
LRRK2 Targeting Strategies as Potential Treatment of Parkinson’s Disease



Abstract
:1. Introduction
2. LRRK2: Structure, Function, and Role in PD
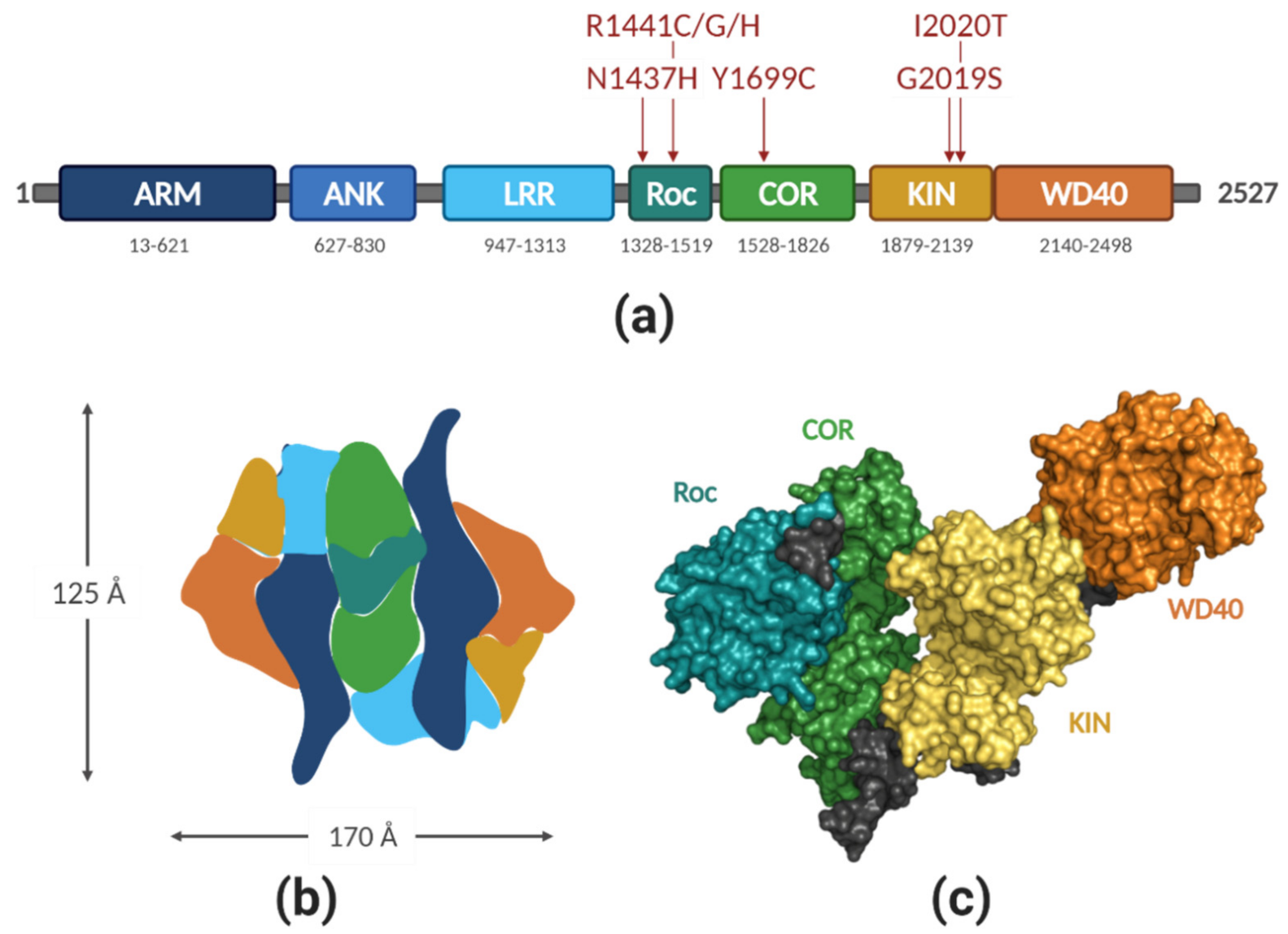
2.1. LRRK2 Structure
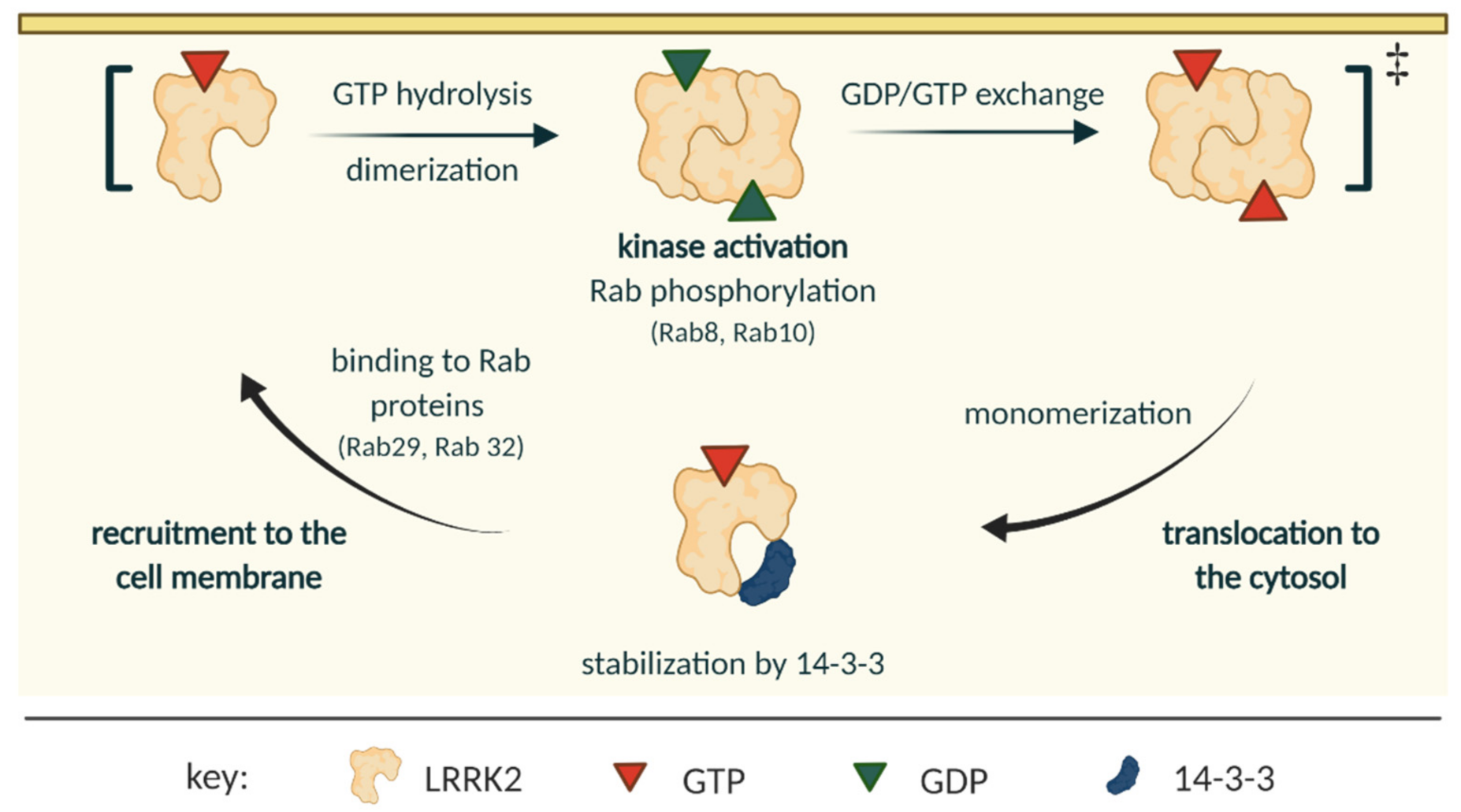
2.2. LRRK2 Activation and Cycle
2.3. LRRK2 Activation in Parkinson’s Disease
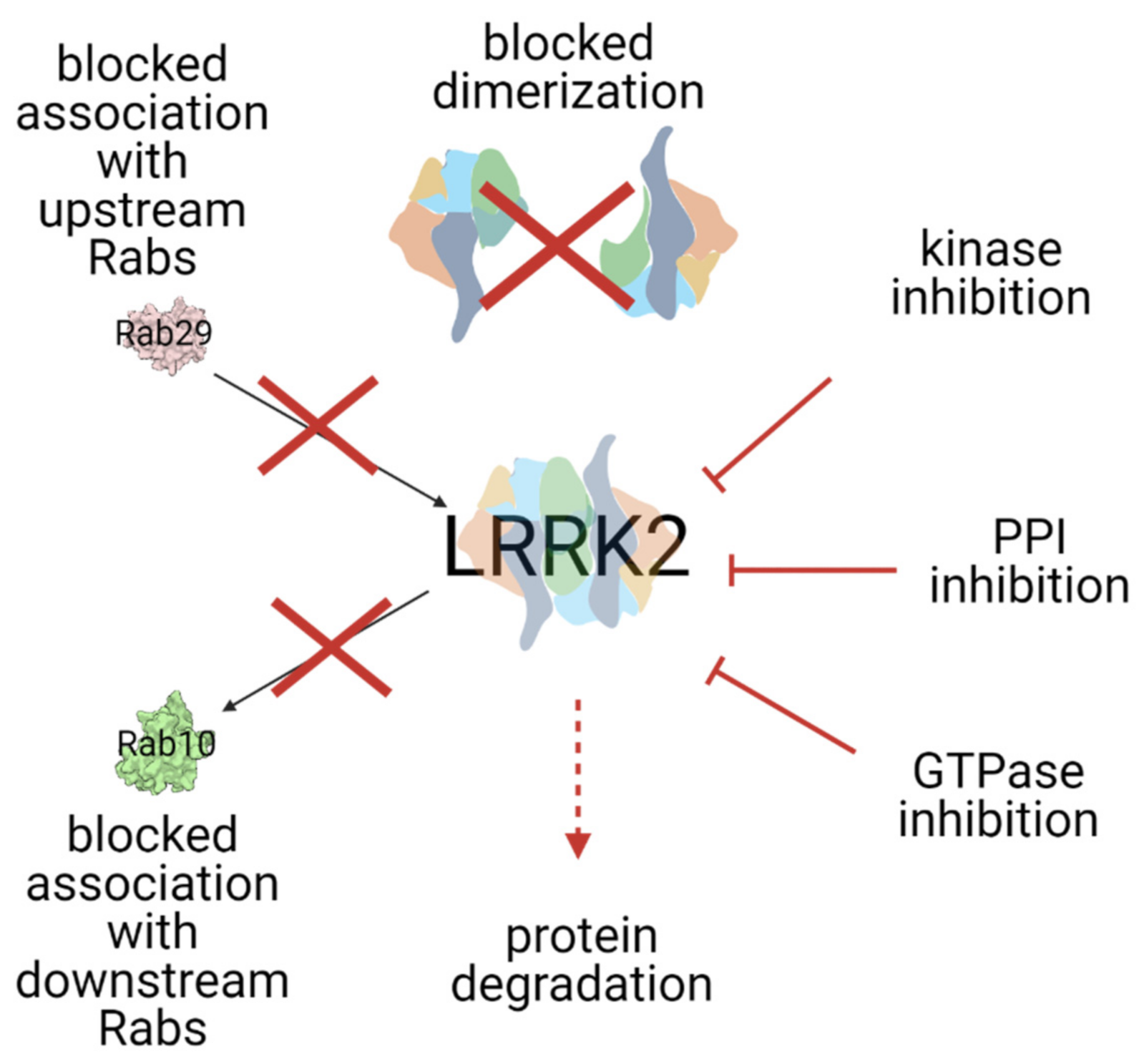
3. Modes of LRRK2 Inhibition
3.1. Kinase Inhibitors
3.1.1. Early Safety Concerns Regarding Kinase Inhibition
3.1.2. Rational Design of Improved ATP-Competitive Kinase Inhibitors
3.1.3. Allosteric LRRK2 Inhibitors Targeting the Kinase Domain
3.2. GTPase Modulators
4. Downregulating LRRK2 Protein Levels
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloemd, B.R. The emerging evidence of the Parkinson pandemic. J. Parkinsons. Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hustad, E.; Aasly, J.O. Clinical and Imaging Markers of Prodromal Parkinson’s Disease. Front. Neurol. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139, 318–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Postuma, R.B.; Aarsland, D.; Barone, P.; Burn, D.J.; Hawkes, C.H.; Oertel, W.; Ziemssen, T. Identifying prodromal Parkinson’s disease: Pre-Motor disorders in Parkinson’s disease. Mov. Disord. 2012, 27, 617–626. [Google Scholar] [CrossRef]
- Dickson, D.W. Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med. 2012, 2, a009258. [Google Scholar] [CrossRef] [Green Version]
- Monfrini, E.; Di Fonzo, A. Leucine-Rich Repeat Kinase (LRRK2) Genetics and Parkinson’s Disease. In Leucine-Rich Repeat Kinase 2 (LRRK2); Rideout, H.J., Ed.; Springer: New York, NY, USA, 2017; pp. 3–30. ISBN 978-3-319-49969-7. [Google Scholar]
- Li, D.; Mastaglia, F.L.; Fletcher, S.; Wilton, S.D. Progress in the molecular pathogenesis and nucleic acid therapeutics for Parkinson’s disease in the precision medicine era. Med. Res. Rev. 2020, 40, 2650–2681. [Google Scholar] [CrossRef]
- Kumari, U.; Tan, E.K. LRRK2 in Parkinson’s disease: Genetic and clinical studies from patients. FEBS J. 2009, 276, 6455–6463. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 2018, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Fava, V.M.; Xu, Y.Z.; Lettre, G.; Van Thuc, N.; Orlova, M.; Thai, V.H.; Tao, S.; Croteau, N.; Eldeeb, M.A.; MacDougall, E.J.; et al. Pleiotropic effects for Parkin and LRRK2 in leprosy type-1 reactions and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 15616–15624. [Google Scholar] [CrossRef] [Green Version]
- Hui, K.Y.; Fernandez-Hernandez, H.; Hu, J.; Schaffner, A.; Pankratz, N.; Hsu, N.Y.; Chuang, L.S.; Carmi, S.; Villaverde, N.; Li, X.; et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci. Transl. Med. 2018, 10, 7795. [Google Scholar] [CrossRef] [Green Version]
- Umeno, J.; Asano, K.; Matsushita, T.; Matsumoto, T.; Kiyohara, Y.; Iida, M.; Nakamura, Y.; Kamatani, N.; Kubo, M. Meta-analysis of published studies identified eight additional common susceptibility loci for Crohnʼs disease and ulcerative colitis. Inflamm. Bowel Dis. 2011, 17, 2407–2415. [Google Scholar] [CrossRef]
- Gardet, A.; Benita, Y.; Li, C.; Sands, B.E.; Ballester, I.; Stevens, C.; Korzenik, J.R.; Rioux, J.D.; Daly, M.J.; Xavier, R.J.; et al. LRRK2 Is Involved in the IFN-γ Response and Host Response to Pathogens. J. Immunol. 2010, 185, 5577–5585. [Google Scholar] [CrossRef] [Green Version]
- Hakimi, M.; Selvanantham, T.; Swinton, E.; Padmore, R.F.; Tong, Y.; Kabbach, G.; Venderova, K.; Girardin, S.E.; Bulman, D.E.; Scherzer, C.R.; et al. Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J. Neural Transm. 2011, 118, 795–808. [Google Scholar] [CrossRef] [Green Version]
- Giasson, B.I.; Covy, J.P.; Bonini, N.M.; Hurtig, H.I.; Farrer, M.J.; Trojanowski, J.Q.; Van Deerlin, V.M. Biochemical and pathological characterization of Lrrk2. Ann. Neurol. 2006, 59, 315–322. [Google Scholar] [CrossRef] [PubMed]
- West, A.B. Achieving neuroprotection with LRRK2 kinase inhibitors in Parkinson disease. Exp. Neurol. 2017, 298, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Wallings, R.; Manzoni, C.; Bandopadhyay, R. Cellular processes associated with LRRK2 function and dysfunction. FEBS J. 2015, 282, 2806–2826. [Google Scholar] [CrossRef]
- Kuwahara, T.; Iwatsubo, T. The Emerging Functions of LRRK2 and Rab GTPases in the Endolysosomal System. Front. Neurosci. 2020, 14, 227. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.R. LRRK2 and Rab GTPases. Biochem. Soc. Trans. 2018, 46, 1707–1712. [Google Scholar] [CrossRef] [PubMed]
