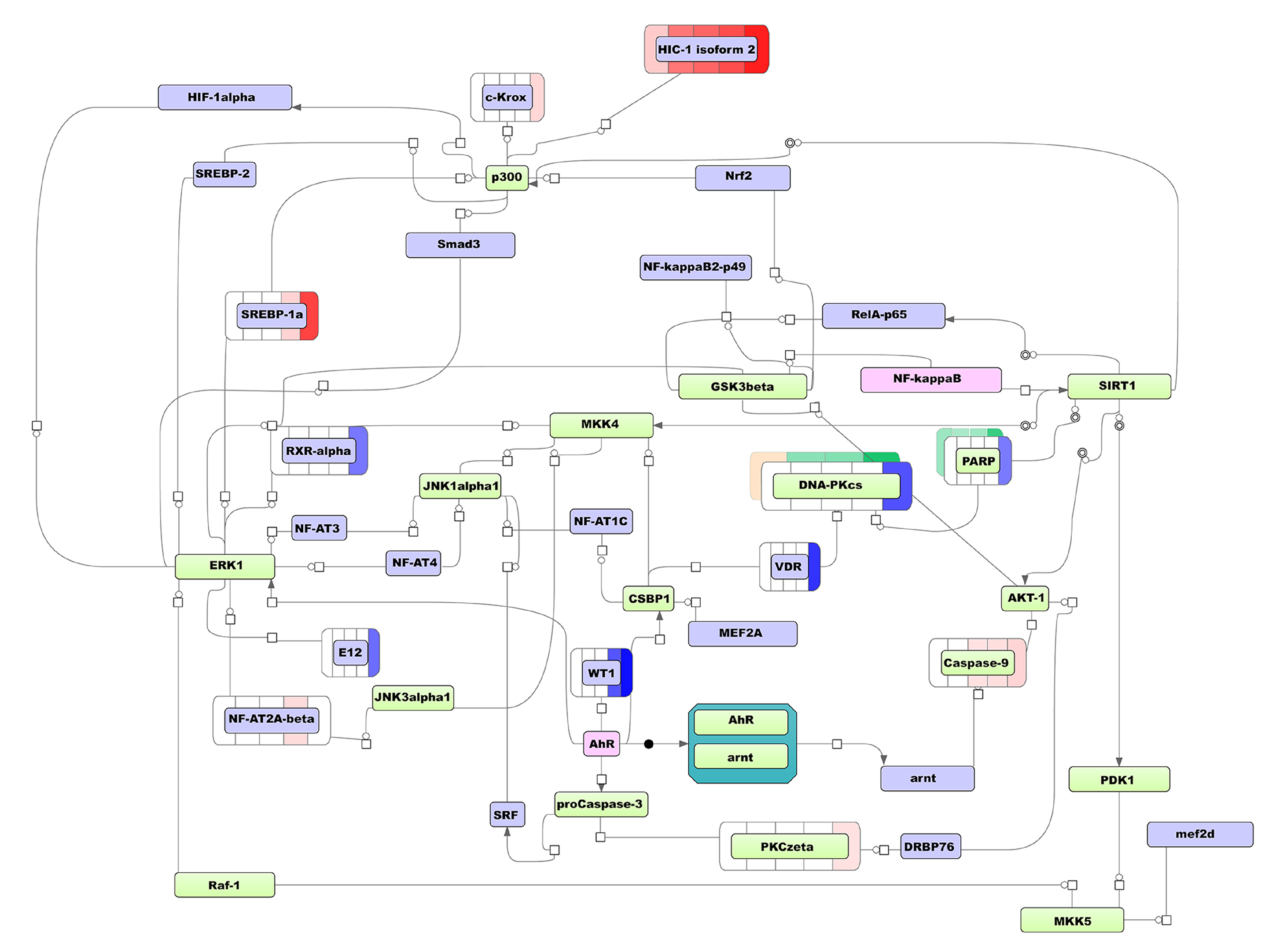
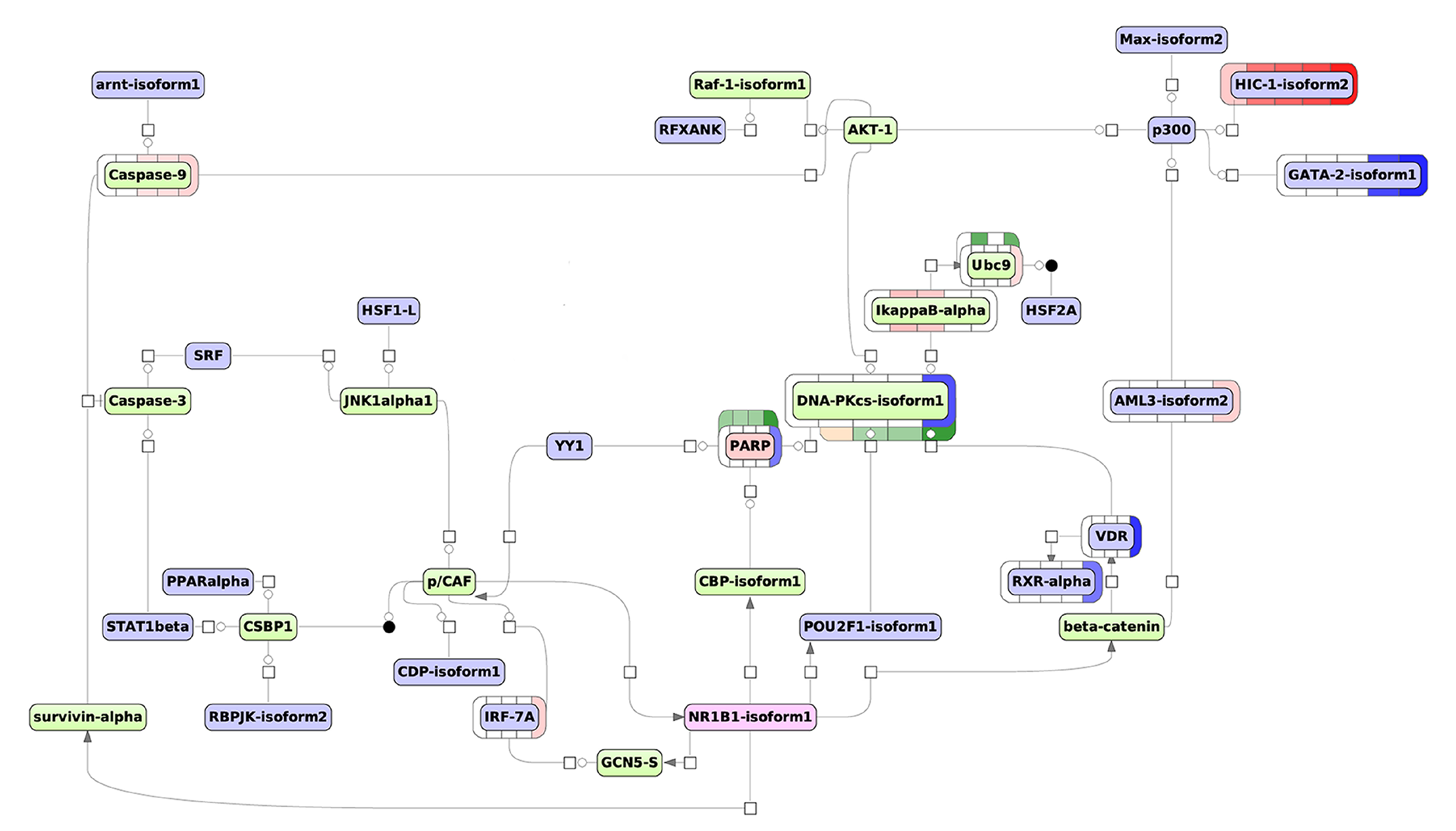
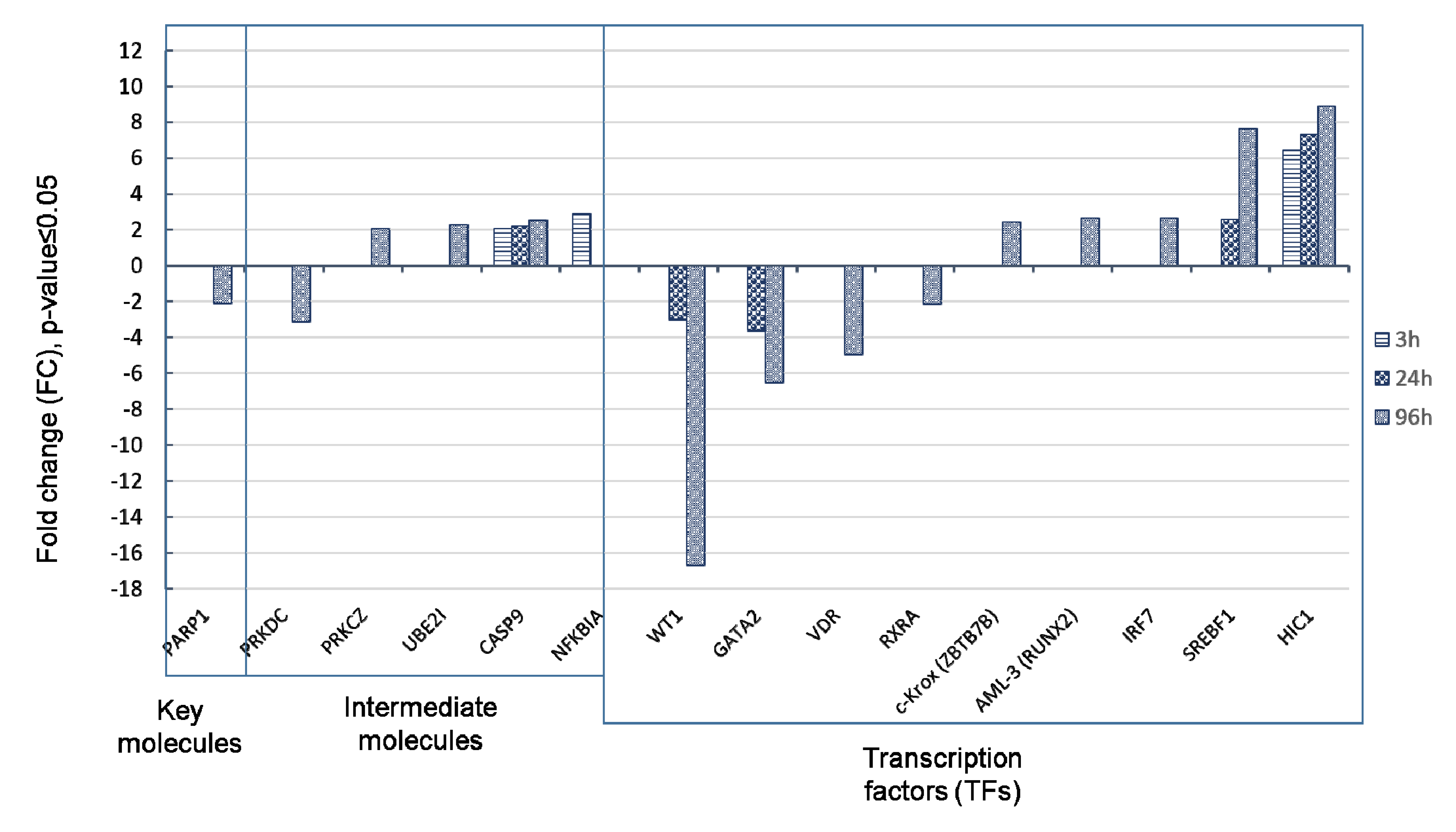
3.3.2. The Proteome-Based Modeling Pathway
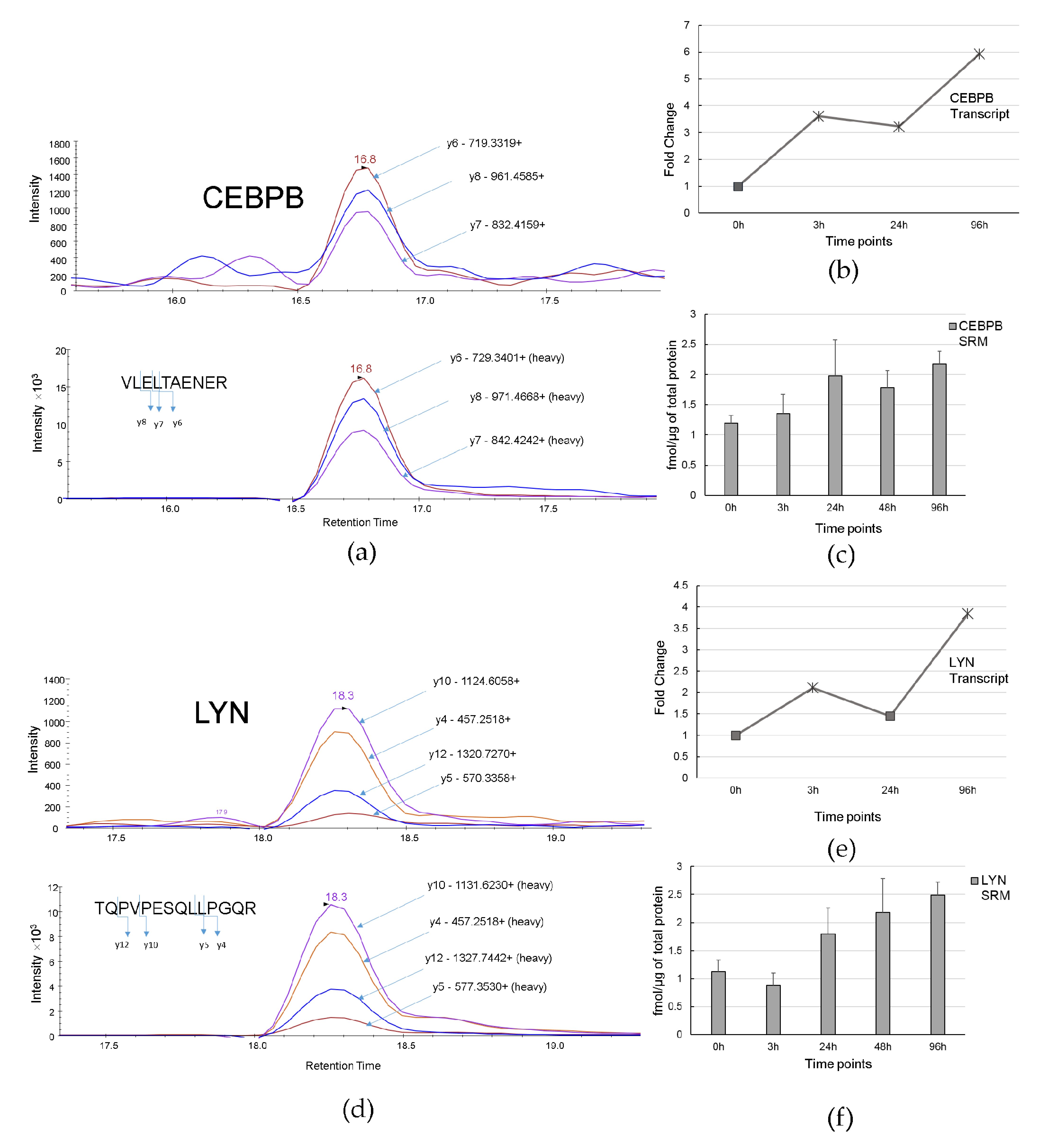
3.4. Verification of Protein Levels of HIC1, PARP1, CEBPB, and LYN During ATRA-Induced Differentiation by SRM Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kambhampati, S.; Verma, A.; Li, Y.; Parmar, S.; Sassano, A.; Platanias, L. Signalling Pathways Activated by All- trans -Retinoic Acid in Acute Promyelocytic Leukemia Cells. Leuk. Lymphoma 2004, 45, 2175–2185. [Google Scholar] [CrossRef]
- Jimenez, J.J.; Chale, R.S.; Abad, A.C.; Schally, A.V. Acute promyelocytic leukemia (APL): A review of the literature. Oncotarget 2020, 11, 992–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, P.Z.; Wang, K.K.; Zhang, Q.Y.; Huang, Q.H.; Du, Y.Z.; Zhang, Q.H.; Xiao, D.K.; Shen, S.H.; Imbeaud, S.; Eveno, E.; et al. Systems analysis of transcriptome and proteome in retinoic acid/arsenic trioxide-induced cell differentiation apoptosis of promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mengeling, B.J.; Phan, T.Q.; Goodson, M.L.; Privalsky, M.L. Aberrant corepressor interactions implicated in PML-RARα and PLZF-RARα leukemogenesis reflect an altered recruitment and release of specific NCoR and SMRT splice variants. J. Biol. Chem. 2011. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.J.; Sternsdorf, T.; Tini, M.; Evans, R.M. Transcriptional regulation in acute promyelocytic leukemia. Oncogene 2001, 20, 7204–7215. [Google Scholar] [CrossRef]
- Isakson, P.; Bjørås, M.; Bøe, S.O.; Simonsen, A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010. [Google Scholar] [CrossRef] [PubMed]
