



Single-Stranded DNA Binding Proteins and Their Identification Using Machine Learning-Based Approaches

Abstract
:1. Introduction
2. SSB Structure, Binding Specificity and Function
2.1. Structural Folds of ssDNA Binding Domains
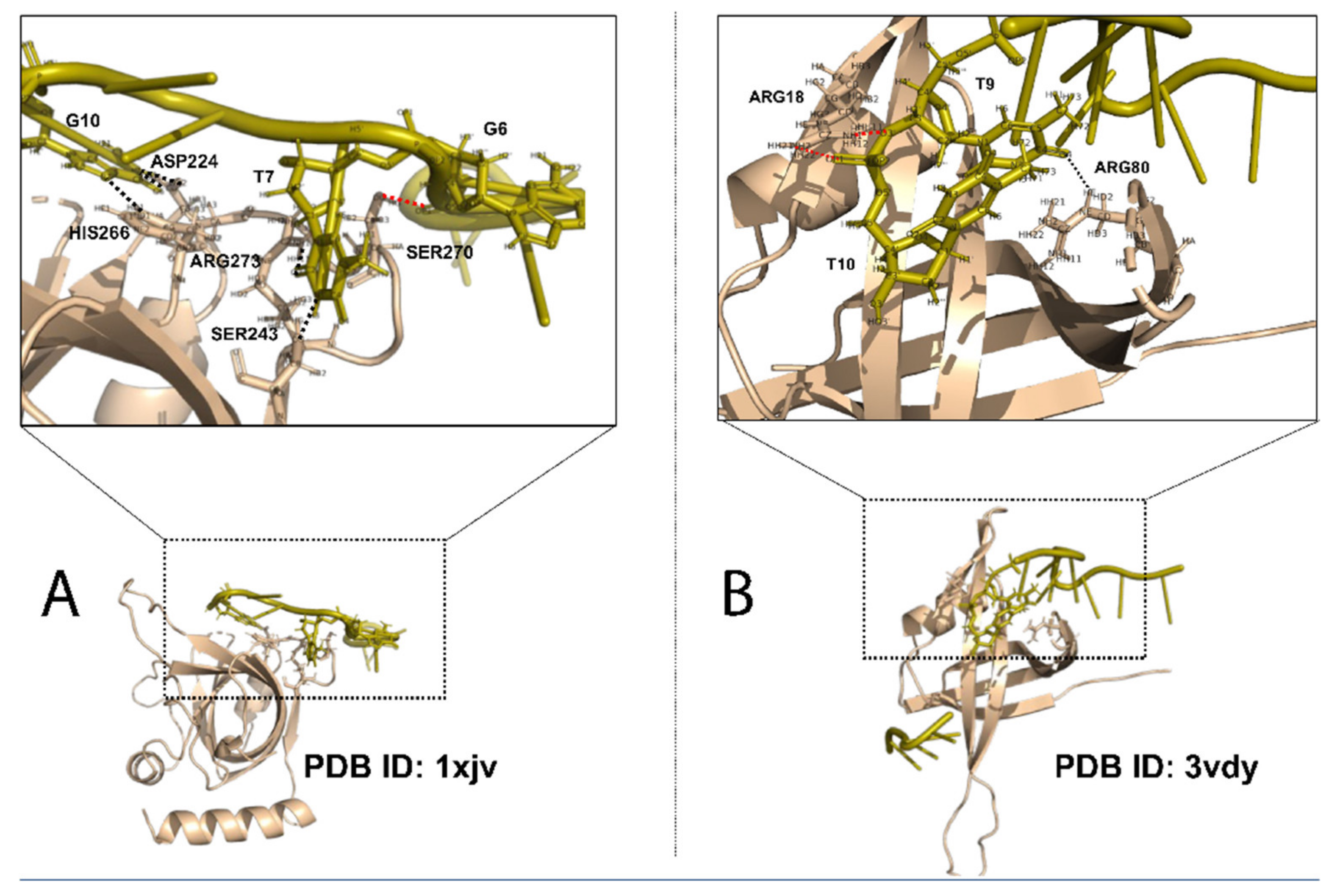
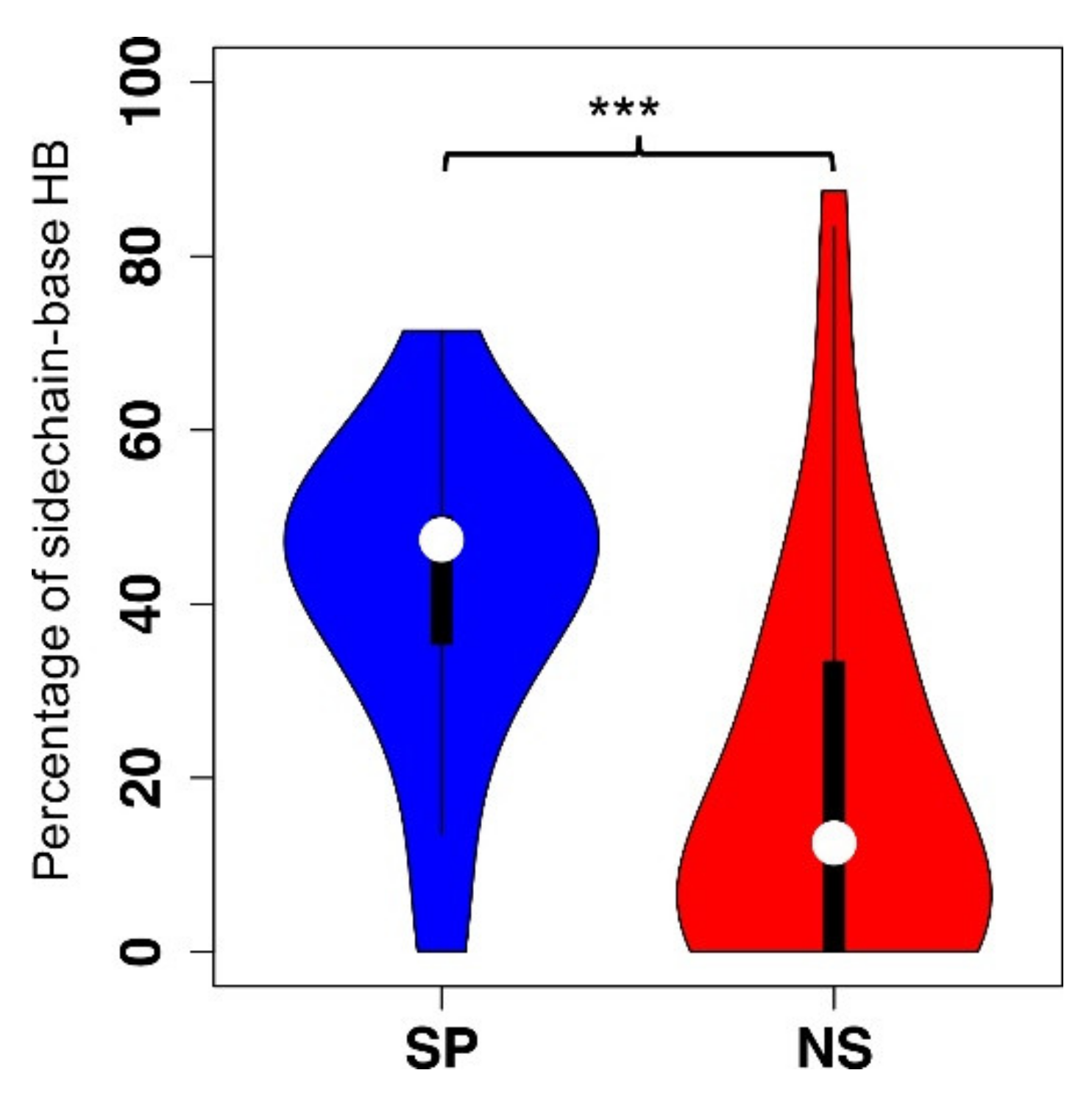
2.2. Structural Features in SSB Binding Specificity
2.3. Role of ssDNA and SSBs in Transcription Regulation
2.4. SSBs and Diseases
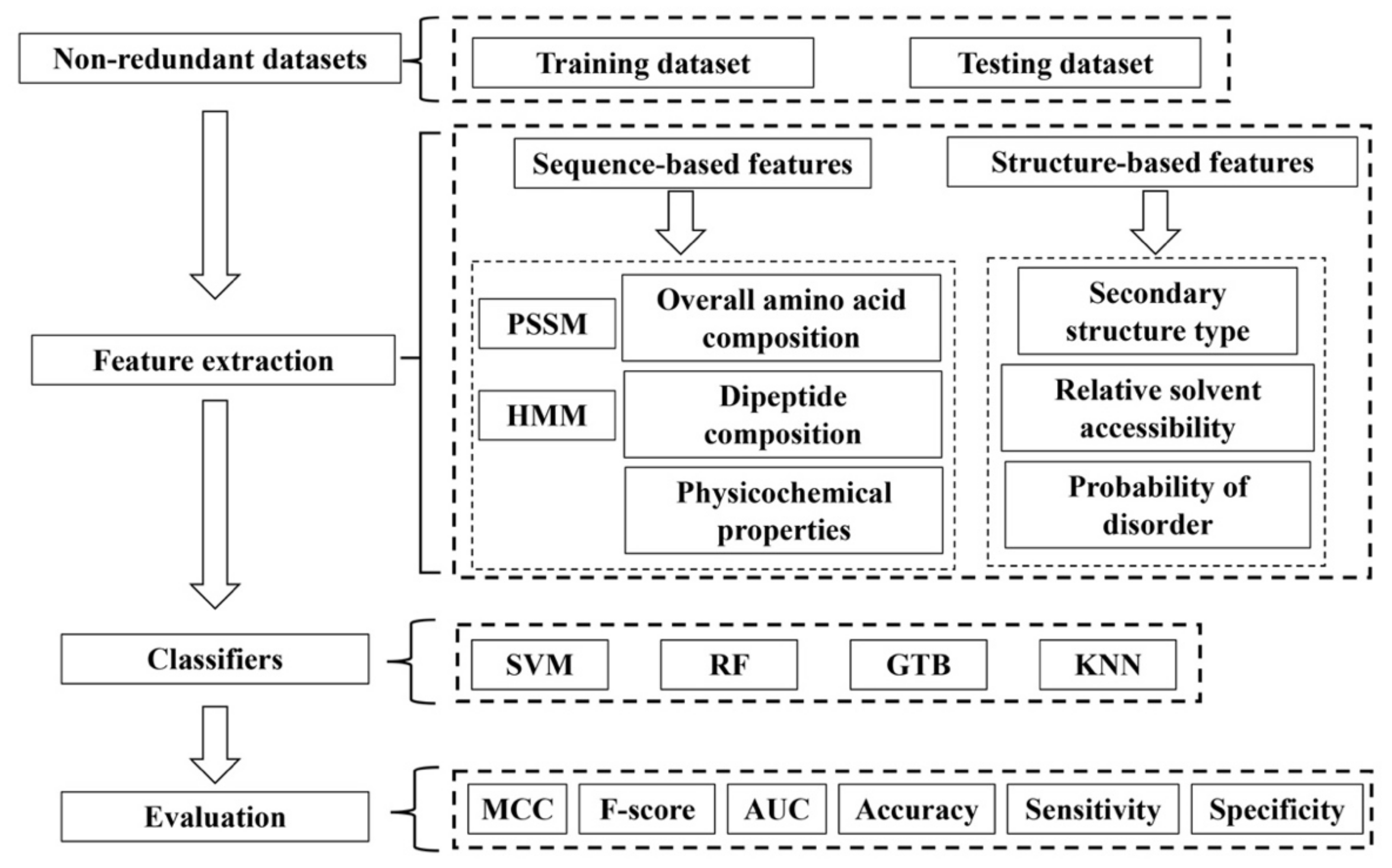
3. Machine Learning-Based Methods for SSB Prediction
3.1. Datasets
3.2. Features
3.3. Classification Models
3.4. Performance Evaluation
4. Challenges and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Dickey, T.H.; Altschuler, S.E.; Wuttke, D.S. Single-stranded DNA-binding proteins: Multiple domains for multiple functions. Structure 2013, 21, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Mishra, G.; Levy, Y. Molecular determinants of the interactions between proteins and ssDNA. Proc. Natl. Acad. Sci. USA 2015, 112, 5033–5038. [Google Scholar] [CrossRef] [PubMed]
- Eoff, R.L.; Raney, K.D. A catch and release program for single-stranded DNA. J. Biol. Chem. 2017, 292, 13085–13086. [Google Scholar] [CrossRef]
- Ashton, N.W.; Bolderson, E.; Cubeddu, L.; O’Byrne, K.J.; Richard, D.J. Human single- stranded DNA binding proteins are essential for maintaining genomic stability. BMC Mol. Biol. 2013, 14, 9. [Google Scholar] [CrossRef]
- Mortusewicz, O.; Evers, B.; Helleday, T. PC4 promotes genome stability and DNA repair through binding of ssDNA at DNA damage sites. Oncogene 2016, 35, 761–770. [Google Scholar] [CrossRef]
- Croft, L.V.; Bolderson, E.; Adams, M.N.; El-Kamand, S.; Kariawasam, R.; Cubeddu, L.; Gamsjaeger, R.; Richard, D.J. Human single-stranded DNA binding protein 1 (hSSB1, OBFC2B), a critical component of the DNA damage response. Semin. Cell Dev. Biol. 2018, 86, 121–128. [Google Scholar] [CrossRef]
- Croy, J.E.; Wuttke, D.S. Themes in ssDNA recognition by telomere-end protection proteins. Trends Biochem. Sci. 2006, 31, 516–525. [Google Scholar] [CrossRef]
- Lloyd, N.R.; Dickey, T.H.; Hom, R.A.; Wuttke, D.S. Tying up the Ends: Plasticity in the Recognition of Single-Stranded DNA at Telomeres. Biochemistry 2016, 55, 5326–5340. [Google Scholar] [CrossRef]
- Alberts, B.M.; Frey, L. T4 bacteriophage gene 32: A structural protein in the replication and recombination of DNA. Nature 1970, 227, 1313–1318. [Google Scholar] [CrossRef]