- Seol, W.; Nam, D.; Son, I. Rab GTPases as physiological substrates of LRRK2 kinase. Exp. Neurobiol. 2019, 28, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Bonet-Ponce, L.; Beilina, A.; Williamson, C.D.; Lindberg, E.; Kluss, J.H.; Saez-Atienzar, S.; Landeck, N.; Kumaran, R.; Mamais, A.; Bleck, C.K.E.; et al. LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Cookson, M.R. Cellular functions of LRRK2 implicate vesicular trafficking pathways in Parkinson’s disease. Biochem. Soc. Trans. 2016, 44, 1603–1610. [Google Scholar] [CrossRef]
- Tang, B.L. Sec16 in conventional and unconventional exocytosis: Working at the interface of membrane traffic and secretory autophagy? J. Cell. Physiol. 2017, 232, 3234–3243. [Google Scholar] [CrossRef]
- Wallings, R.L.; Herrick, M.K.; Tansey, M.G. LRRK2 at the Interface Between Peripheral and Central Immune Function in Parkinson’s. Front. Neurosci. 2020, 14. [Google Scholar]
- Malik, A.U.; Karapetsas, A.; Nirujogi, R.S.; Mathea, S.; Chatterjee, D.; Pal, P.; Lis, P.; Taylor, M.; Purlyte, E.; Gourlay, R.; et al. Deciphering the LRRK code: LRRK1 and LRRK2 phosphorylate distinct Rab proteins and are regulated by diverse mechanisms. Biochem. J. 2021, 478, 553–578. [Google Scholar] [CrossRef] [PubMed]
- Deniston, C.K.; Salogiannis, J.; Mathea, S.; Snead, D.M.; Lahiri, I.; Matyszewski, M.; Donosa, O.; Watanabe, R.; Böhning, J.; Shiau, A.K.; et al. Structure of LRRK2 in Parkinson’s disease and model for microtubule interaction. Nature 2020, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Guaitoli, G.; Raimondi, F.; Gilsbach, B.K.; Gómez-Llorente, Y.; Deyaert, E.; Renzi, F.; Li, X.; Schaffner, A.; Jagtap, P.K.A.; Boldt, K.; et al. Structural model of the dimeric Parkinson’s protein LRRK2 reveals a compact architecture involving distant interdomain contacts. Proc. Natl. Acad. Sci. USA 2016, 113, E4357–E4366. [Google Scholar] [CrossRef] [Green Version]
- Sejwal, K.; Chami, M.; Rémigy, H.; Vancraenenbroeck, R.; Sibran, W.; Sütterlin, R.; Baumgartner, P.; McLeod, R.; Chartier-Harlin, M.C.; Baekelandt, V.; et al. Cryo-EM analysis of homodimeric full-length LRRK2 and LRRK1 protein complexes. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, R.; Buschauer, R.; Böhning, J.; Audagnotto, M.; Lasker, K.; Wen Lu, T.; Boassa, D.; Taylor, S.S.; Villa, E. The In situ Structure of Parkinson’s Disease-Linked LRRK2. Biophys. J. 2019. [Google Scholar] [CrossRef]
- Myasnikov, A.; Zhu, H.; Hixson, P.; Xie, B.; Yu, K.; Pitre, A.; Peng, J.; Sun, J. Structural analysis of the full-length human LRRK2. Cell 2021, 184, 3519–3527.e10. [Google Scholar] [CrossRef]
- Jorgensen, N.D.; Peng, Y.; Ho, C.C.Y.; Rideout, H.J.; Petrey, D.; Liu, P.; Dauer, W.T. The WD40 domain is required for LRRK2 neurotoxicity. PLoS ONE 2009, 4. [Google Scholar] [CrossRef]
- Piccoli, G.; Onofri, F.; Cirnaru, M.D.; Kaiser, C.J.O.; Jagtap, P.; Kastenmuller, A.; Pischedda, F.; Marte, A.; von Zweydorf, F.; Vogt, A.; et al. Leucine-Rich Repeat Kinase 2 Binds to Neuronal Vesicles through Protein Interactions Mediated by Its C-Terminal WD40 Domain. Mol. Cell. Biol. 2014, 34, 2147–2161. [Google Scholar] [CrossRef] [Green Version]
- Biosa, A.; Trancikova, A.; Civiero, L.; Glauser, L.; Bubacco, L.; Greggio, E.; Moore, D.J. GTPase activity regulates kinase activity and cellular phenotypes of parkinson’s disease-associated LRRK2. Hum. Mol. Genet. 2013, 22, 1140–1156. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.; Alessi, D.R. Advances in elucidating the function of leucine-rich repeat protein kinase-2 in normal cells and Parkinson’s disease. Curr. Opin. Cell Biol. 2020, 63, 102–113. [Google Scholar] [CrossRef]
- Mattea, S.; Baptista, M.; Reichert, P.; Spinale, A.; Wu, J.; Allaire, M.; Fiske, B.; Knapp, S. Crystallizing the Parkinson’s Disease Protein LRRK2 Under Microgravity Conditions. bioRxiv 2018, 259655. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Fan, Y.; Ru, H.; Wang, L.; Magupalli, V.G.; Taylor, S.S.; Alessi, D.R.; Wu, H. Crystal structure of the WD40 domain dimer of LRRK2. Proc. Natl. Acad. Sci. USA 2019, 116, 1579–1584. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Lewis, P.A.; Greggio, E.; Sluch, E.; Beilina, A.; Cookson, M.R. Structure of the ROC domain from the Parkinson’s disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc. Natl. Acad. Sci. USA 2008, 105, 1499–1504. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-X.; Liao, J.; Park, Y.; Hoang, N.C.; Engel, V.A.; Wan, L.; Oh, M.; Sanishvili, R.; Takagi, Y.; Johnson, S.M.; et al. A revised 1.6 A structure of the GTPase domain of the Parkinson’s disease-associated protein LRRK2 provides insights into mechanisms. bioRxiv 2019, 676627. [Google Scholar] [CrossRef]