- Coombs, C.C.; Tavakkoli, M.; Tallman, M.S. Acute promyelocytic Leukemia: Where did we start, where are we now, and the future. Blood Cancer J. 2015, 5, e304. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Pregartner, G.; Rücker, F.G.; Heitzer, E.; Zebisch, A.; Bullinger, L.; Berghold, A.; Döhner, K.; Sill, H. Functional classification of tp53 mutations in acute myeloid leukemia. Cancers 2020, 12, 637. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.E.; Redner, R.L. An ATRActive future for differentiation therapy in AML. Blood Rev. 2015. [Google Scholar] [CrossRef] [Green Version]
- Dalton, W.T.; Ahearn, M.J.; McCredie, K.B.; Freireich, E.J.; Stass, S.A.; Trujillo, J.M. HL-60 cell line was derived from a patient with FAB-M2 and not FAB-M3. Blood 1988, 71, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Birnie, G.D. The HL60 cell line: A model system for studying human myeloid cell differentiation. Br. J. Cancer. Suppl. 1988, 9, 41–45. [Google Scholar] [PubMed]
- Wolf, D.; Rotter, V. Major deletions in the gene encoding the p53 tumor antigen cause lack of p53 expression in HL-60 cells. Proc. Natl. Acad. Sci. USA 1985. [Google Scholar] [CrossRef] [Green Version]
- Shima, H.; Nakayasu, M.; Aonuma, S.; Sugimura, T.; Nagao, M. Loss of the MYC gene amplified in human HL-60 cells after treatment with inhibitors of poly(ADP-ribose) polymerase or with dimethyl sulfoxide. Proc. Natl. Acad. Sci. USA 1989. [Google Scholar] [CrossRef] [Green Version]