- Sigal, N.; Delius, H.; Kornberg, T.; Gefter, M.L.; Alberts, B. A DNA-unwinding protein isolated from Escherichia coli: Its interaction with DNA and with DNA polymerases. Proc. Natl. Acad. Sci. USA 1972, 69, 3537–3541. [Google Scholar] [CrossRef]
- Overman, L.B.; Lohman, T.M. Linkage of pH, anion and cation effects in protein-nucleic acid equilibria. Escherichia coli SSB protein-single stranded nucleic acid interactions. J. Mol. Biol. 1994, 236, 165–178. [Google Scholar] [CrossRef]
- Wobbe, C.R.; Weissbach, L.; Borowiec, J.A.; Dean, F.B.; Murakami, Y.; Bullock, P.; Hurwitz, J. Replication of simian virus 40 origin-containing DNA in vitro with purified proteins. Proc. Natl. Acad. Sci. USA 1987, 84, 1834–1838. [Google Scholar] [CrossRef]
- Wold, M.S.; Kelly, T. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc. Natl. Acad. Sci. USA 1988, 85, 2523–2527. [Google Scholar] [CrossRef]
- Fairman, M.P.; Stillman, B. Cellular factors required for multiple stages of SV40 DNA replication in vitro. EMBO J. 1988, 7, 1211–1218. [Google Scholar] [CrossRef]
- Dean, F.B.; Bullock, P.; Murakami, Y.; Wobbe, C.R.; Weissbach, L.; Hurwitz, J. Simian virus 40 (SV40) DNA replication: SV40 large T antigen unwinds DNA containing the SV40 origin of replication. Proc. Natl. Acad. Sci. USA 1987, 84, 16–20. [Google Scholar] [CrossRef]
- Wu, Y.; Lu, J.; Kang, T. Human single-stranded DNA binding proteins: Guardians of genome stability. Acta Biochim. Biophys. Sin. 2016, 48, 671–677. [Google Scholar] [CrossRef]
- Richard, D.J.; Bolderson, E.; Cubeddu, L.; Wadsworth, R.I.; Savage, K.; Sharma, G.G.; Nicolette, M.L.; Tsvetanov, S.; McIlwraith, M.J.; Pandita, R.K.; et al. Single-stranded DNA-binding protein hSSB1 is critical for genomic stability. Nature 2008, 453, 677–681. [Google Scholar] [CrossRef]
- Bunch, J.T.; Bae, N.S.; Leonardi, J.; Baumann, P. Distinct requirements for Pot1 in limiting telomere length and maintaining chromosome stability. Mol. Cell Biol. 2005, 25, 5567–5578. [Google Scholar] [CrossRef]
- Veldman, T.; Etheridge, K.T.; Counter, C.M. Loss of hPot1 function leads to telomere instability and a cut-like phenotype. Curr. Biol. 2004, 14, 2264–2270. [Google Scholar] [CrossRef] [Green Version]
- Murzin, A.G. OB (oligonucleotide/oligosaccharide binding)-fold: Common structural and functional solution for non-homologous sequences. EMBO J. 1993, 12, 861–867. [Google Scholar] [CrossRef]