- Liu, Z.; Galemmo, R.A.; Fraser, K.B.; Moehle, M.S.; Sen, S.; Volpicelli-Daley, L.A.; DeLucas, L.J.; Ross, L.J.; Valiyaveettil, J.; Moukha-Chafiq, O.; et al. Unique functional and structural properties of the LRRK2 protein ATP-binding pocket. J. Biol. Chem. 2014, 289, 32937–32951. [Google Scholar] [CrossRef] [Green Version]
- Luzón-Toro, B.; de la Torre, E.R.; Delgado, A.; Pérez-Tur, J.; Hilfiker, S. Mechanistic insight into the dominant mode of the Parkinson’s disease-associated G2019S LRRK2 mutation. Hum. Mol. Genet. 2007, 16, 2031–2039. [Google Scholar] [CrossRef] [Green Version]
- Mills, R.D.; Liang, L.Y.; Lio, D.S.S.; Mok, Y.F.; Mulhern, T.D.; Cao, G.; Griffin, M.; Kenche, V.B.; Culvenor, J.G.; Cheng, H.C. The Roc-COR tandem domain of leucine-rich repeat kinase 2 forms dimers and exhibits conventional Ras-like GTPase properties. J. Neurochem. 2018, 147, 409–428. [Google Scholar] [CrossRef] [Green Version]
- Vancraenenbroeck, R.; Lobbestael, E.; Weeks, S.D.; Strelkov, S.V.; Baekelandt, V.; Taymans, J.M.; De Maeyer, M. Expression, purification and preliminary biochemical and structural characterization of the leucine rich repeat namesake domain of leucine rich repeat kinase 2. Biochim. Biophys. Acta Proteins Proteomics 2012, 1824, 450–460. [Google Scholar] [CrossRef]
- Mills, R.D.; Mulhern, T.D.; Cheng, H.C.; Culvenor, J.G. Analysis of LRRK2 accessory repeat domains: Prediction of repeat length, number and sites of Parkinson’s disease mutations. Biochem. Soc. Trans. 2012, 40, 1086–1089. [Google Scholar] [CrossRef] [Green Version]
- Mills, R.D.; Mulhern, T.D.; Liu, F.; Culvenor, J.G.; Cheng, H.C. Prediction of the Repeat Domain Structures and Impact of Parkinsonism-Associated Variations on Structure and Function of all Functional Domains of Leucine-Rich Repeat Kinase 2 (LRRK2). Hum. Mutat. 2014, 35, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, K.; Weyand, M.; Kortholt, A.; Van Haastert, P.J.M.; Wittinghofer, A. Structure of the Roc-COR domain tandem of C. tepidum, a prokaryotic homologue of the human LRRK2 Parkinson kinase. EMBO J. 2008, 27, 2239–2249. [Google Scholar] [CrossRef] [PubMed]
- Deyaert, E.; Leemans, M.; Singh, R.K.; Gallardo, R.; Steyaert, J.; Kortholt, A.; Lauer, J.; Versées, W. Structure and nucleotide-induced conformational dynamics of the Chlorobium tepidum Roco protein. Biochem. J. 2019, 476, 51–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terheyden, S.; Ho, F.Y.; Gilsba, B.K.; Wittinghofer, A.; Kortholt, A. Revisiting the Roco G-protein cycle. Biochem. J. 2015, 465, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Gilsbach, B.K.; Ho, F.Y.; Vetter, I.R.; Van Haastert, P.J.M.; Wittinghofer, A.; Kortholt, A. Roco kinase structures give insights into the mechanism of Parkinson disease-related leucine-rich-repeat kinase 2 mutations. Proc. Natl. Acad. Sci. USA 2012, 109, 10322–10327. [Google Scholar] [CrossRef] [Green Version]
- Deyaert, E.; Wauters, L.; Guaitoli, G.; Konijnenberg, A.; Leemans, M.; Terheyden, S.; Petrovic, A.; Gallardo, R.; Nederveen-Schippers, L.M.; Athanasopoulos, P.S.; et al. A homologue of the Parkinson’s disease-associated protein LRRK2 undergoes a monomer-dimer transition during GTP turnover. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Wauters, L.; Terheyden, S.; Gilsbach, B.K.; Leemans, M.; Athanasopoulos, P.S.; Guaitoli, G.; Wittinghofer, A.; Gloeckner, C.J.; Versées, W.; Kortholt, A. Biochemical and kinetic properties of the complex Roco G-protein cycle. Biol. Chem. 2018, 399, 1447–1456. [Google Scholar] [CrossRef]
- Wauters, L.; Versées, W.; Kortholt, A. Roco proteins: GTPases with a baroque structure and mechanism. Int. J. Mol. Sci. 2019, 20, 147. [Google Scholar] [CrossRef] [Green Version]
- Gilsbach, B.K.; Messias, A.C.; Ito, G.; Sattler, M.; Alessi, D.R.; Wittinghofer, A.; Kortholt, A. Structural characterization of LRRK2 inhibitors. J. Med. Chem. 2015, 58, 3751–3756. [Google Scholar] [CrossRef]
- Berger, Z.; Smith, K.A.; Lavoie, M.J. Membrane localization of LRRK2 is associated with increased formation of the highly active lrrk2 dimer and changes in its phosphorylation. Biochemistry 2010, 49, 5511–5523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, Q.J.; Pan, N.; Lee, S.; Zhao, Y.; Chait, B.T.; Yue, Z. Phosphorylation-Dependent 14-3-3 Binding to LRRK2 Is Impaired by Common Mutations of Familial Parkinson’s Disease. PLoS ONE 2011, 6, e17153. [Google Scholar] [CrossRef]
- Nichols, R.J.; Dzamko, N.; Morrice, N.A.; Campbell, D.G.; Deak, M.; Ordureau, A.; Macartney, T.; Tong, Y.; Shen, J.; Prescott, A.R.; et al. 14-3-3 Binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem. J. 2010, 430, 393–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalogeropulou, A.F.; Freemantle, J.B.; Lis, P.; Vides, E.G.; Polinski, N.K.; Alessi, D.R. Endogenous Rab29 does not impact basal or stimulated LRRK2 pathway activity. Biochem. J. 2020, 477, 4397–4423. [Google Scholar] [CrossRef]
- Gomez, R.C.; Wawro, P.; Lis, P.; Alessi, D.R.; Pfeffer, S.R. Membrane association but not identity is required for LRRK2 activation and phosphorylation of Rab GTPases. J. Cell Biol. 2019, 218, 4157–4170. [Google Scholar] [CrossRef] [Green Version]
- Gasper, R.; Meyer, S.; Gotthardt, K.; Sirajuddin, M.; Wittinghofer, A. It takes two to tango: Regulation of G proteins by dimerization. Nat. Rev. Mol. Cell Biol. 2009, 10, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Terheyden, S.; Nederveen-Schippers, L.M.; Kortholt, A. The unconventional G-protein cycle of LRRK2 and Roco proteins. Biochem. Soc. Trans. 2016, 44, 1611–1616. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R. Mechanisms of mutant LRRK2 neurodegeneration. In Advances in Neurobiology; Rideout, H., Ed.; Springer: New York, NY, USA, 2017; pp. 227–239. Volume 14, ISBN 978-3-319-49969-7. [Google Scholar]
- Greggio, E.; Cookson, M.R. Leucine-Rich Repeat Kinase 2 Mutations and Parkinson’s Disease: Three Questions. ASN Neuro 2009, 1, AN20090007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon-Abell, J.; Berwick, D.C.; Harvey, K. L’RRK de Triomphe: A solution for LRRK2 GTPase activity? Biochem. Soc. Trans. 2016, 44, 1625–1634. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Q.; Tan, L.; Yu, J.T. The role of the LRRK2 gene in Parkinsonism. Mol. Neurodegener. 2014, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- Jaleel, M.; Nichols, R.J.; Deak, M.; Campbell, D.G.; Gillardon, F.; Knebel, A.; Alessi, D.R. LRRK2 phosphorylates moesin at threonine-558: Characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J. 2007, 405, 307–317. [Google Scholar] [CrossRef]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.; Zhang, S.; Bustos, D.; Kleinheinz, T.; Le Pichon, C.E.; Dominguez, S.L.; Solanoy, H.O.; Drummond, J.; Zhang, X.; Ding, X.; et al. Ser1292 Autophosphorylation Is an Indicator of LRRK2 Kinase Activity and Contributes to the Cellular Effects of PD Mutations. Sci. Transl. Med. 2012, 4, 161–164. [Google Scholar] [CrossRef]
- Cardona, F.; Tormos-Pérez, M.; Pérez-Tur, J. Structural and functional in silico analysis of LRRK2 missense substitutions. Mol. Biol. Rep. 2014, 41, 2529–2542. [Google Scholar] [CrossRef]
- Li, Y.; Dunn, L.; Greggio, E.; Krumm, B.; Jackson, G.S.; Cookson, M.R.; Lewis, P.A.; Deng, J. The R1441C mutation alters the folding properties of the ROC domain of LRRK2. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 1194–1197. [Google Scholar] [CrossRef] [Green Version]
- Nixon-Abell, J.; Berwick, D.C.; Grannó, S.; Spain, V.A.; Blackstone, C.; Harvey, K. Protective LRRK2 R1398H variant enhances GTPase and Wnt signaling activity. Front. Mol. Neurosci. 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Sen, S.; Webber, P.J.; West, A.B. Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. J. Biol. Chem. 2009, 284, 36346–36356. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Yang, D.; Zhong, S.; Thomas, J.M.; Xue, F.; Liu, J.; Kong, L.; Voulalas, P.; Hassan, H.E.; Park, J.S.; et al. Novel LRRK2 GTP-binding inhibitors reduced degeneration in Parkinson’s disease cell and mouse models. Hum. Mol. Genet. 2014, 23, 6212–6222. [Google Scholar] [CrossRef] [Green Version]
- Bhayye, S.S.; Roy, K.; Saha, A. Molecular dynamics simulation study reveals polar nature of pathogenic mutations responsible for stabilizing active conformation of kinase domain in leucine-rich repeat kinase II. Struct. Chem. 2018, 29, 657–666. [Google Scholar] [CrossRef]
- Ho, D.H.; Jang, J.; Joe, E.H.; Son, I.; Seo, H.; Seol, W. G2385R and I2020T Mutations Increase LRRK2 GTPase Activity. Biomed. Res. Int. 2016. [Google Scholar] [CrossRef] [Green Version]