- Tasseff, R.; Jensen, H.A.; Congleton, J.; Dai, D.; Rogers, K.V.; Sagar, A.; Bunaciu, R.P.; Yen, A.; Varner, J.D. An Effective Model of the Retinoic Acid Induced HL-60 Differentiation Program. Sci. Rep. 2017. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-H.; Chang, M.-Y.; Ahn, J.-I.; Yu, D.-H.; Jung, S.-S.; Choi, J.-H.; Noh, Y.-H.; Lee, Y.-S.; Ahn, M.-J. Differential gene expression in retinoic acid-induced differentiation of acute promyelocytic leukemia cells, NB4 and HL-60 cells. Biochem. Biophys. Res. Commun. 2002, 296, 1125–1133. [Google Scholar] [CrossRef]
- Mollinedo, F.; López-Pérez, R.; Gajate, C. Differential gene expression patterns coupled to commitment and acquisition of phenotypic hallmarks during neutrophil differentiation of human leukaemia HL-60 cells. Gene 2008, 419, 16–26. [Google Scholar] [CrossRef]
- Wang, J.; Fong, C.-C.; Tzang, C.-H.; Xiao, P.; Han, R.; Yang, M. Gene expression analysis of human promyelocytic leukemia HL-60 cell differentiation and cytotoxicity induced by natural and synthetic retinoids. Life Sci. 2009, 84, 576–583. [Google Scholar] [CrossRef]
- Harris, M.N.; Ozpolat, B.; Abdi, F.; Gu, S.; Legler, A.; Mawuenyega, K.G.; Tirado-Gomez, M.; Lopez-Berestein, G.; Chen, X. Comparative proteomic analysis of all-trans-retinoic acid treatment reveals systematic posttranscriptional control mechanisms in acute promyelocytic leukemia. Blood 2004, 104, 1314–1323. [Google Scholar] [CrossRef]
- Hofmann, A.; Gerrits, B.; Schmidt, A.; Bock, T.; Bausch-Fluck, D.; Aebersold, R.; Wollscheid, B. Proteomic cell surface phenotyping of differentiating acute myeloid leukemia cells. Blood 2010, 116, e26–e34. [Google Scholar] [CrossRef] [PubMed]
- Kel, A.; Voss, N.; Valeev, T.; Stegmaier, P.; Kel-Margoulis, O.; Wingender, E. ExPlain: Finding upstream drug targets in disease gene regulatory networks. SAR QSAR Environ. Res. 2008, 19, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Fleck, R.A.; Athwal, H.; Bygraves, J.A.; Hockley, D.J.; Feavers, I.M.; Stacey, G.N. Optimization of NB-4 and HL-60 differentiation for use in opsonophagocytosis assays. Vitr. Cell. Dev. Biol. Anim. 2003. [Google Scholar] [CrossRef]