- Gamsjaeger, R.; Kariawasam, R.; Gimenez, A.X.; Touma, C.; McIlwain, E.; Bernardo, R.E.; Shepherd, N.E.; Ataide, S.F.; Dong, Q.; Richard, D.J.; et al. The structural basis of DNA binding by the single-stranded DNA-binding protein from Sulfolobus solfataricus. Biochem. J. 2015, 465, 337–346. [Google Scholar] [CrossRef]
- Corona, R.I.; Guo, J.T. Statistical analysis of structural determinants for protein-DNA-binding specificity. Proteins 2016, 84, 1147–1161. [Google Scholar] [CrossRef]
- Rohs, R.; Jin, X.; West, S.M.; Joshi, R.; Honig, B.; Mann, R.S. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010, 79, 233–269. [Google Scholar] [CrossRef] [PubMed]
- Corona, R.I.; Sudarshan, S.; Aluru, S.; Guo, J.T. An SVM-based method for assessment of transcription factor-DNA complex models. BMC Bioinform. 2018, 19, 506. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Guo, J.T. New insights into protein-DNA binding specificity from hydrogen bond based comparative study. Nucleic Acids Res. 2019, 47, 11103–11113. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Malik, F.K.; Guo, J.T. A comparative study of protein-ssDNA interactions. NAR Genom. Bioinform. 2021, 3, lqab006. [Google Scholar] [CrossRef]
- Malik, F.K.; Guo, J.T. Insights into protein-DNA interactions from hydrogen bond energy-based comparative protein-ligand analyses. Proteins 2022, 90, 1303–1314. [Google Scholar] [CrossRef]
- Angarica, V.E.; Perez, A.G.; Vasconcelos, A.T.; Collado-Vides, J.; Contreras-Moreira, B. Prediction of TF target sites based on atomistic models of protein-DNA complexes. BMC Bioinform. 2008, 9, 436. [Google Scholar] [CrossRef]
- Luscombe, N.M.; Laskowski, R.A.; Thornton, J.M. Amino acid-base interactions: A three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 2001, 29, 2860–2874. [Google Scholar] [CrossRef] [Green Version]
- Seeman, N.C.; Rosenberg, J.M.; Rich, A. Sequence-specific recognition of double helical nucleic acids by proteins. Proc. Natl. Acad. Sci. USA 1976, 73, 804–808. [Google Scholar] [CrossRef]
- Lei, M.; Podell, E.R.; Cech, T.R. Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat. Struct. Mol. Biol. 2004, 11, 1223–1229. [Google Scholar] [CrossRef]
- Bochkarev, A.; Pfuetzner, R.A.; Edwards, A.M.; Frappier, L. Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature 1997, 385, 176–181. [Google Scholar] [CrossRef]
- Yadav, T.; Carrasco, B.; Myers, A.R.; George, N.P.; Keck, J.L.; Alonso, J.C. Genetic recombination in Bacillus subtilis: A division of labor between two single-strand DNA-binding proteins. Nucleic Acids Res. 2012, 40, 5546–5559. [Google Scholar] [CrossRef]
- Cernooka, E.; Rumnieks, J.; Tars, K.; Kazaks, A. Structural Basis for DNA Recognition of a Single-stranded DNA-binding Protein from Enterobacter Phage Enc34. Sci. Rep. 2017, 7, 15529. [Google Scholar] [CrossRef]
- Crichlow, G.V.; Zhou, H.; Hsiao, H.H.; Frederick, K.B.; Debrosse, M.; Yang, Y.; Folta-Stogniew, E.J.; Chung, H.J.; Fan, C.; De la Cruz, E.M.; et al. Dimerization of FIR upon FUSE DNA binding suggests a mechanism of c-myc inhibition. EMBO J. 2008, 27, 277–289. [Google Scholar] [CrossRef]
- Myers, J.C.; Shamoo, Y. Human UP1 as a model for understanding purine recognition in the family of proteins containing the RNA recognition motif (RRM). J. Mol. Biol. 2004, 342, 743–756. [Google Scholar] [CrossRef]
- Soufari, H.; Mackereth, C.D. Conserved binding of GCAC motifs by MEC-8, couch potato, and the RBPMS protein family. RNA 2017, 23, 308–316. [Google Scholar] [CrossRef]
- Amrane, S.; Rebora, K.; Zniber, I.; Dupuy, D.; Mackereth, C.D. Backbone-independent nucleic acid binding by splicing factor SUP-12 reveals key aspects of molecular recognition. Nat. Commun. 2014, 5, 4595. [Google Scholar] [CrossRef]
- Joshi, R.; Passner, J.M.; Rohs, R.; Jain, R.; Sosinsky, A.; Crickmore, M.A.; Jacob, V.; Aggarwal, A.K.; Honig, B.; Mann, R.S. Functional specificity of a Hox protein mediated by the recognition of minor groove structure. Cell 2007, 131, 530–543. [Google Scholar] [CrossRef] [Green Version]
- Rohs, R.; West, S.M.; Liu, P.; Honig, B. Nuance in the double-helix and its role in protein-DNA recognition. Curr. Opin. Struct. Biol. 2009, 19, 171–177. [Google Scholar] [CrossRef]
- Rohs, R.; West, S.M.; Sosinsky, A.; Liu, P.; Mann, R.S.; Honig, B. The role of DNA shape in protein-DNA recognition. Nature 2009, 461, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Orenstein, Y.; Jolma, A.; Yin, Y.; Taipale, J.; Shamir, R.; Rohs, R. Transcription factor family-specific DNA shape readout revealed by quantitative specificity models. Mol. Syst. Biol. 2017, 13, 910. [Google Scholar] [CrossRef]
- Luscombe, N.M.; Thornton, J.M. Protein-DNA interactions: Amino acid conservation and the effects of mutations on binding specificity. J. Mol. Biol. 2002, 320, 991–1009. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Berman, H.M.; Christie, C.; Duarte, J.M.; Feng, Z.; Westbrook, J.; Young, J.; Zardecki, C. RCSB Protein Data Bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci. 2018, 27, 316–330. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Wang, W.; Liu, J.; Sun, L. Surface shapes and surrounding environment analysis of single- and double-stranded DNA-binding proteins in protein-DNA interface. Proteins 2016, 84, 979–989. [Google Scholar] [CrossRef]
- Swamynathan, S.K.; Nambiar, A.; Guntaka, R.V. Role of single-stranded DNA regions and Y-box proteins in transcriptional regulation of viral and cellular genes. FASEB J. 1998, 12, 515–522. [Google Scholar] [CrossRef]
- Duncan, R.; Bazar, L.; Michelotti, G.; Tomonaga, T.; Krutzsch, H.; Avigan, M.; Levens, D. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 1994, 8, 465–480. [Google Scholar] [CrossRef] [Green Version]
- Tomonaga, T.; Levens, D. Activating transcription from single stranded DNA. Proc. Natl. Acad. Sci. USA 1996, 93, 5830–5835. [Google Scholar] [CrossRef]
- Gupta, M.; Sueblinvong, V.; Raman, J.; Jeevanandam, V.; Gupta, M.P. Single-stranded DNA-binding proteins PURalpha and PURbeta bind to a purine-rich negative regulatory element of the alpha-myosin heavy chain gene and control transcriptional and translational regulation of the gene expression. Implications in the repression of alpha-myosin heavy chain during heart failure. J. Biol. Chem. 2003, 278, 44935–44948. [Google Scholar]
- Thakur, S.; Nakamura, T.; Calin, G.; Russo, A.; Tamburrino, J.F.; Shimizu, M.; Baldassarre, G.; Battista, S.; Fusco, A.; Wassell, R.P.; et al. Regulation of BRCA1 transcription by specific single-stranded DNA binding factors. Mol. Cell Biol. 2003, 23, 3774–3787. [Google Scholar] [CrossRef]
- Phillips, B.W.; Sharma, R.; Leco, P.A.; Edwards, D.R. A sequence-selective single-strand DNA-binding protein regulates basal transcription of the murine tissue inhibitor of metalloproteinases-1 (Timp-1) gene. J. Biol. Chem. 1999, 274, 22197–22207. [Google Scholar] [CrossRef]
- Ko, J.L.; Loh, H.H. Single-stranded DNA-binding complex involved in transcriptional regulation of mouse mu-opioid receptor gene. J. Biol. Chem. 2001, 276, 788–795. [Google Scholar] [CrossRef]
- Desveaux, D.; Despres, C.; Joyeux, A.; Subramaniam, R.; Brisson, N. PBF-2 is a novel single-stranded DNA binding factor implicated in PR-10a gene activation in potato. Plant. Cell 2000, 12, 1477–1489. [Google Scholar] [CrossRef]
- Boyle, B.; Brisson, N. Repression of the defense gene PR-10a by the single-stranded DNA binding protein SEBF. Plant. Cell 2001, 13, 2525–2537. [Google Scholar] [CrossRef]
- Desveaux, D.; Allard, J.; Brisson, N.; Sygusch, J. A new family of plant transcription factors displays a novel ssDNA-binding surface. Nat. Struct. Biol. 2002, 9, 512–517. [Google Scholar] [CrossRef]
- Grabowski, E.; Miao, Y.; Mulisch, M.; Krupinska, K. Single-stranded DNA-binding protein Whirly1 in barley leaves is located in plastids and the nucleus of the same cell. Plant Physiol. 2008, 147, 1800–1804. [Google Scholar] [CrossRef]
- Richard, D.J.; Bell, S.D.; White, M.F. Physical and functional interaction of the archaeal single-stranded DNA-binding protein SSB with RNA polymerase. Nucleic Acids Res. 2004, 32, 1065–1074. [Google Scholar] [CrossRef]
- Liu, J.; Kouzine, F.; Nie, Z.; Chung, H.J.; Elisha-Feil, Z.; Weber, A.; Zhao, K.; Levens, D. The FUSE/FBP/FIR/TFIIH system is a molecular machine programming a pulse of c-myc expression. EMBO J. 2006, 25, 2119–2130. [Google Scholar] [CrossRef] [Green Version]
- Michelotti, E.F.; Michelotti, G.A.; Aronsohn, A.I.; Levens, D. Heterogeneous nuclear ribonucleoprotein K is a transcription factor. Mol. Cell Biol. 1996, 16, 2350–2360. [Google Scholar] [CrossRef]
- Yoo, H.H.; Kwon, C.; Lee, M.M.; Chung, I.K. Single-stranded DNA binding factor AtWHY1 modulates telomere length homeostasis in Arabidopsis. Plant J. 2007, 49, 442–451. [Google Scholar] [CrossRef]
- Wang, Y.; Putnam, C.D.; Kane, M.F.; Zhang, W.; Edelmann, L.; Russell, R.; Carrion, D.V.; Chin, L.; Kucherlapati, R.; Kolodner, R.D.; et al. Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability and cancer in mice. Nat. Genet. 2005, 37, 750–755. [Google Scholar] [CrossRef]
- Shi, W.; Bain, A.L.; Schwer, B.; Al-Ejeh, F.; Smith, C.; Wong, L.; Chai, H.; Miranda, M.S.; Ho, U.; Kawaguchi, M.; et al. Essential developmental, genomic stability, and tumour suppressor functions of the mouse orthologue of hSSB1/NABP2. PLoS Genet. 2013, 9, e1003298. [Google Scholar] [CrossRef]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar] [CrossRef]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef]
- Thorslund, T.; McIlwraith, M.J.; Compton, S.A.; Lekomtsev, S.; Petronczki, M.; Griffith, J.D.; West, S.C. The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1263–1265. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Tumour suppressor mechanisms in the control of chromosome stability: Insights from BRCA2. Mol. Cells 2014, 37, 95–99. [Google Scholar] [CrossRef]
- Zamborszky, J.; Szikriszt, B.; Gervai, J.Z.; Pipek, O.; Poti, A.; Krzystanek, M.; Ribli, D.; Szalai-Gindl, J.M.; Csabai, I.; Szallasi, Z.; et al. Loss of BRCA1 or BRCA2 markedly increases the rate of base substitution mutagenesis and has distinct effects on genomic deletions. Oncogene 2017, 36, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Shuck, S.C.; Turchi, J.J. Targeted inhibition of Replication Protein A reveals cytotoxic activity, synergy with chemotherapeutic DNA-damaging agents, and insight into cellular function. Cancer Res. 2010, 70, 3189–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kustatscher, G.; Collins, T.; Gingras, A.C.; Guo, T.; Hermjakob, H.; Ideker, T.; Lilley, K.S.; Lundberg, E.; Marcotte, E.M.; Ralser, M.; et al. Understudied proteins: Opportunities and challenges for functional proteomics. Nat. Methods 2022, 19, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Kustatscher, G.; Collins, T.; Gingras, A.C.; Guo, T.; Hermjakob, H.; Ideker, T.; Lilley, K.S.; Lundberg, E.; Marcotte, E.M.; Ralser, M.; et al. An open invitation to the Understudied Proteins Initiative. Nat. Biotechnol. 2022, 40, 815–817. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M. Nature of the protein universe. Proc. Natl. Acad Sci. USA 2009, 106, 11079–11084. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Koonin, E.V. ‘Conserved hypothetical’ proteins: Prioritization of targets for experimental study. Nucleic Acids Res. 2004, 32, 5452–5463. [Google Scholar] [CrossRef]
- Shumilin, I.A.; Cymborowski, M.; Chertihin, O.; Jha, K.N.; Herr, J.C.; Lesley, S.A.; Joachimiak, A.; Minor, W. Identification of unknown protein function using metabolite cocktail screening. Structure 2012, 20, 1715–1725. [Google Scholar] [CrossRef]
- Ellens, K.W.; Christian, N.; Singh, C.; Satagopam, V.P.; May, P.; Linster, C.L. Confronting the catalytic dark matter encoded by sequenced genomes. Nucleic Acids Res. 2017, 45, 11495–11514. [Google Scholar] [CrossRef]
- AlQuraishi, M. Machine learning in protein structure prediction. Curr. Opin. Chem. Biol. 2021, 65, 1–8. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Wang, S.; Sun, S.; Li, Z.; Zhang, R.; Xu, J. Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model. PLoS Comput. Biol. 2017, 13, e1005324. [Google Scholar] [CrossRef]
- Xu, J. Distance-based protein folding powered by deep learning. Proc. Natl. Acad. Sci. USA 2019, 116, 16856–16865. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.K.; Pugalenthi, G.; Suganthan, P.N. DNA-Prot: Identification of DNA binding proteins from protein sequence information using random forest. J. Biomol. Struct. Dyn. 2009, 26, 679–686. [Google Scholar] [CrossRef]
- Kumar, M.; Gromiha, M.M.; Raghava, G.P. Identification of DNA-binding proteins using support vector machines and evolutionary profiles. BMC Bioinform. 2007, 8, 463. [Google Scholar] [CrossRef]
- Qiu, J.; Bernhofer, M.; Heinzinger, M.; Kemper, S.; Norambuena, T.; Melo, F.; Rost, B. ProNA2020 predicts protein-DNA, protein-RNA, and protein-protein binding proteins and residues from sequence. J. Mol. Biol. 2020, 432, 2428–2443. [Google Scholar] [CrossRef]
- Xu, R.; Zhou, J.; Wang, H.; He, Y.; Wang, X.; Liu, B. Identifying DNA-binding proteins by combining support vector machine and PSSM distance transformation. BMC Syst. Biol. 2015, 9 (Suppl. S1), S10. [Google Scholar] [CrossRef]
- Ali, F.; Ahmed, S.; Swati, Z.N.K.; Akbar, S. DP-BINDER: Machine learning model for prediction of DNA-binding proteins by fusing evolutionary and physicochemical information. J. Comput. Aided. Mol. Des. 2019, 33, 645–658. [Google Scholar] [CrossRef]
- Hu, S.; Ma, R.; Wang, H. An improved deep learning method for predicting DNA- binding proteins based on contextual features in amino acid sequences. PLoS ONE 2019, 14, e0225317. [Google Scholar] [CrossRef]
- Lou, W.; Wang, X.; Chen, F.; Chen, Y.; Jiang, B.; Zhang, H. Sequence based prediction of DNA-binding proteins based on hybrid feature selection using random forest and Gaussian naive Bayes. PLoS ONE 2014, 9, e86703. [Google Scholar] [CrossRef]
- Ma, X.; Guo, J.; Sun, X. DNABP: Identification of DNA-Binding Proteins Based on Feature Selection Using a Random Forest and Predicting Binding Residues. PLoS ONE 2016, 11, e0167345. [Google Scholar] [CrossRef]
- Mishra, A.; Pokhrel, P.; Hoque, M.T. StackDPPred: A stacking based prediction of DNA-binding protein from sequence. Bioinformatics 2019, 35, 433–441. [Google Scholar] [CrossRef]
- Motion, G.B.; Howden, A.J.; Huitema, E.; Jones, S. DNA-binding protein prediction using plant specific support vector machines: Validation and application of a new genome annotation tool. Nucleic Acids Res. 2015, 43, e158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.H.; Yu, H.; Gong, X.J.; Xu, J.H.; Lee, H.S. On the prediction of DNA-binding proteins only from primary sequences: A deep learning approach. PLoS ONE 2017, 12, e0188129. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Du, X.; Li, X.; Zou, L.; Zhang, G.; Wu, Z. Prediction of DNA binding proteins using local features and long-term dependencies with primary sequences based on deep learning. PeerJ 2021, 9, e11262. [Google Scholar] [CrossRef] [PubMed]
- Zaitzeff, A.; Leiby, N.; Motta, F.C.; Haase, S.B.; