
Role of Purinergic Signalling in Endothelial Dysfunction and Thrombo-Inflammation in Ischaemic Stroke and Cerebral Small Vessel Disease


 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Endothelial Dysfunction
Emerging Role of Erythrocytes in Endothelial Dysfunction
3. Thrombo-Inflammation in Atherothrombotic Stroke
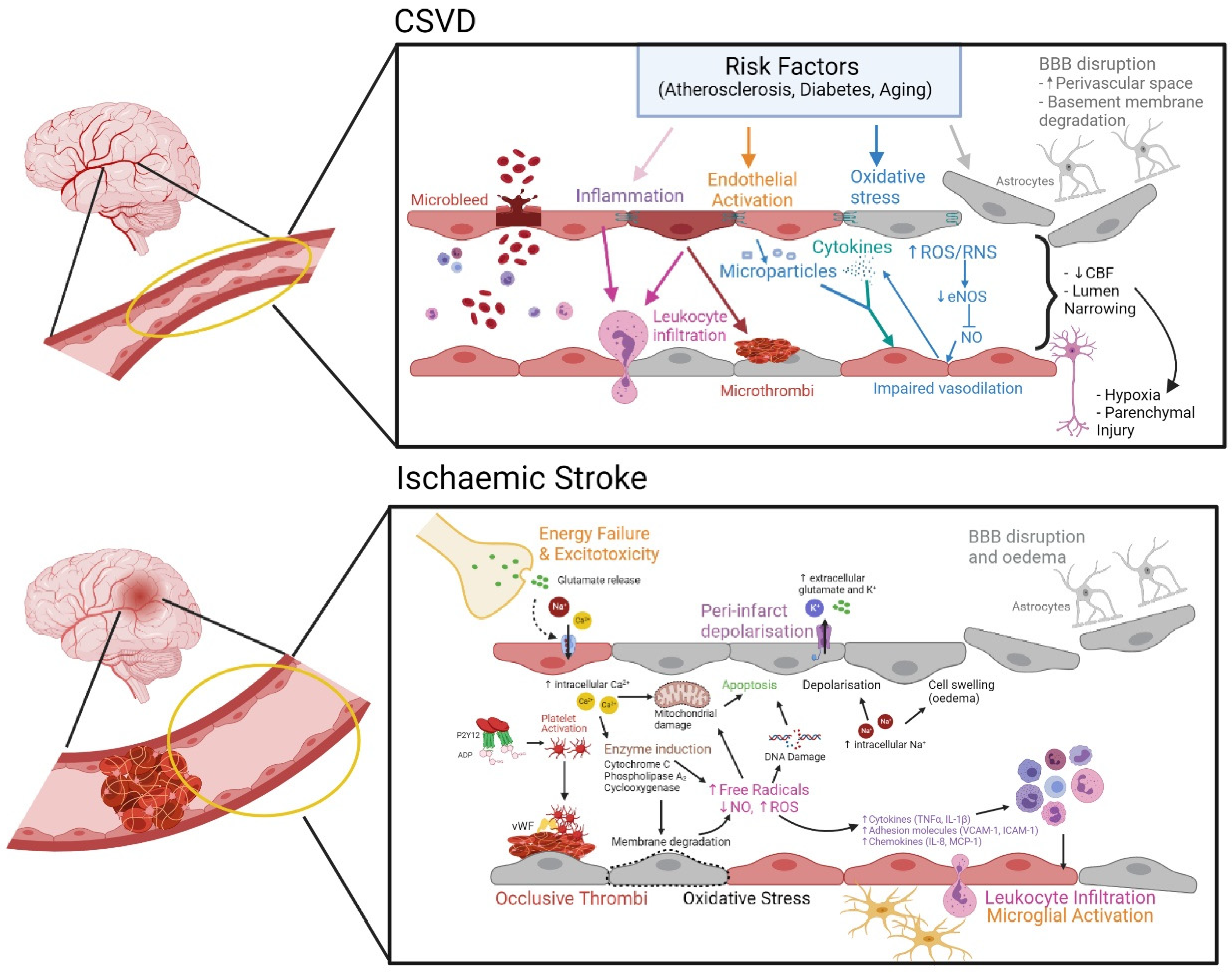
3.1. Cerebral Ischemia Leading to Stroke
Mechanism of Endothelial Dysfunction in Ischemic Stroke
3.2. Cerebral Small Vessel Disease (CSVD)
Evolving Pathomechanism of CSVD
4. Involvements of Purinergic Signaling
4.1. Purinergic Signaling
4.2. Role of Endothelial CD39 in Preventing Thrombo-Inflammation
4.3. Involvement of Adenosine as Potential Innate Neuroprotection
5. Therapeutic Potential of CD39
6. Targeted CD39 Therapeutics for Ischemic Stroke
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Bassenge, E. Endothelial function in different organs. Prog. Cardiovasc. Dis. 1996, 39, 209–228. [Google Scholar] [CrossRef]
- Yano, K.; Gale, D.; Massberg, S.; Cheruvu, P.K.; Monahan-Earley, R.; Morgan, E.S.; Haig, D.; Von Andrian, U.H.; Dvorak, A.M.; Aird, W.C. Phenotypic heterogeneity is an evolutionarily conserved feature of the endothelium. Blood 2007, 109, 613–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumbria, R.; Fisher, M. Chapter 8—Endothelium. In Primer on Cerebrovascular Diseases, 2nd ed.; Caplan, L.R., Biller, J., Leary, M.C., Lo, E.H., Thomas, A.J., Yenari, M., Zhang, J.H., Eds.; Academic Press: San Diego, CA, USA, 2017; pp. 47–51. [Google Scholar]
- Sandoval, K.E.; Witt, K.A. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 2008, 32, 200–219. [Google Scholar] [CrossRef]
- Taddei, S.; Ghiadoni, L.; Virdis, A.; Versari, D.; Salvetti, A. Mechanisms of endothelial dysfunction: Clinical significance and preventive non-pharmacological therapeutic strategies. Curr. Pharm. Des. 2003, 9, 2385–2402. [Google Scholar] [CrossRef]
- Chien, S. Determinants of blood viscosity and red cell deformability. Scand. J. Clin. Lab. Investig. 1981, 41, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Lazari, D.; Leal, J.K.F.; Brock, R.; Bosman, G. The Relationship Between Aggregation and Deformability of Red Blood Cells in Health and Disease. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef]
- Chien, S.; Usami, S.; Dellenback, R.J.; Gregersen, M.I.; Nanninga, L.B.; Guest, M.M. Blood viscosity: Influence of erythrocyte aggregation. Science 1967, 157, 829–831. [Google Scholar] [CrossRef]
- Baskurt, O.K.; Yalcin, O.; Ozdem, S.; Armstrong, J.K.; Meiselman, H.J. Modulation of endothelial nitric oxide synthase expression by red blood cell aggregation. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H222–H229. [Google Scholar] [CrossRef] [Green Version]
- Mannini, L.; Cecchi, E.; Fatini, C.; Marcucci, R.; Liotta, A.A.; Matucci-Cerinic, M.; Abbate, R.; Gensini, G.F. Clinical haemorheology and microcirculation. Ann. Ist. Super. Sanita 2007, 43, 144. [Google Scholar]
- Shevkoplyas, S.S.; Yoshida, T.; Gifford, S.C.; Bitensky, M.W. Direct measurement of the impact of impaired erythrocyte deformability on microvascular network perfusion in a microfluidic device. Lab Chip 2006, 6, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Watts, T.; Barigou, M.; Nash, G.B. Comparative rheology of the adhesion of platelets and leukocytes from flowing blood: Why are platelets so small? Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1483–H1494. [Google Scholar] [CrossRef] [PubMed]
- Cowan, A.Q.; Cho, D.J.; Rosenson, R.S. Importance of blood rheology in the pathophysiology of atherothrombosis. Cardiovasc. Drugs Ther. 2012, 26, 339–348. [Google Scholar] [CrossRef]
- Kensey, K.R. The mechanistic relationships between hemorheological characteristics and cardiovascular disease. Curr. Med. Res. Opin. 2003, 19, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Malek, A.M.; Alper, S.L.; Izumo, S. Hemodynamic shear stress and its role in atherosclerosis. Jama 1999, 282, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Forconi, S.; Gori, T. Endothelium and hemorheology. Clin. Hemorheol. Microcirc. 2013, 53, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Sumino, H.; Aoki, T.; Tsunekawa, K.; Araki, O.; Kimura, T.; Nara, M.; Ogiwara, T.; Murakami, M. Impaired blood rheology is associated with endothelial dysfunction in patients with coronary risk factors. Clin. Hemorheol. Microcirc. 2016, 62, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloner, R.A. No-reflow phenomenon: Maintaining vascular integrity. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.J.; Popel, A.S.; Intaglietta, M.; Johnson, P.C. Effects of erythrocyte aggregation and venous network geometry on red blood cell axial migration. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H939–H950. [Google Scholar] [CrossRef]
- Fahraeus, R. The influence of the rouleau formation of the erythrocytes on the rheology of the blood. Acta Med. Scand. 1958, 161, 151–165. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Schulz, R.; Rassaf, T.; Lauer, T.; Dejam, A.; Jax, T.; Kumara, I.; Gharini, P.; Kabanova, S.; Ozüyaman, B. Red blood cells express a functional endothelial nitric oxide synthase. Blood 2006, 107, 2943–2951. [Google Scholar] [CrossRef] [Green Version]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III-27–III-32. [Google Scholar] [CrossRef] [Green Version]
- Lyons, C.R. The role of nitric oxide in inflammation. Adv. Immunol. 1995, 60, 323–371. [Google Scholar]
- Hathcock, J.J. Flow effects on coagulation and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1729–1737. [Google Scholar] [CrossRef]
- Peyrou, V.; Lormeau, J.; Herault, J.; Gaich, C.; Pfliegger, A.; Herbert, J. Contribution of erythrocytes to thrombin generation in whole blood. Thromb. Haemost. 1999, 81, 400–406. [Google Scholar] [CrossRef]
- Whelihan, M.F.; Mann, K.G. The role of the red cell membrane in thrombin generation. Thromb. Res. 2013, 131, 377–382. [Google Scholar] [CrossRef]
- Horne III, M.K.; Cullinane, A.M.; Merryman, P.K.; Hoddeson, E.K. The effect of red blood cells on thrombin generation. Br. J. Haematol. 2006, 133, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.T.; Armstrong, J.K.; Tripette, J.; Meiselman, H.J.; Cloutier, G. A local increase in red blood cell aggregation can trigger deep vein thrombosis: Evidence based on quantitative cellular ultrasound imaging. J. Thromb. Haemost. 2011, 9, 481–488. [Google Scholar] [CrossRef]
- Wautier, J.-L.; Wautier, M.-P. Molecular basis of erythrocyte adhesion to endothelial cells in diseases. Clin. Hemorheol. Microcirc. 2013, 53, 11–21. [Google Scholar] [CrossRef]
- Popel, A.S.; Johnson, P.C. Microcirculation and Hemorheology. Annu. Rev. Fluid Mech. 2005, 37, 43–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becatti, M.; Marcucci, R.; Gori, A.; Mannini, L.; Grifoni, E.; Alessandrello Liotta, A.; Sodi, A.; Tartaro, R.; Taddei, N.; Rizzo, S. Erythrocyte oxidative stress is associated with cell deformability in patients with retinal vein occlusion. J. Thromb. Haemost. 2016, 14, 2287–2297. [Google Scholar] [CrossRef] [Green Version]
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef]
- Hu, X.; De Silva, T.M.; Chen, J.; Faraci, F.M. Cerebral vascular disease and neurovascular injury in ischemic stroke. Circ. Res. 2017, 120, 449–471. [Google Scholar] [CrossRef] [Green Version]
- Enache, S.; Zaharia, B.; Georgescu, C.V.; Ţenovici, M. Study of cerebral vascular structures in hypertensive intracerebral haemorrhage. Rom. J. Morphol. Embryol. 2005, 46, 249–256. [Google Scholar]
- Fisher, C. The arterial lesions underlying lacunes. Acta Neuropathol. 1969, 12, 1–15. [Google Scholar] [CrossRef]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Dirnagl, U. Pathobiology of injury after stroke: The neurovascular unit and beyond. Ann. N. Y. Acad. Sci. 2012, 1268, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Jones, D.; Adams, R.; Carnethon, M.; De Simone, G.; Ferguson, T.B.; Flegal, K.; Ford, E.; Furie, K.; Go, A.; Greenlund, K. Heart disease and stroke statistics—2009 update a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2009, 119, e21–e181. [Google Scholar] [PubMed] [Green Version]
- Durukan, A.; Tatlisumak, T. Acute ischemic stroke: Overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol. Biochem. Behav. 2007, 87, 179–197. [Google Scholar] [CrossRef]
- Nakagomi, T.; Kassell, N.; Sasaki, T.; Fujiwara, S.; Lehman, R.; Torner, J. Impairment of endothelium-dependent vasodilation induced by acetylcholine and adenosine triphosphate following experimental subarachnoid hemorrhage. Stroke 1987, 18, 482–489. [Google Scholar] [CrossRef]
- Srivastava, K.; Bath, P.M.; Bayraktutan, U. Current therapeutic strategies to mitigate the eNOS dysfunction in ischaemic stroke. Cell. Mol. Neurobiol. 2012, 32, 319–336. [Google Scholar] [CrossRef]
- Ivacko, J.; Szaflarski, J.; Malinak, C.; Flory, C.; Warren, J.S.; Silverstein, F.S. Hypoxic-ischemic injury induces monocyte chemoattractant protein-1 expression in neonatal rat brain. J. Cereb. Blood Flow Metab. 1997, 17, 759–770. [Google Scholar] [CrossRef]
- Yamasaki, Y.; Matsuura, N.; Shozuhara, H.; Onodera, H.; Itoyama, Y.; Kogure, K. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke 1995, 26, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Del Zoppo, G.J.; Hallenbeck, J.M. Advances in the vascular pathophysiology of ischemic stroke. Thromb. Res. 2000, 98, 73–81. [Google Scholar] [CrossRef]
- El Amki, M.; Glück, C.; Binder, N.; Middleham, W.; Wyss, M.T.; Weiss, T.; Meister, H.; Luft, A.; Weller, M.; Weber, B. Neutrophils Obstructing Brain Capillaries Are a Major Cause of No-Reflow in Ischemic Stroke. Cell Rep. 2020, 33, 108260. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Förstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.-L.; Chopp, M.; Chen, H.; Garcia, J.H. Temporal profile of ischemic tissue damage, neutrophil response, and vascular plugging following permanent and transient (2H) middle cerebral artery occlusion in the rat. J. Neurol. Sci. 1994, 125, 3–10. [Google Scholar] [CrossRef]
- Seal, J.B.; Gewertz, B.L. Vascular dysfunction in ischemia-reperfusion injury. Ann. Vasc. Surg. 2005, 19, 572–584. [Google Scholar] [CrossRef]
- Smith, E.E. Clinical presentations and epidemiology of vascular dementia. Clin. Sci. 2017, 131, 1059–1068. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Smith, E.E.; Biessels, G.J.; Cordonnier, C.; Fazekas, F.; Frayne, R.; Lindley, R.I.; T O’Brien, J.; Barkhof, F.; Benavente, O.R. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013, 12, 822–838. [Google Scholar] [CrossRef] [Green Version]
- Lampe, L.; Kharabian-Masouleh, S.; Kynast, J.; Arelin, K.; Steele, C.J.; Löffler, M.; Witte, A.V.; Schroeter, M.L.; Villringer, A.; Bazin, P.-L. Lesion location matters: The relationships between white matter hyperintensities on cognition in the healthy elderly. J. Cereb. Blood Flow Metab. 2019, 39, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Pantoni, L. Cerebral small vessel disease: From pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010, 9, 689–701. [Google Scholar] [CrossRef]
- Caunca, M.R.; Leon-Benedetti, D.; Latour, L.; Leigh, R.; Wright, C.B. Neuroimaging of cerebral small vessel disease and age-related cognitive changes. Front. Aging Neurosci. 2019, 11, 145. [Google Scholar] [CrossRef] [PubMed]
- Akoudad, S.; Portegies, M.L.; Koudstaal, P.J.; Hofman, A.; Van der Lugt, A.; Ikram, M.A.; Vernooij, M.W. Cerebral microbleeds are associated with an increased risk of stroke: The Rotterdam Study. Circulation 2015, 132, 509–516. [Google Scholar] [CrossRef]
- Xiong, Y.; Mok, V. Age-related white matter changes. J. Aging Res. 2011, 2011, 617927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Yang, Y.; Reis, C.; Tao, T.; Li, W.; Li, X.; Zhang, J.H. Cerebral small vessel disease. Cell Transplant. 2018, 27, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Bours, M.; Swennen, E.; Di Virgilio, F.; Cronstein, B.; Dagnelie, P. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 2006, 112, 358–404. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yao, Y.; Sumi, Y.; Li, A.; To, U.K.; Elkhal, A.; Inoue, Y.; Woehrle, T.; Zhang, Q.; Hauser, C. Purinergic signaling: A fundamental mechanism in neutrophil activation. Sci. Signal. 2010, 3. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Kukulski, F.; Lévesque, S.A.; Sévigny, J. Impact of ectoenzymes on p2 and p1 receptor signaling. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 2011; Volume 61, pp. 263–299. [Google Scholar]
- Zhao, H.; Bo, C.; Kang, Y.; Li, H. What else can CD39 tell us? Front. Immunol. 2017, 8, 727. [Google Scholar] [CrossRef] [Green Version]
- Hechler, B.; Gachet, C. Purinergic receptors in thrombosis and inflammation. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2307–2315. [Google Scholar] [CrossRef] [Green Version]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Yegutkin, G.G. Nucleotide-and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2008, 1783, 673–694. [Google Scholar] [CrossRef] [Green Version]
- Kogure, K.; Alonso, O.F. A pictorial representation of endogenous brain ATP by a bioluminescent method. Brain Res. 1978, 154, 273–284. [Google Scholar] [CrossRef]
- Pedata, F.; Dettori, I.; Coppi, E.; Melani, A.; Fusco, I.; Corradetti, R.; Pugliese, A.M. Purinergic signalling in brain ischemia. Neuropharmacology 2016, 104, 105–130. [Google Scholar] [CrossRef]
- Imura, Y.; Morizawa, Y.; Komatsu, R.; Shibata, K.; Shinozaki, Y.; Kasai, H.; Moriishi, K.; Moriyama, Y.; Koizumi, S. Microglia release ATP by exocytosis. Glia 2013, 61, 1320–1330. [Google Scholar] [CrossRef]
- Hamilton, N.B.; Attwell, D. Do astrocytes really exocytose neurotransmitters? Nat. Rev. Neurosci. 2010, 11, 227–238. [Google Scholar] [CrossRef]
- Melani, A.; Corti, F.; Stephan, H.; Müller, C.E.; Donati, C.; Bruni, P.; Vannucchi, M.G.; Pedata, F. Ecto-ATPase inhibition: ATP and adenosine release under physiological and ischemic in vivo conditions in the rat striatum. Exp. Neurol. 2012, 233, 193–204. [Google Scholar] [CrossRef]
- Cisneros-Mejorado, A.; Pérez-Samartín, A.; Gottlieb, M.; Matute, C. ATP signaling in brain: Release, excitotoxicity and potential therapeutic targets. Cell. Mol. Neurobiol. 2015, 35, 1–6. [Google Scholar] [CrossRef]
- Rozalski, M.; Nocun, M.; Watala, C. Adenosine diphosphate receptors on blood platelets: Potential new targets for antiplatelet therapy. Acta Biochim. Pol. 2005, 52, 411–415. [Google Scholar] [PubMed]
- Linden, J. Molecular approach to adenosine receptors: Receptor-mediated mechanisms of tissue protection. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Németh, Z.H.; Leibovich, S.J.; Deitch, E.A.; Sperlágh, B.; Virág, L.; Vizi, E.S.; Szabó, C.; Haskó, G. Adenosine stimulates CREB activation in macrophages via a p38 MAPK-mediated mechanism. Biochem. Biophys. Res. Commun. 2003, 312, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef] [PubMed]
- Haskó, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef]
- Grenz, A.; Homann, D.; Eltzschig, H.K. Extracellular adenosine: A safety signal that dampens hypoxia-induced inflammation during ischemia. Antioxid. Redox Signal. 2011, 15, 2221–2234. [Google Scholar] [CrossRef] [Green Version]
- Eckle, T.; Krahn, T.; Grenz, A.; Kohler, D.; Mittelbronn, M.; Ledent, C.; Jacobson, M.A.; Osswald, H.; Thompson, L.F.; Unertl, K. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation 2007, 115, 1581. [Google Scholar] [CrossRef] [Green Version]
- Grenz, A.; Zhang, H.; Eckle, T.; Mittelbronn, M.; Wehrmann, M.; Köhle, C.; Kloor, D.; Thompson, L.F.; Osswald, H.; Eltzschig, H.K. Protective role of ecto-5′-nucleotidase (CD73) in renal ischemia. J. Am. Soc. Nephrol. 2007, 18, 833–845. [Google Scholar] [CrossRef] [Green Version]
- Grenz, A.; Zhang, H.; Hermes, M.; Eckle, T.; Klingel, K.; Huang, D.Y.; Müller, C.E.; Robson, S.C.; Osswald, H.; Eltzschig, H.K. Contribution of E-NTPDase1 (CD39) to renal protection from ischemia-reperfusion injury. FASEB J. 2007, 21, 2863–2873. [Google Scholar] [CrossRef]
- Hart, M.L.; Gorzolla, I.C.; Schittenhelm, J.; Robson, S.C.; Eltzschig, H.K. SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J. Immunol. 2010, 184, 4017–4024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, M.L.; Henn, M.; Kohler, D.; Kloor, D.; Mittelbronn, M.; Gorzolla, I.C.; Stahl, G.L.; Eltzschig, H.K. Role of extracellular nucleotide phosphohydrolysis in intestinal ischemia-reperfusion injury. FASEB J. 2008, 22, 2784–2797. [Google Scholar] [CrossRef] [Green Version]
- Hart, M.L.; Much, C.; Gorzolla, I.C.; Schittenhelm, J.; Kloor, D.; Stahl, G.L.; Eltzschig, H.K. Extracellular adenosine production by ecto-5′-nucleotidase protects during murine hepatic ischemic preconditioning. Gastroenterology 2008, 135, 1739–1750.e3. [Google Scholar] [CrossRef] [PubMed]
- Köhler, D.; Eckle, T.; Faigle, M.; Grenz, A.; Mittelbronn, M.; Laucher, S.; Hart, M.L.; Robson, S.C.; Müller, C.E.; Eltzschig, H.K. CD39/ectonucleoside triphosphate diphosphohydrolase 1 provides myocardial protection during cardiac ischemia/reperfusion injury. Circulation 2007, 116, 1784–1794. [Google Scholar] [CrossRef] [Green Version]
- Koshiba, M.; Rosin, D.L.; Hayashi, N.; Linden, J.; Sitkovsky, M.V. Patterns of A2A extracellular adenosine receptor expression in different functional subsets of human peripheral T cells. Flow cytometry studies with anti-A2A receptor monoclonal antibodies. Mol. Pharmacol. 1999, 55, 614–624. [Google Scholar] [PubMed]
- Takahashi-Sato, K.; Murakawa, M.; Kimura, J.; Ito, M.-a.; Matsuoka, I. Loss of ectonucleotidases from the coronary vascular bed after ischemia-reperfusion in isolated rat heart. BMC Cardiovasc. Disord. 2013, 13, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helenius, M.H.; Vattulainen, S.; Orcholski, M.; Aho, J.; Komulainen, A.; Taimen, P.; Wang, L.; De Jesus Perez, V.A.; Koskenvuo, J.W.; Alastalo, T.-P. Suppression of endothelial CD39/ENTPD1 is associated with pulmonary vascular remodeling in pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1046–L1057. [Google Scholar] [CrossRef] [PubMed]
- Beldi, G.; Wu, Y.; Sun, X.; Imai, M.; Enjyoji, K.; Csizmadia, E.; Candinas, D.; Erb, L.; Robson, S.C. Regulated catalysis of extracellular nucleotides by vascular CD39/ENTPD1 is required for liver regeneration. Gastroenterology 2008, 135, 1751–1760. [Google Scholar] [CrossRef] [Green Version]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic signaling during inflammation. N. Engl. J. Med. 2012, 367, 2322–2333. [Google Scholar] [CrossRef] [Green Version]
- Friedman, D.J.; Rennke, H.G.; Csizmadia, E.; Enjyoji, K.; Robson, S.C. The vascular ectonucleotidase ENTPD1 is a novel renoprotective factor in diabetic nephropathy. Diabetes 2007, 56, 2371–2379. [Google Scholar] [CrossRef] [Green Version]
- Enjyoji, K.; Sévigny, J.; Lin, Y.; Frenette, P.S.; Christie, P.D.; Am Esch II, J.S.; Imai, M.; Edelberg, J.M.; Rayburn, H.; Lech, M. Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat. Med. 1999, 5, 1010. [Google Scholar] [CrossRef]
- Banz, Y.; Beldi, G.; Wu, Y.; Atkinson, B.; Usheva, A.; Robson, S.C. CD39 is incorporated into plasma microparticles where it maintains functional properties and impacts endothelial activation. Br. J. Haematol. 2008, 142, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Huttinger, Z.M.; Milks, M.W.; Nickoli, M.S.; Aurand, W.L.; Long, L.C.; Wheeler, D.G.; Dwyer, K.M.; d’Apice, A.J.; Robson, S.C.; Cowan, P.J. Ectonucleotide triphosphate diphosphohydrolase-1 (CD39) mediates resistance to occlusive arterial thrombus formation after vascular injury in mice. Am. J. Pathol. 2012, 181, 322–333. [Google Scholar] [CrossRef] [Green Version]
- Covarrubias, R.; Chepurko, E.; Reynolds, A.; Huttinger, Z.M.; Huttinger, R.; Stanfill, K.; Wheeler, D.G.; Novitskaya, T.; Robson, S.C.; Dwyer, K.M. Role of the CD39/CD73 purinergic pathway in modulating arterial thrombosis in mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1809–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwyer, K.M.; Robson, S.C.; Nandurkar, H.H.; Campbell, D.J.; Gock, H.; Murray-Segal, L.J.; Fisicaro, N.; Mysore, T.B.; Kaczmarek, E.; Cowan, P.J. Thromboregulatory manifestations in human CD39 transgenic mice and the implications for thrombotic disease and transplantation. J. Clin. Investig. 2004, 113, 1440–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, B.; Dwyer, K.; Enjyoji, K.; Robson, S.C. Ecto-nucleotidases of the CD39/NTPDase family modulate platelet activation and thrombus formation: Potential as therapeutic targets. Blood Cells Mol. Dis. 2006, 36, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Kanthi, Y.M.; Sutton, N.R.; Pinsky, D.J. CD39: Interface between vascular thrombosis and inflammation. Curr. Atheroscler. Rep. 2014, 16, 425. [Google Scholar] [CrossRef] [PubMed]
- Salmi, M.; Jalkanen, S. Cell-surface enzymes in control of leukocyte trafficking. Nat. Rev. Immunol. 2005, 5, 760–771. [Google Scholar] [CrossRef]
- Dwyer, K.M.; Deaglio, S.; Gao, W.; Friedman, D.; Strom, T.B.; Robson, S.C. CD39 and control of cellular immune responses. Purinergic Signal. 2007, 3, 171. [Google Scholar] [CrossRef] [Green Version]
- Eltzschig, H.K.; Thompson, L.F.; Karhausen, J.; Cotta, R.J.; Ibla, J.C.; Robson, S.C.; Colgan, S.P. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: Coordination by extracellular nucleotide metabolism. Blood 2004, 104, 3986–3992. [Google Scholar] [CrossRef]
- Pinsky, D.J.; Broekman, M.J.; Peschon, J.J.; Stocking, K.L.; Fujita, T.; Ramasamy, R.; Connolly, E.S.; Huang, J.; Kiss, S.; Zhang, Y. Elucidation of the thromboregulatory role of CD39/ectoapyrase in the ischemic brain. J. Clin. Investig. 2002, 109, 1031–1040. [Google Scholar] [CrossRef]
- Kitagawa, H.; Mori, A.; Shimada, J.; Mitsumoto, Y.; Kikuchi, T. Intracerebral adenosine infusion improves neurological outcome after transient focal ischemia in rats. Neurol. Res. 2002, 24, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Pignataro, G.; Simon, R.P.; Boison, D. Transgenic overexpression of adenosine kinase aggravates cell death in ischemia. J. Cereb. Blood Flow Metab. 2007, 27, 1–5. [Google Scholar] [CrossRef]
- Laghi Pasini, F.; Guideri, F.; Picano, E.; Parenti, G.F.; Peterson, C.; Varga, A.; Di Perri, T. Increase in plasma adenosine during brain ischemia in man: A study during ischemic attacks and stroke. Brain Res. Bull. 2000, 51, 327–330. [Google Scholar] [CrossRef]
- Williams-Karnesky, R.L.; Stenzel-Poore, M.P. Adenosine and stroke: Maximizing the therapeutic potential of adenosine as a prophylactic and acute neuroprotectant. Curr. Neuropharmacol. 2009, 7, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maestroni, G.J. Dendritic cell migration controlled by α1b-adrenergic receptors. J. Immunol. 2000, 165, 6743–6747. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.N.; Youkey, R.; Liu, X.; Jia, L.; Blatt, R.; Day, Y.-J.; Sullivan, G.W.; Linden, J.; Tucker, A.L. A1 adenosine receptor activation promotes angiogenesis and release of VEGF from monocytes. Circ. Res. 2007, 101, 1130–1138. [Google Scholar] [CrossRef] [Green Version]
- Mukerji, S.S.; Rainey, R.N.; Rhodes, J.L.; Hall, A.K. Delayed activin A administration attenuates tissue death after transient focal cerebral ischemia and is associated with decreased stress-responsive kinase activation. J. Neurochem. 2009, 111, 1138–1148. [Google Scholar] [CrossRef] [Green Version]
- Buchthal, B.; Weiss, U.; Bading, H. Post-injury nose-to-brain delivery of activin A and SerpinB2 reduces brain damage in a mouse stroke model. Mol. Ther. 2018, 26, 2357–2365. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Li, X.; Turner, R.C.; Logsdon, A.F.; Lucke-Wold, B.; DiPasquale, K.; Jeong, S.S.; Chen, R.; Huber, J.D.; Rosen, C.L. Combination treatment of r-tPA and an optimized human apyrase reduces mortality rate and hemorrhagic transformation 6 h after ischemic stroke in aged female rats. Eur. J. Pharmacol. 2014, 738, 368–373. [Google Scholar] [CrossRef] [Green Version]
- Hot, A.; Lavocat, F.; Lenief, V.; Miossec, P. Simvastatin inhibits the pro-inflammatory and pro-thrombotic effects of IL-17 and TNF-α on endothelial cells. Ann. Rheum. Dis. 2013, 72, 754–760. [Google Scholar] [CrossRef]
- Kaneider, N.C.; Egger, P.; Dunzendorfer, S.; Noris, P.; Balduini, C.L.; Gritti, D.; Ricevuti, G.; Wiedermann, C.J. Reversal of thrombin-induced deactivation of CD39/ATPDase in endothelial cells by HMG-CoA reductase inhibition: Effects on Rho-GTPase and adenosine nucleotide metabolism. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 894–900. [Google Scholar] [CrossRef] [Green Version]
- Chróinín, D.N.; Asplund, K.; Åsberg, S.; Callaly, E.; Cuadrado-Godia, E.; Díez-Tejedor, E.; Di Napoli, M.; Engelter, S.T.; Furie, K.L.; Giannopoulos, S. Statin therapy and outcome after ischemic stroke. Stroke 2013, 44, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Hong, K.-S.; Lee, J.S. Statins in acute ischemic stroke: A systematic review. J. Stroke 2015, 17, 282. [Google Scholar] [CrossRef] [PubMed]
- Synnestvedt, K.; Furuta, G.T.; Comerford, K.M.; Louis, N.; Karhausen, J.; Eltzschig, H.K.; Hansen, K.R.; Thompson, L.F.; Colgan, S.P. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J. Clin. Investig. 2002, 110, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.F.; Eltzschig, H.K.; Ibla, J.C.; Van De Wiele, C.J.; Resta, R.; Morote-Garcia, J.C.; Colgan, S.P. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J. Exp. Med. 2004, 200, 1395–1405. [Google Scholar] [CrossRef]
- Bönner, F.; Borg, N.; Burghoff, S.; Schrader, J. Resident cardiac immune cells and expression of the ectonucleotidase enzymes CD39 and CD73 after ischemic injury. PLoS ONE 2012, 7, e34730. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, G.R.; Huang, L.; Jaworska, K.; Khutsishvili, K.; Becker, D.A.; Ye, H.; Lobo, P.I.; Okusa, M.D. Autocrine adenosine signaling promotes regulatory T cell–mediated renal protection. J. Am. Soc. Nephrol. 2012, 23, 1528–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, M.; Huttinger, Z.M.; He, H.; Zhang, W.; Li, F.; Goodman, L.A.; Wheeler, D.G.; Druhan, L.J.; Zweier, J.L.; Dwyer, K.M.; et al. Transgenic over expression of ectonucleotide triphosphate diphosphohydrolase-1 protects against murine myocardial ischemic injury. J. Mol. Cell. Cardiol. 2011, 51, 927–935. [Google Scholar] [CrossRef] [Green Version]
- Crikis, S.; Lu, B.; Murray-Segal, L.; Selan, C.; Robson, S.C.; D’Apice, A.J.; Nandurkar, H.H.; Cowan, P.J.; Dwyer, K.M. Transgenic overexpression of CD39 protects against renal ischemia-reperfusion and transplant vascular injury. Am. J. Transplant. 2010, 10, 2586–2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommey, S.; Lu, B.; McRae, J.; Stagg, J.; Hill, P.; Salvaris, E.; Robson, S.C.; d’Apice, A.J.; Cowan, P.J.; Dwyer, K.M. Liver grafts from CD39-overexpressing rodents are protected from ischemia reperfusion injury due to reduced numbers of resident CD4+ T cells. Hepatology 2013, 57, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Baek, A.E.; Sutton, N.R.; Petrovic-Djergovic, D.; Liao, H.; Ray, J.J.; Park, J.; Kanthi, Y.; Pinsky, D.J. Ischemic cerebroprotection conferred by myeloid lineage-restricted or global CD39 transgene expression. Circulation 2017, 135, 2389–2402. [Google Scholar] [CrossRef]
- Straub, A.; Krajewski, S.; Hohmann, J.D.; Westein, E.; Jia, F.; Bassler, N.; Selan, C.; Kurz, J.; Wendel, H.P.; Dezfouli, S. Evidence of platelet activation at medically used hypothermia and mechanistic data indicating ADP as a key mediator and therapeutic target. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1607–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohmann, J.D.; Wang, X.; Krajewski, S.; Selan, C.; Haller, C.A.; Straub, A.; Chaikof, E.L.; Nandurkar, H.H.; Hagemeyer, C.E.; Peter, K. Delayed targeting of CD39 to activated platelet GPIIb/IIIa via a single-chain antibody: Breaking the link between antithrombotic potency and bleeding? Blood 2013, 121, 3067–3075. [Google Scholar] [CrossRef] [Green Version]
- Marcus, A.J.; Broekman, M.J.; Drosopoulos, J.H.; Islam, N.; Pinsky, D.J.; Sesti, C.; Levi, R. Metabolic control of excessive extracellular nucleotide accumulation by CD39/ecto-nucleotidase-1: Implications for ischemic vascular diseases. J. Pharmacol. Exp. Ther. 2003, 305, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sashindranath, M.; Dwyer, K.M.; Dezfouli, S.; Selan, C.; Crikis, S.; Lu, B.; Yuan, Y.; Hickey, M.J.; Peter, K.; Robson, S.C. Development of a novel strategy to target CD39 antithrombotic activity to the endothelial-platelet microenvironment in kidney ischemia–reperfusion injury. Purinergic Signal. 2017, 13, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Degen, H.; Borst, O.; Ziegler, M.; Mojica Munoz, A.K.; Jamasbi, J.; Walker, B.; Göbel, S.; Fassbender, J.; Adler, K.; Brandl, R. ADPase CD39 Fused to Glycoprotein VI-Fc Boosts local antithrombotic effects at vascular lesions. J. Am. Heart Assoc. 2017, 6, e005991. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, N.T.; Ong, L.K.; Gyawali, P.; Nassir, C.M.N.C.M.; Mustapha, M.; Nandurkar, H.H.; Sashindranath, M. Role of Purinergic Signalling in Endothelial Dysfunction and Thrombo-Inflammation in Ischaemic Stroke and Cerebral Small Vessel Disease. Biomolecules 2021, 11, 994. https://doi.org/10.3390/biom11070994
Lee NT, Ong LK, Gyawali P, Nassir CMNCM, Mustapha M, Nandurkar HH, Sashindranath M. Role of Purinergic Signalling in Endothelial Dysfunction and Thrombo-Inflammation in Ischaemic Stroke and Cerebral Small Vessel Disease. Biomolecules. 2021; 11(7):994. https://doi.org/10.3390/biom11070994
Chicago/Turabian StyleLee, Natasha Ting, Lin Kooi Ong, Prajwal Gyawali, Che Mohd Nasril Che Mohd Nassir, Muzaimi Mustapha, Harshal H. Nandurkar, and Maithili Sashindranath. 2021. "Role of Purinergic Signalling in Endothelial Dysfunction and Thrombo-Inflammation in Ischaemic Stroke and Cerebral Small Vessel Disease" Biomolecules 11, no. 7: 994. https://doi.org/10.3390/biom11070994