- Zgoda, V.G.; Kopylov, A.T.; Tikhonova, O.V.; Moisa, A.A.; Pyndyk, N.V.; Farafonova, T.E.; Novikova, S.E.; Lisitsa, A.V.; Ponomarenko, E.A.; Poverennaya, E.V.; et al. Chromosome 18 transcriptome profiling and targeted proteome mapping in depleted plasma, liver tissue and HepG2 cells. J. Proteome Res. 2013, 12. [Google Scholar] [CrossRef] [PubMed]
- Kolker, E.; Higdon, R.; Morgan, P.; Sedensky, M.; Welch, D.; Bauman, A.; Stewart, E.; Haynes, W.; Broomall, W.; Kolker, N. SPIRE: Systematic protein investigative research environment. J. Proteomics 2011, 75, 122–126. [Google Scholar] [CrossRef]
- Goedhart, J.; Luijsterburg, M.S. VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci. Rep. 2020. [Google Scholar] [CrossRef]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. TRANSFAC(R) and its module TRANSCompel(R): Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, C.A.; Fuentes, G.; Patel, H.; Page, K.; Menakuru, M.; Park, J.H. Fast conventional Fmoc solid-phase peptide synthesis with HCTU. J. Pept. Sci. 2008, 14, 97–101. [Google Scholar] [CrossRef] [Green Version]
- Fekkes, D. State-of-the-art of high-performance liquid chromatographic analysis of amino acids in physiological samples. J. Chromatogr. B Biomed. Appl. 1996, 682, 3–22. [Google Scholar] [CrossRef]
- Congleton, J.; Jiang, H.; Malavasi, F.; Lin, H.; Yen, A. ATRA-induced HL-60 myeloid leukemia cell differentiation depends on the CD38 cytosolic tail needed for membrane localization, but CD38 enzymatic activity is unnecessary. Exp. Cell Res. 2011, 317, 910–919. [Google Scholar] [CrossRef] [Green Version]
- De The, H.; Marchio, A.; Tiollais, P.; Dejean, A. Differential expression and ligand regulation of the retinoic acid receptor alpha and beta genes. EMBO J. 1989, 8, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Pavri, R.; Lewis, B.; Kim, T.-K.; Dilworth, F.J.; Erdjument-Bromage, H.; Tempst, P.; de Murcia, G.; Evans, R.; Chambon, P.; Reinberg, D. PARP-1 determines specificity in a retinoid signaling pathway via direct modulation of mediator. Mol. Cell 2005, 18, 83–96. [Google Scholar] [CrossRef]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef]
- Hoffman, B.; Liebermann, D.A. Apoptotic signaling by c-MYC. Oncogene 2008, 27, 6462–6472. [Google Scholar] [CrossRef] [Green Version]
- Collins, S.J. The HL-60 promyelocytic leukemia cell line: Proliferation, differentiation, and cellular oncogene expression. Blood 1987, 70, 1233–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Baruch, N.; Reinhold, W.C.; Alexandrova, N.; Ichinose, I.; Blake, M.; Trepel, J.B.; Zajac-Kaye, M. c-myc Down-regulation in suramin-treated HL60 cells precedes growth inhibition but does not trigger differentiation. Mol. Pharmacol. 1994, 46, 73–78. [Google Scholar]
- Amati, B.; Land, H. Myc-Max-Mad: A transcription factor network controlling cell cycle progression, differentiation and death. Curr. Opin. Genet. Dev. 1994. [Google Scholar] [CrossRef]
- Williams, A.B.; Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.H.; Na, H.; Kim, E.J.; Lee, H.W.; Lee, M.O. Poly(ADP-ribosyl)ation of p53 induces gene-specific transcriptional repression of MTA1. Oncogene 2012, 31, 5099–5107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beneke, R.; Geisen, C.; Zevnik, B.; Bauch, T.; Müller, W.U.; Küpper, J.H.; Möröy, T. DNA