



Portability of a Small-Molecule Binding Site between Disordered Proteins

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Myc353–437, MaxRH, Max, and Myc402–412 Purification
2.2. Preparation, Characterization and pKa Determination of 34RH
2.3. Tyrosine Fluorescence Quenching Assay
2.4. Circular Dichroism (CD)
3. Results
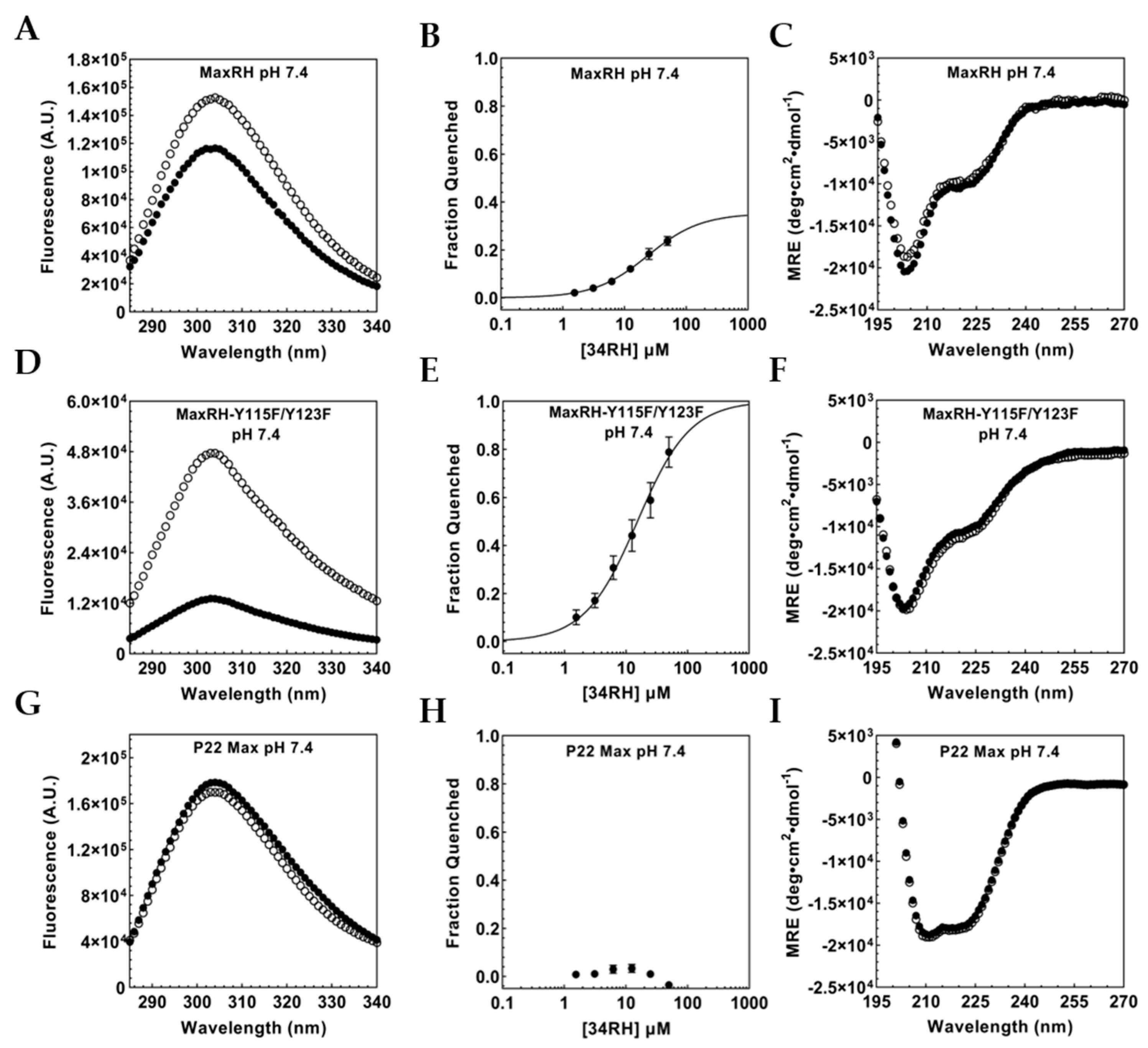
3.1. Binding of the Small Molecule 34RH to the Myc Target Site
3.2. Binding of 34RH to the Myc402–412 Peptide
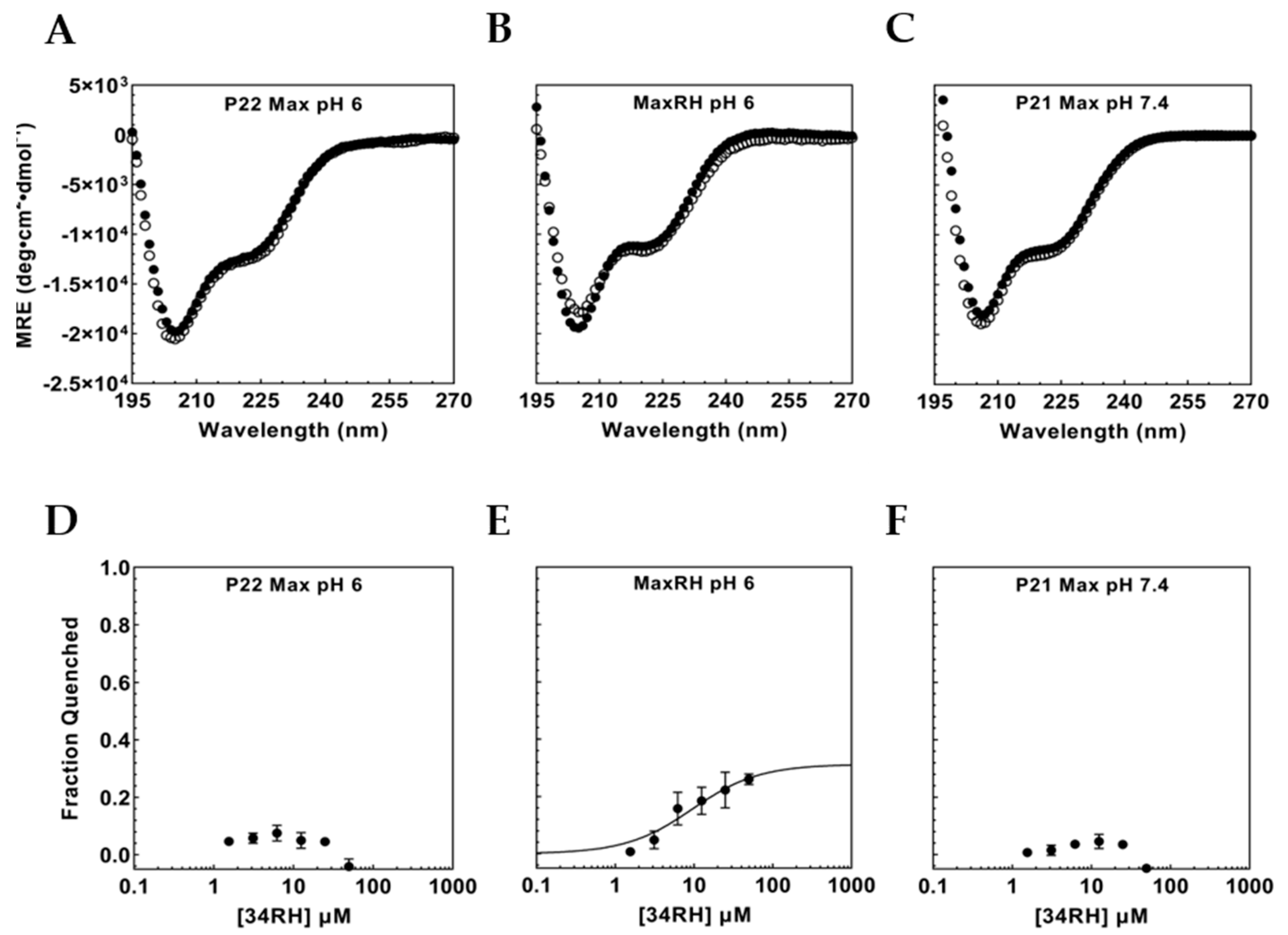
3.3. Portability of the Small-Molecule IDP Binding Site
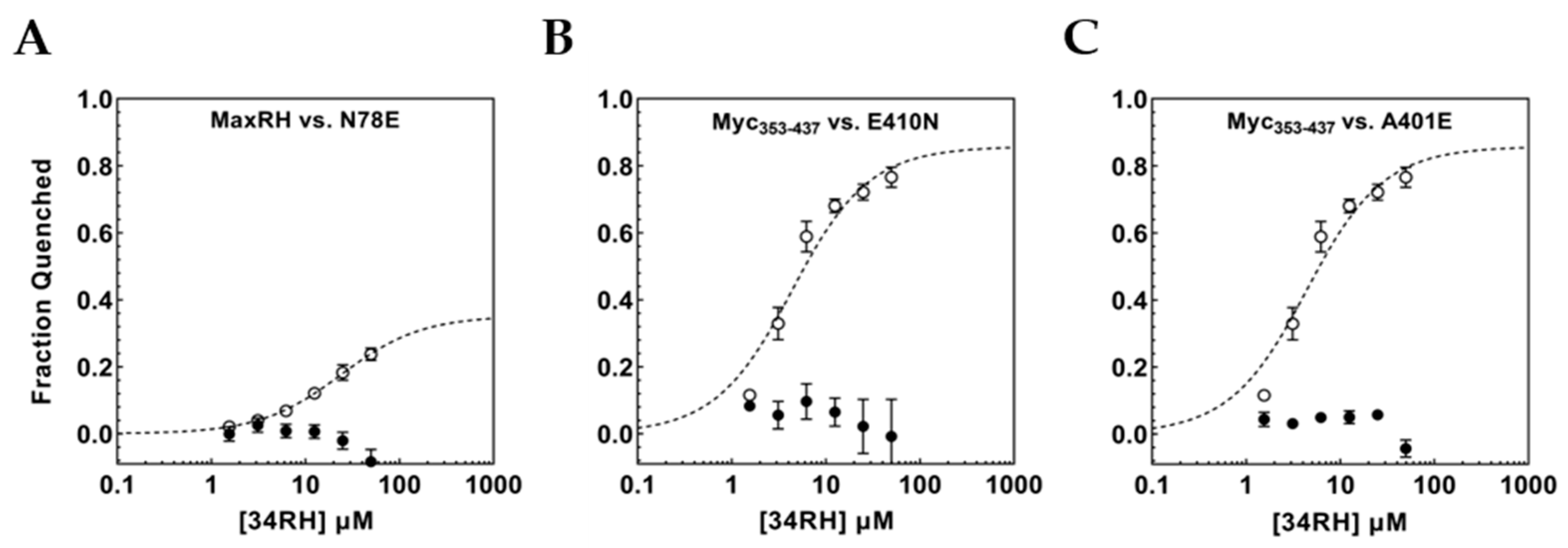
3.4. Flanking Residues Modulate 34RH Binding
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [Green Version]
- Fisher, C.K.; Stultz, C.M. Protein Structure along the Order-Disorder Continuum. J. Am. Chem. Soc. 2011, 133, 10022–10025. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically Disordered Proteins and Their “Mysterious” (Meta) Physics. Front. Phys. 2019, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenwick, R.B.; Esteban-Martin, S.; Salvatella, X. Understanding biomolecular motion, recognition, and allostery by use of conformational ensembles. Eur. Biophys. J. Biophy. 2011, 40, 1339–1355. [Google Scholar] [CrossRef] [Green Version]
- Turoverov, K.K.; Kuznetsova, I.M.; Uversky, V.N. The protein kingdom extended: Ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog. Biophys. Mol. Bio. 2010, 102, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galea, C.A.; High, A.A.; Obenauer, J.C.; Mishra, A.; Park, C.G.; Punta, M.; Schllessinger, A.; Ma, J.; Rost, B.; Slaughter, C.A.; et al. Large-Scale Analysis of Thermostable, Mammalian Proteins Provides Insights into the Intrinsically Disordered Proteome. J. Proteome Res. 2009, 8, 211–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.G.; Perumal, N.B.; Oldfield, C.J.; Su, E.W.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in transcription factors. Biochemistry 2006, 45, 6873–6888. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Babu, M.M.; van der Lee, R.; de Groot, N.S.; Gsponer, J. Intrinsically disordered proteins: Regulation and disease. Curr. Opin. Struc. Biol. 2011, 21, 432–440. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Tompa, P.; Simon, I.; Uversky, V.N.; Hansen, J.C.; Asturias, F.J. Malleable machines take shape in eukaryotic transcriptional regulation. Nat. Chem. Biol. 2008, 4, 728–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Neduva, V.; Russell, R.B. Linear motifs: Evolutionary interaction switches. Febs. Lett. 2005, 579, 3342–3345. [Google Scholar] [CrossRef] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Bio. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Roden, C.; Gladfelter, A.S. RNA contributions to the form and function of biomolecular condensates. Nat. Rev. Mol. Cell Biol. 2021, 22, 183–195. [Google Scholar] [CrossRef]
- Weber, S.C.; Brangwynne, C.P. Getting RNA and Protein in Phase. Cell 2012, 149, 1188–1191. [Google Scholar] [CrossRef] [Green Version]
- Lyon, A.S.; Peeples, W.B.; Rosen, M.K. A framework for understanding the functions of biomolecular condensates across scales. Nat. Rev. Mol. Cell Biol. 2021, 22, 215–235. [Google Scholar] [CrossRef]
- Shammas, S.L.; Crabtree, M.D.; Dahal, L.; Wicky, B.I.M.; Clarke, J. Insights into Coupled Folding and Binding Mechanisms from Kinetic Studies. J. Biol. Chem. 2016, 291, 6689–6695. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.E.; Dyson, H.J. Linking folding and binding. Curr. Opin. Struc. Biol. 2009, 19, 31–38. [Google Scholar] [CrossRef]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; LeGall, T.; Oldfield, C.J.; Mueller, J.P.; Van, Y.Y.; Romero, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 2006, 24, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Malhis, N.; Gsponer, J. Computational identification of MoRFs in protein sequences. Bioinformatics 2015, 31, 1738–1744. [Google Scholar] [CrossRef] [Green Version]
- Disfani, F.M.; Hsu, W.L.; Mizianty, M.J.; Oldfield, C.J.; Xue, B.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics 2012, 28, I75–I83. [Google Scholar] [CrossRef] [Green Version]
- Burgi, J.; Xue, B.; Uversky, V.N.; van der Goot, F.G. Intrinsic Disorder in Transmembrane Proteins: Roles in Signaling and Topology Prediction. PLoS ONE 2016, 11, e0158594. [Google Scholar] [CrossRef] [Green Version]
- Davey, N.E.; Van Roey, K.; Weatheritt, R.J.; Toedt, G.; Uyar, B.; Altenberg, B.; Budd, A.; Diella, F.; Dinkel, H.; Gibson, T.J. Attributes of short linear motifs. Mol. Biosyst. 2012, 8, 268–281. [Google Scholar] [CrossRef]
- Davey, N.E. The functional importance of structure in unstructured protein regions. Curr. Opin. Struc. Biol. 2019, 56, 155–163. [Google Scholar] [CrossRef]
- Berg, T.; Cohen, S.B.; Desharnais, J.; Sonderegger, C.; Maslyar, D.J.; Goldberg, J.; Boger, D.L.; Vogt, P.K. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Nat. Acad. Sci. USA 2002, 99, 3830–3835. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Giap, C.; Lazo, J.S.; Prochownik, E.V. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 2003, 22, 6151–6159. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitfield, J.R.; Soucek, L. The long journey to bring a Myc inhibitor to the clinic. J. Cell Biol. 2021, 220, e202103090. [Google Scholar] [CrossRef] [PubMed]
- Llombart, V.; Mansour, M.R. Therapeutic targeting of “undruggable” MYC. Ebiomedicine 2022, 75, 103756. [Google Scholar] [CrossRef] [PubMed]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef]
- Metallo, S.J. Intrinsically disordered proteins are potential drug targets. Curr. Opin. Chem. Biol. 2010, 14, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Ruan, H.; Sun, Q.; Zhang, W.L.; Liu, Y.; Lai, L.H. Targeting intrinsically disordered proteins at the edge of chaos. Drug Discov. Today 2019, 24, 217–227. [Google Scholar] [CrossRef]
- Santofimia-Castano, P.; Rizzuti, B.; Xia, Y.; Abian, O.; Peng, L.; Velazquez-Campoy, A.; Neira, J.L.; Iovanna, J. Targeting intrinsically disordered proteins involved in cancer. Cell Mol. Life Sci. 2020, 77, 1695–1707. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.L.; Liu, X.R.; Chen, J.H. Targeting Intrinsically Disordered Proteins through Dynamic Interactions. Biomolecules 2020, 10, 743. [Google Scholar] [CrossRef]
- Biesaga, M.; Frigole-Vivas, M.; Salvatella, X. Intrinsically disordered proteins and biomolecular condensates as drug targets. Curr. Opin. Chem. Biol. 2021, 62, 90–100. [Google Scholar] [CrossRef]
- Benz, C.; Ali, M.; Krystkowiak, I.; Simonetti, L.; Sayadi, A.; Mihalic, F.; Kliche, J.; Andersson, E.; Jemth, P.; Davey, N.E.; et al. Proteome-scale mapping of binding sites in the unstructured regions of the human proteome. Mol. Syst. Biol. 2022, 18, e10584. [Google Scholar] [CrossRef] [PubMed]
- Hammoudeh, D.I.; Follis, A.V.; Prochownik, E.V.; Metallo, S.J. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J. Am. Chem. Soc. 2009, 131, 7390–7401. [Google Scholar] [CrossRef]
- Studier, F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expres. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hammoudeh, D.I.; Follis, A.V.; Reese, B.E.; Lazo, J.S.; Metallo, S.J.; Prochownik, E.V. Improved low molecular weight Myc-Max inhibitors. Mol. Cancer Ther. 2007, 6, 2399–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luiz, F.C.L.; Louro, S.R.W. Acid-base equilibrium of drugs in time-resolved fluorescence measurements: Theoretical aspects and expressions for apparent pK(a) shifts. J. Photoch. Photobio. A 2011, 222, 10–15. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: New York, NY, USA, 2006. [Google Scholar]
- Dobrev, V.S.; Fred, L.M.; Gerhart, K.P.; Metallo, S.J. Characterization of the Binding of Small Molecules to Intrinsically Disordered Proteins. Method Enzym. 2018, 611, 677–702. [Google Scholar] [CrossRef]
- Jarmoskaite, I.; AlSadhan, I.; Vaidyanathan, P.P.; Herschlag, D. How to measure and evaluate binding affinities. eLife 2020, 9, e57264. [Google Scholar] [CrossRef]
- Follis, A.V.; Hammoudeh, D.I.; Wang, H.B.; Prochownik, E.V.; Metallo, S.J. Structural Rationale for the Coupled Binding and Unfolding of the c-Myc Oncoprotein by Small Molecules. Chem. Biol. 2008, 15, 1149–1155. [Google Scholar] [CrossRef] [Green Version]
- Heller, G.T.; Aprile, F.A.; Bonomi, M.; Camilloni, C.; De Simone, A.; Vendruscolo, M. Sequence Specificity in the Entropy-Driven Binding of a Small Molecule and a Disordered Peptide. J. Mol. Biol. 2017, 429, 2772–2779. [Google Scholar] [CrossRef]
- Panova, S.; Cliff, M.J.; Macek, P.; Blackledge, M.; Jensen, M.R.; Nissink, J.W.M.; Embrey, K.J.; Davies, R.; Waltho, J.P. Mapping Hidden Residual Structure within the Myc bHLH-LZ Domain Using Chemical Denaturant Titration. Structure 2019, 27, 1537–1546. [Google Scholar] [CrossRef]
- Macek, P.; Cliff, M.J.; Embrey, K.J.; Holdgate, G.A.; Nissink, J.W.M.; Panova, S.; Waltho, J.P.; Davies, R.A. Myc phosphorylation in its basic helix-loop-helix region destabilizes transient -helical structures, disrupting Max and DNA binding. J. Biol. Chem. 2018, 293, 9301–9310. [Google Scholar] [CrossRef] [Green Version]
- Sammak, S.; Hamdani, N.; Gorrec, F.; Allen, M.D.; Freund, S.M.V.; Bycroft, M.; Zinzalla, G. Crystal Structures and Nuclear Magnetic Resonance Studies of the Apo Form of the c-MYC:MAX bHLHZip Complex Reveal a Helical Basic Region in the Absence of DNA. Biochemistry 2019, 58, 3144–3154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.K.; Burley, S.K. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 2003, 112, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pursglove, S.E.; Fladvad, M.; Bellanda, M.; Moshref, A.; Henriksson, M.; Carey, J.; Sunnerhagen, M. Biophysical properties of regions flanking the bHLH-Zip motif in the p22 Max protein. Biochem. Bioph. Res. Com. 2004, 323, 750–759. [Google Scholar] [CrossRef]
- Brownlie, P.; Ceska, T.A.; Lamers, M.; Romier, C.; Stier, G.; Teo, H.; Suck, D. The crystal structure of an intact human Max-DNA complex: New insights into mechanisms of transcriptional control. Structure 1997, 5, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Naud, J.F.; McDuff, F.O.; Sauve, S.; Montagne, M.; Webb, B.A.; Smith, S.P.; Chabot, B.; Lavigne, P. Structural and thermodynamical characterization of the complete p21 gene product of Max. Biochemistry 2005, 44, 12746–12758. [Google Scholar] [CrossRef] [PubMed]
- Tchan, M.C.; Weiss, A.S. Asn(78) and His(81) form a destabilizing locus within the Max HLH-LZ homodimer. Febs. Lett. 2001, 509, 177–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Fan, S.J.; Prochownik, E.V. Distinct roles for MAX protein isoforms in proliferation and apoptosis. J. Biol. Chem. 1997, 272, 17416–17424. [Google Scholar] [CrossRef] [Green Version]
- Kizilsavas, G.; Ledolter, K.; Kurzbach, D. Hydrophobic Collapse of the Intrinsically Disordered Transcription Factor Myc Associated Factor X. Biochemistry 2017, 56, 5365–5372. [Google Scholar] [CrossRef] [Green Version]
- Crabtree, M.D.; Borcherds, W.; Poosapati, A.; Shammas, S.L.; Daughdrill, G.W.; Clarke, J. Conserved Helix-Flanking Prolines Modulate Intrinsically Disordered Protein:Target Affinity by Altering the Lifetime of the Bound Complex. Biochemistry 2017, 56, 2379–2384. [Google Scholar] [CrossRef]
- Das, R.K.; Crick, S.L.; Pappu, R.V. N-Terminal Segments Modulate the alpha-Helical Propensities of the Intrinsically Disordered Basic Regions of bZIP Proteins. J. Mol. Biol. 2012, 416, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaiprashad, R.; De Silva, S.R.; Fred Lucena, L.M.; Meyer, E.; Metallo, S.J. Portability of a Small-Molecule Binding Site between Disordered Proteins. Biomolecules 2022, 12, 1887. https://doi.org/10.3390/biom12121887
Jaiprashad R, De Silva SR, Fred Lucena LM, Meyer E, Metallo SJ. Portability of a Small-Molecule Binding Site between Disordered Proteins. Biomolecules. 2022; 12(12):1887. https://doi.org/10.3390/biom12121887
Chicago/Turabian StyleJaiprashad, Rajesh, Sachith Roch De Silva, Lisette M. Fred Lucena, Ella Meyer, and Steven J. Metallo. 2022. "Portability of a Small-Molecule Binding Site between Disordered Proteins" Biomolecules 12, no. 12: 1887. https://doi.org/10.3390/biom12121887
APA StyleJaiprashad, R., De Silva, S. R., Fred Lucena, L. M., Meyer, E., & Metallo, S. J. (2022). Portability of a Small-Molecule Binding Site between Disordered Proteins. Biomolecules, 12(12), 1887. https://doi.org/10.3390/biom12121887