- Kluss, J.H.; Mamais, A.; Cookson, M.R. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 2019, 47, 651–661. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–376. [Google Scholar] [CrossRef]
- Hu, Y.; Kunimoto, R.; Bajorath, J. Mapping of inhibitors and activity data to the human kinome and exploring promiscuity from a ligand and target perspective. Chem. Biol. Drug Des. 2017, 89, 834–845. [Google Scholar] [CrossRef]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.R.; Castro, S.; Drolet, R.; Hatcher, N.G.; Yao, L.; Smith, S.M.; Keeney, M.T.; Di Maio, R.; Kofler, J.; et al. LRRK2 inhibition prevents endolysosomal deficits seen in human Parkinson’s disease. Neurobiol. Dis. 2020, 134, 104626. [Google Scholar] [CrossRef]
- Fell, M.J.; Mirescu, C.; Basu, K.; Cheewatrakoolpong, B.; DeMong, D.E.; Ellis, J.M.; Hyde, L.A.; Lin, Y.; Markgraf, C.G.; Mei, H.; et al. MLi-2, a potent, selective, and centrally active compound for exploring the therapeutic potential and safety of LRRK2 kinase inhibition. J. Pharmacol. Exp. Ther. 2015, 355, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.D.; DeMong, D.E.; Greshock, T.J.; Basu, K.; Dai, X.; Harris, J.; Hruza, A.; Li, S.W.; Lin, S.I.; Liu, H.; et al. Discovery of a 3-(4-Pyrimidinyl) Indazole (MLi-2), an Orally Available and Selective Leucine-Rich Repeat Kinase 2 (LRRK2) Inhibitor that Reduces Brain Kinase Activity. J. Med. Chem. 2017, 60, 2983–2992. [Google Scholar] [CrossRef]
- Henderson, J.L.; Kormos, B.L.; Hayward, M.M.; Coffman, K.J.; Jasti, J.; Kurumbail, R.G.; Wager, T.T.; Verhoest, P.R.; Noell, G.S.; Chen, Y.; et al. Discovery and preclinical profiling of 3-[4-(morpholin-4-yl)-7H-pyrrolo [2,3-d]pyrimidin-5-yl]benzonitrile (PF-06447475), a highly potent, selective, brain penetrant, and in vivo active LRRK2 kinase inhibitor. J. Med. Chem. 2015, 58, 419–432. [Google Scholar] [CrossRef]
- Andersen, M.A.; Wegener, K.M.; Larsen, S.; Badolo, L.; Smith, G.P.; Jeggo, R.; Jensen, P.H.; Sotty, F.; Christensen, K.V.; Thougaard, A. PFE-360-induced LRRK2 inhibition induces reversible, non-adverse renal changes in rats. Toxicology 2018, 395, 15–22. [Google Scholar] [CrossRef]
- Estrada, A.A.; Chan, B.K.; Baker-Glenn, C.; Beresford, A.; Burdick, D.J.; Chambers, M.; Chen, H.; Dominguez, S.L.; Dotson, J.; Drummond, J.; et al. Discovery of highly potent, selective, and brain-penetrant aminopyrazole Leucine-rich repeat kinase 2 (LRRK2) small molecule inhibitors. J. Med. Chem. 2014, 57, 921–936. [Google Scholar] [CrossRef]
- Estrada, A.A.; Liu, X.; Baker-Glenn, C.; Beresford, A.; Burdick, D.J.; Chambers, M.; Chan, B.K.; Chen, H.; Ding, X.; Dipasquale, A.G.; et al. Discovery of highly potent, selective, and brain-penetrable leucine-rich repeat kinase 2 (LRRK2) small molecule inhibitors. J. Med. Chem. 2012, 55, 9416–9433. [Google Scholar] [CrossRef]
- Reith, A.D.; Bamborough, P.; Jandu, K.; Andreotti, D.; Mensah, L.; Dossang, P.; Choi, H.G.; Deng, X.; Zhang, J.; Alessi, D.R.; et al. GSK2578215A.; A potent and highly selective 2-arylmethyloxy-5-substitutent- N-arylbenzamide LRRK2 kinase inhibitor. Bioorganic Med. Chem. Lett. 2012, 22, 5625–5629. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.G.; Zhang, J.; Deng, X.; Hatcher, J.M.; Patricelli, M.P.; Zhao, Z.; Alessi, D.R.; Gray, N.S. Brain penetrant LRRK2 inhibitor. ACS Med. Chem. Lett. 2012, 3, 658–662. [Google Scholar] [CrossRef]
- Deng, X.; Dzamko, N.; Prescott, A.; Davies, P.; Liu, Q.; Yang, Q.; Lee, J.D.; Patricelli, M.P.; Nomanbhoy, T.K.; Alessi, D.R.; et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol. 2011, 7, 203–205. [Google Scholar] [CrossRef] [Green Version]
- Fuji, R.N.; Flagella, M.; Baca, M.; Baptista, M.A.S.; Brodbeck, J.; Chan, B.K.; Fiske, B.K.; Honigberg, L.; Jubb, A.M.; Katavolos, P.; et al. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci. Transl. Med. 2015, 7, 273. [Google Scholar] [CrossRef]
- Baptista, M.A.S.; Merchant, K.; Barrett, T.; Bhargava, S.; Bryce, D.K.; Ellis, J.M.; Estrada, A.A.; Fell, M.J.; Fiske, B.K.; Fuji, R.N.; et al. LRRK2 inhibitors induce reversible changes in nonhuman primate lungs without measurable pulmonary deficits. Sci. Transl. Med. 2020, 12, eaav0820. [Google Scholar] [CrossRef]
- Lobbestael, E.; Civiero, L.; De Wit, T.; Taymans, J.-M.; Greggio, E.; Baekelandt, V. Pharmacological LRRK2 kinase inhibition induces LRRK2 protein destabilization and proteasomal degradation. Sci. Rep. 2016, 6, 33897. [Google Scholar] [CrossRef] [Green Version]
- Baptista, M.A.S.; Dave, K.D.; Frasier, M.A.; Sherer, T.B.; Greeley, M.; Beck, M.J.; Varsho, J.S.; Parker, G.A.; Moore, C.; Churchill, M.J.; et al. Loss of Leucine-Rich Repeat Kinase 2 (LRRK2) in Rats Leads to Progressive Abnormal Phenotypes in Peripheral Organs. PLoS ONE 2013, 8, e80705. [Google Scholar] [CrossRef] [Green Version]
- Whiffin, N.; Armean, I.M.; Kleinman, A.; Marshall, J.L.; Minikel, E.V.; Goodrich, J.K.; Quaife, N.M.; Cole, J.B.; Wang, Q.; Karczewski, K.J.; et al. The effect of LRRK2 loss-of-function variants in humans. Nat. Med. 2020, 26, 869–877. [Google Scholar] [CrossRef]
- Bhayye, S.S.; Roy, K.; Saha, A. Exploring structural requirement, pharmacophore modeling, and de novo design of LRRK2 inhibitors using homology modeling approach. Med. Chem. Res. 2014, 23, 3705–3713. [Google Scholar] [CrossRef]
- Chen, H.; Chan, B.K.; Drummond, J.; Estrada, A.A.; Gunzner-Toste, J.; Liu, X.; Liu, Y.; Moffat, J.; Shore, D.; Sweeney, Z.K.; et al. Discovery of selective LRRK2 inhibitors guided by computational analysis and molecular modeling. J. Med. Chem. 2012, 55, 5536–5545. [Google Scholar] [CrossRef] [PubMed]
- Gancia, E.; De Groot, M.; Burton, B.; Clark, D.E. Discovery of LRRK2 inhibitors by using an ensemble of virtual screening methods. Bioorganic Med. Chem. Lett. 2017, 27, 2520–2527. [Google Scholar] [CrossRef]
- Garofalo, A.W.; Bright, J.; De Lombaert, S.; Toda, A.M.A.; Zobel, K.; Andreotti, D.; Beato, C.; Bernardi, S.; Budassi, F.; Caberlotto, L.; et al. Selective Inhibitors of G2019S-LRRK2 Kinase Activity. J. Med. Chem. 2020, 63, 14821–14839. [Google Scholar] [CrossRef] [PubMed]
- Greshock, T.J.; Sanders, J.M.; Drolet, R.E.; Rajapakse, H.A.; Chang, R.K.; Kim, B.; Rada, V.L.; Tiscia, H.E.; Su, H.; Lai, M.T.; et al. Potent, selective and orally bioavailable leucine-rich repeat kinase 2 (LRRK2) inhibitors. Bioorganic Med. Chem. Lett. 2016, 26, 2631–2635. [Google Scholar] [CrossRef]
- Smith, G.P.; Badolo, L.; Chell, V.; Chen, I.J.; Christensen, K.V.; David, L.; Daechsel, J.A.; Hentzer, M.; Herzig, M.C.; Mikkelsen, G.K.; et al. The design and SAR of a novel series of 2-aminopyridine based LRRK2 inhibitors. Bioorganic Med. Chem. Lett. 2017, 27, 4500–4505. [Google Scholar] [CrossRef]
- Williamson, D.S.; Smith, G.P.; Acheson-Dossang, P.; Bedford, S.T.; Chell, V.; Chen, I.J.; Daechsel, J.C.A.; Daniels, Z.; David, L.; Dokurno, P.; et al. Design of Leucine-Rich Repeat Kinase 2 (LRRK2) Inhibitors Using a Crystallographic Surrogate Derived from Checkpoint Kinase 1 (CHK1). J. Med. Chem. 2017, 60, 8945–8962. [Google Scholar] [CrossRef]
- Williamson, D.S.; Smith, G.P.; Mikkelsen, G.K.; Jensen, T.; Acheson-Dossang, P.; Badolo, L.; Bedford, S.T.; Chell, V.; Chen, I.-J.; Dokurno, P.; et al. Design and Synthesis of Pyrrolo[2,3- d ]pyrimidine-Derived Leucine-Rich Repeat Kinase 2 (LRRK2) Inhibitors Using a Checkpoint Kinase 1 (CHK1)-Derived Crystallographic Surrogate. J. Med. Chem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.A.; Ray, S.S.; Liu, M.; Singh, A.K.; Cuny, G.D. Discovery of LRRK2 inhibitors using sequential in silico joint pharmacophore space (JPS) and ensemble docking. Bioorganic Med. Chem. Lett. 2015, 25, 2713–2719. [Google Scholar] [CrossRef]
- Kelly, K.; Wang, S.; Boddu, R.; Liu, Z.; Moukha-Chafiq, O.; Augelli-Szafran, C.; West, A.B. The G2019S mutation in LRRK2 imparts resiliency to kinase inhibition. Exp. Neurol. 2018, 309, 1–13. [Google Scholar] [CrossRef]
- Panicker, R.C.; Chattopadhaya, S.; Coyne, A.G.; Srinivasan, R. Allosteric small-molecule serine/threonine kinase inhibitors. In Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; Volume 1163, pp. 253–278. [Google Scholar]
- Yueh, C.; Rettenmaier, J.; Xia, B.; Hall, D.R.; Alekseenko, A.; Porter, K.A.; Barkovich, K.; Keseru, G.; Whitty, A.; Wells, J.A.; et al. Kinase atlas: Druggability analysis of potential allosteric sites in kinases. J. Med. Chem. 2019, 62, 6512–6524. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, A.; Li, X.; Gomez-Llorente, Y.; Leandrou, E.; Memou, A.; Clemente, N.; Yao, C.; Afsari, F.; Zhi, L.; Pan, N.; et al. Vitamin B 12 modulates Parkinson’s disease LRRK2 kinase activity through allosteric regulation and confers neuroprotection. Cell Res. 2019, 29, 313–329. [Google Scholar] [CrossRef]
- Taymans, J.-M.; Vancraenenbroeck, R.; Ollikainen, P.; Beilina, A.; Lobbestael, E.; De Maeyer, M.; Baekelandt, V.; Cookson, M.R. LRRK2 Kinase Activity Is Dependent on LRRK2 GTP Binding Capacity but Independent of LRRK2 GTP Binding. PLoS ONE 2011, 6, e23207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsika, E.; Moore, D.J. Contribution of GTPase activity to LRRK2-associated Parkinson disease. Small GTPases 2013, 4, 163–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; He, X.; Thomas, J.M.; Yang, D.; Zhong, S.; Xue, F.; Smith, W.W. A Novel GTP-Binding Inhibitor, FX2149, Attenuates LRRK2 Toxicity in Parkinson’s Disease Models. PLoS ONE 2015, 10, e0122461. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.M.; Li, T.; Yang, W.; Xue, F.; Fishman, P.S.; Smith, W.W. 68 and FX2149 attenuate mutant LRRK2-R1441C-induced neural transport impairment. Front. Aging Neurosci. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Thomas, J.M.; Wang, X.; Gongbo Guo, |; Li, T.; Dai, B.; Leslie, |; Nucifora, G.; Frederick, |; Nucifora, C.; Liu, Z.; et al. GTP-binding inhibitors increase LRRK2-linked ubiquitination and Lewy body-like inclusions. J. Cell Physiol 2020. [Google Scholar] [CrossRef] [PubMed]
- Nucifora, F.C.; Nucifora, L.G.; Ng, C.H.; Arbez, N.; Guo, Y.; Roby, E.; Shani, V.; Engelender, S.; Wei, D.; Wang, X.F.; et al. Ubiqutination via K27 and K29 chains signals aggregation and neuronal protection of LRRK2 by WSB1. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Ohtake, F.; Tsuchiya, H.; Saeki, Y.; Tanaka, K. K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc. Natl. Acad. Sci. USA 2018, 115, E1401–E1408. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Ning, B.; Kong, L.; Dai, B.; He, X.; Thomas, J.M.; Sawa, A.; Ross, C.A.; Smith, W.W. A LRRK2 GTP Binding Inhibitor, 68, Reduces LPS-Induced Signaling Events and TNF-α Release in Human Lymphoblasts. Cells 2021, 10, 480. [Google Scholar] [CrossRef]
- Blanca Ramírez, M.; Ordóñez, A.J.L.; Fdez, E.; Madero-Pérez, J.; Gonnelli, A.; Drouyer, M.; Chartier-Harlin, M.-C.; Taymans, J.-M.; Bubacco, L.; Greggio, E.; et al. GTP binding regulates cellular localization of Parkinson’s disease-associated LRRK2. Hum. Mol. Genet. 2017, 26, 2747–2767. [Google Scholar] [CrossRef] [Green Version]
- Tomoshige, S.; Ishikawa, M. PROTACs and Other Chemical Protein Degradation Technologies for the Treatment of Neurodegenerative Disorders. Angew Chem. Int. Ed. Engl. 2021, 60, 3346–3354. [Google Scholar] [CrossRef] [PubMed]
- de Yñigo-Mojado, L.; Martín-Ruíz, I.; Sutherland, J.D. Efficient Allele-Specific Targeting of LRRK2 R1441 Mutations Mediated by RNAi. PLoS ONE 2011, 6, e21352. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of Antisense Drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [Google Scholar] [CrossRef]
- Rigo, F.; Chun, S.J.; Norris, D.A.; Hung, G.; Lee, S.; Matson, J.; Fey, R.A.; Gaus, H.; Hua, Y.; Grundy, J.S.; et al. Pharmacology of a Central Nervous System Delivered 2′-O-Methoxyethyl–Modified Survival of Motor Neuron Splicing Oligonucleotide in Mice and Nonhuman Primates. J. Pharmacol. Exp. Ther. 2014, 350, 46–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.T.; John, N.; Delic, V.; Ikeda-Lee, K.; Kim, A.; Weihofen, A.; Swayze, E.E.; Kordasiewicz, H.B.; West, A.B.; Volpicelli-Daley, L.A. LRRK2 Antisense Oligonucleotides Ameliorate α-Synuclein Inclusion Formation in a Parkinson’s Disease Mouse Model. Mol. Ther. Nucleic. Acids 2017, 8, 508–519. [Google Scholar] [CrossRef] [Green Version]
- Korecka, J.A.; Thomas, R.; Hinrich, A.J.; Moskites, A.M.; Macbain, Z.K.; Hallett, P.J.; Isacson, O.; Hastings, M.L. Splice-Switching Antisense Oligonucleotides Reduce LRRK2 Kinase Activity in Human LRRK2 Transgenic Mice. Mol. Ther. Nucleic. Acids 2020, 21, 623–635. [Google Scholar] [CrossRef]
- Vlachakis, D.; Labrou, N.E.; Iliopoulos, C.; Hardy, J.; Lewis, P.A.; Rideout, H.; Trabzuni, D. Insights into the influence of specific splicing events on the structural organization of LRRK2. Int. J. Mol. Sci. 2018, 19, 2784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef]
- Gao, H.; Sun, X.; Rao, Y. PROTAC Technology: Opportunities and Challenges. ACS Med. Chem. Lett. 2020, 11, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Ma, D.; Wang, Y. The PROTAC technology in drug development. Cell Biochem. Funct. 2019, 37, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kargbo, R.B. Degradation of LRRK2 in the Treatment of Parkinson’s Disease. ACS Med. Chem. Lett. 2020, 11, 2070–2071. [Google Scholar] [CrossRef]
- Denali press release 14 January 2020. Available online: https://denalitherapeutics.com/investors/press-release?id=7361 (accessed on 26 May 2020).
- Denali press release 6 August 2020. Available online: https://www.denalitherapeutics.com/investors/press-release?id=7661 (accessed on 20 December 2020).
- Denali press release 8 January 2021. Available online: https://www.denalitherapeutics.com/investors/press-release?id=7881 (accessed on 18 March 2021).
- Soliman, A.; Cankara, F.N.; Kortholt, A. Allosteric inhibition of LRRK2, where are we now. Biochem. Soc. Trans. 2020. [Google Scholar] [CrossRef]
- Berndsen, K.; Lis, P.; Yeshaw, W.M.; Wawro, P.S.; Nirujogi, R.S.; Wightman, M.; Macartney, T.; Dorward, M.; Knebel, A.; Tonelli, F.; et al. PPM1H phosphatase counteracts LRRK2 signaling by selectively dephosphorylating Rab proteins. Elife 2019, 8, e50416. [Google Scholar] [CrossRef]
- Fan, Y.; Tonelli, F.; Padmanabhan, S.; Baptista, M.A.S.; Riley, L.; Smith, D.; Marras, C.; Howden, A.; Alessi, D.R.; Sammler, E. Human peripheral blood neutrophil isolation for interrogating the parkinson’s associated LRRK2 kinase pathway by assessing RAB10 phosphorylation. J. Vis. Exp. 2020, 2020. [Google Scholar] [CrossRef]
- Nirujogi, R.S.; Tonelli, F.; Taylor, M.; Lis, P.; Zimprich, A.; Sammler, E.; Alessi, D.R. Development of a multiplexed targeted mass spectrometry assay for LRRK2-phosphorylated Rabs and Ser910/Ser935 biomarker sites. Biochem. J. 2021, 478, 299–326. [Google Scholar] [CrossRef] [PubMed]
- Virreira Winter, S.; Karayel, O.; Strauss, M.T.; Padmanabhan, S.; Surface, M.; Merchant, K.; Alcalay, R.N.; Mann, M. Urinary proteome profiling for stratifying patients with familial Parkinson’s disease. EMBO Mol. Med. 2021, 13, e13257. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Mutation | Domain | Effect on the Kinase | Effect on the GTPase | Probable Mechanism |
|---|---|---|---|---|
| R1441G | Roc | no effect [65] 11–15× ↑ Rab phosph. [66] 2.5× ↑ autophosph. [67] | ↑ GTP binding, ↓ GTP hydrolysis | loss of positive charge that impairs dimerization [68] |
| R1441C | Roc | 4–6× ↑ Rab phosph. [66] 3× ↑ autophosph. [67] | ↑ GTP binding, ↓ GTP hydrolysis [69] | ↓ thermodynamic stability of Roc domain [70]; loss of positive charge that impairs dimerization [68] |
| R1441H | Roc | 9–10× ↑ Rab phosph. [66] | ↑ GTP binding, ↓ GTP hydrolysis [65] | loss of positive charge that impairs dimerization, alteration to tertiary structure of Roc domain [68] |
| Y1699C | COR | 14–18× ↑ Rab phosph. [66] no effect on autophosph. [67] | ↑ GTP binding, ↓ GTP hydrolysis [71] | alteration of electrostatic surface [68] |
| G2019S | kinase | 2–3× ↑ [66] 3.5× ↑ autophosph. [67] | no effect [62] ↑ GTP binding [72] | DYG loop stabilized in active conformation by hydrogen bond [49,68,73] |
| I2020T | kinase | no effect in vitro [74] 6–7× ↑ Rab phosph. [66] 2× ↑ autophosph. [67] | ↑ GTPase activity [74] | affected stability, kinase stabilized in an inactive conformation by hydrogen bond [68,73] |
| Compound Name | Chemical Structure | LRRK2 IC50 [nM] | Brain Permeability | Reference | |
|---|---|---|---|---|---|
| WT | G2019S | ||||
| MLi-2 | 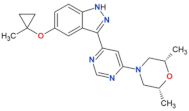 | 0.8 | 0.76 | yes | [80,81] |
| PF-06447475 |  | 3 | 11 | yes | [82] |
| PF-06685360 |  | 2.3 | n.d. | yes | [83] |
| GNE-0877 | 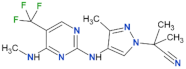 | 3 | n.d. | yes | [84] |
| GNE-7915 |  | 9 | n.d. | yes | [85] |
| GSK2578215A | 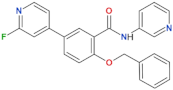 | 10.9 | 8.9 | yes | [86] |
| HG-10-102-1 | 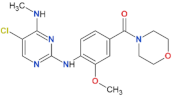 | 20.3 | 3.2 | yes | [87] |
| LRRK2-IN-1 | 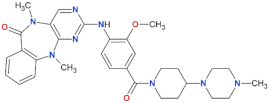 | 13.0 | 6.0 | no | [88] |
| Compound Name | Chemical Structure | Brain Permeability | Reference |
|---|---|---|---|
| 68 | 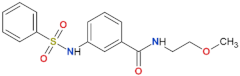 | yes | [72] |
| 70 | 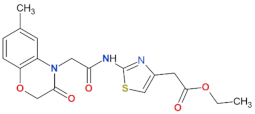 | n.a. | [72] |
| FX2149 | 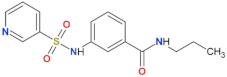 | yes | [109] |
| Clinical Trial | Compound Name | Compound Type | Phase | Funding Body |
|---|---|---|---|---|
| NCT03710707 | DNL201 | kinase inhibitor | Phase Ib (completed) | Denali Therapeutics Inc. (South San Francisco, CA, USA) |
| NCT04056689 | DNL151 | kinase inhibitor | Phase Ib (in progress) | Denali Therapeutics Inc. (South San Francisco, CA, USA) and Biogen (Cambridge, MA, USA) |
| NCT03976349 | BIIB094 | ASO | Phase I (recruiting) | Biogen (Cambridge, MA, USA) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojewska, D.N.; Kortholt, A. LRRK2 Targeting Strategies as Potential Treatment of Parkinson’s Disease. Biomolecules 2021, 11, 1101. https://doi.org/10.3390/biom11081101
Wojewska DN, Kortholt A. LRRK2 Targeting Strategies as Potential Treatment of Parkinson’s Disease. Biomolecules. 2021; 11(8):1101. https://doi.org/10.3390/biom11081101
Chicago/Turabian StyleWojewska, Dominika Natalia, and Arjan Kortholt. 2021. "LRRK2 Targeting Strategies as Potential Treatment of Parkinson’s Disease" Biomolecules 11, no. 8: 1101. https://doi.org/10.3390/biom11081101
APA StyleWojewska, D. N., & Kortholt, A. (2021). LRRK2 Targeting Strategies as Potential Treatment of Parkinson’s Disease. Biomolecules, 11(8), 1101. https://doi.org/10.3390/biom11081101