excision repair and DNA damage-induced apoptosis are linked to Poly(ADP-ribosyl)ation but have different requirements for p53. Mol. Cell. Biol. 2000, 20, 6695–6703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.-M.; Chen, W.; Wang, J. Distinguishing between cancer cell differentiation and resistance induced by all-trans retinoic acid using transcriptional profiles and functional pathway analysis. Sci. Rep. 2014, 4, 5577. [Google Scholar] [CrossRef] [Green Version]
- Ruscetti, T.; Lehnert, B.E.; Halbrook, J.; Le Trong, H.; Hoekstra, M.F.; Chen, D.J.; Peterson, S.R. Stimulation of the DNA-dependent protein kinase by poly(ADP-ribose) polymerase. J. Biol. Chem. 1998, 273, 14461–14467. [Google Scholar] [CrossRef] [Green Version]
- Ting, H.J.; Yasmin-Karim, S.; Yan, S.J.; Hsu, J.W.; Lin, T.H.; Zeng, W.; Messing, J.; Sheu, T.J.; Bao, B.Y.; Li, W.X.; et al. A positive feedback signaling loop between ATM and the vitamin D receptor is critical for cancer chemoprevention by vitamin D. Cancer Res. 2012, 72, 958–968. [Google Scholar] [CrossRef] [Green Version]
- Turk, A.; Wisinski, K.B. PARP Inhibition in BRCA-Mutant Breast Cancer. Cancer 2018, 124, 2498–2506. [Google Scholar] [CrossRef]
- Faraoni, I.; Compagnone, M.; Lavorgna, S.; Angelini, D.F.; Cencioni, M.T.; Piras, E.; Panetta, P.; Ottone, T.; Dolci, S.; Venditti, A.; et al. BRCA1, PARP1 and γH2AX in acute myeloid leukemia: Role as biomarkers of response to the PARP inhibitor olaparib. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Sauve, A.A.; Bai, P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Aspects Med. 2013, 34, 1168–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, A.; Ueda, T.; Nakamura, T. The mechanism of apoptosis induced by anticancer agents in human leukemia HL-60 cells. Rinsho. Ketsueki. 1996, 37, 552–557. [Google Scholar]
- Dos Santos, C.; Demur, C.; Bardet, V.; Prade-Houdellier, N.; Payrastre, B.; Récher, C. A critical role for Lyn in acute myeloid leukemia. Blood 2008, 111, 2269–2279. [Google Scholar] [CrossRef]
- Iriyama, N.; Yuan, B.; Hatta, Y.; Takagi, N.; Takei, M. Lyn, a tyrosine kinase closely linked to the differentiation status of primary acute myeloid leukemia blasts, associates with negative regulation of all-trans retinoic acid (ATRA) and dihydroxyvitamin D3 (VD3)-induced HL-60 cells differentiation. Cancer Cell Int. 2016, 16, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Errico, D.; Yamashita, Y.; Suzuki, R.; Odom, S.; Furumoto, Y.; Yamashita, T.; Rivera, J. Functional analysis of Lyn kinase A and B isoforms reveals redundant and distinct roles in Fc epsilon RI-dependent mast cell activation. J. Immunol. 2010, 184, 5000–5008. [Google Scholar] [CrossRef]
- Tornillo, G.; Knowlson, C.; Kendrick, H.; Buckley, N.E.; Grigoriadis, A.; Smalley Correspondence, M.J. Dual Mechanisms of LYN Kinase Dysregulation Drive Aggressive Behavior in Breast Cancer Cells. CellReports 2018, 25, 3674–3692.e10. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time Point. | Key Molecule Name | Reached from TF Set 1 | Reachable Total 2 | Score 3 | FDR 4 | Z-Score 5 | Ranks Sum 6 |
|---|---|---|---|---|---|---|---|
| 3-24-96 h (MCD) | AhR | 22 | 12488 | 0.34 | 0.011 | 2.31 | 5 |
| arnt | 21 | 8990 | 0.34 | 0.026 | 2.1 | 6 | |
| Nrf2 | 15 | 9200 | 0.24 | 0.033 | 2.61 | 6 | |
| (CKII-α)2:(CKII-β)2 | 22 | 10803 | 0.28 | 0.025 | 2.45 | 8 | |
| NF-kappaB1 | 21 | 10897 | 0.29 | 0.024 | 2.05 | 11 |
| Time Point | Key Molecule Name | Reached from TF Set 1 | Reachable Total 2 | Score 3 | FDR 4 | Z-Score 5 | Ranks Sum 6 |
|---|---|---|---|---|---|---|---|
| Combined 3-24-48-96 h | YY1 | 22 | 32835 | 0.650 | 0.013 | 2.779 | 59 |
| plk1{p} | 22 | 32762 | 0.640 | 0.002 | 2.803 | 63 | |
| PARP1 | 22 | 32360 | 0.607 | 0.006 | 2.950 | 63 | |
| faim | 22 | 32361 | 0.607 | 0.006 | 2.950 | 64 | |
| MKK6 | 22 | 33716 | 0.721 | 0.004 | 2.325 | 88 | |
| NR1B1 (RARA) | 22 | 30223 | 0.505 | 0.018 | 3.007 | 204 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novikova, S.; Tikhonova, O.; Kurbatov, L.; Farafonova, T.; Vakhrushev, I.; Lupatov, A.; Yarygin, K.; Zgoda, V. Omics Technologies to Decipher Regulatory Networks in Granulocytic Cell Differentiation. Biomolecules 2021, 11, 907. https://doi.org/10.3390/biom11060907
Novikova S, Tikhonova O, Kurbatov L, Farafonova T, Vakhrushev I, Lupatov A, Yarygin K, Zgoda V. Omics Technologies to Decipher Regulatory Networks in Granulocytic Cell Differentiation. Biomolecules. 2021; 11(6):907. https://doi.org/10.3390/biom11060907
Chicago/Turabian StyleNovikova, Svetlana, Olga Tikhonova, Leonid Kurbatov, Tatiana Farafonova, Igor Vakhrushev, Alexey Lupatov, Konstantin Yarygin, and Victor Zgoda. 2021. "Omics Technologies to Decipher Regulatory Networks in Granulocytic Cell Differentiation" Biomolecules 11, no. 6: 907. https://doi.org/10.3390/biom11060907
APA StyleNovikova, S., Tikhonova, O., Kurbatov, L., Farafonova, T., Vakhrushev, I., Lupatov, A., Yarygin, K., & Zgoda, V. (2021). Omics Technologies to Decipher Regulatory Networks in Granulocytic Cell Differentiation. Biomolecules, 11(6), 907. https://doi.org/10.3390/biom11060907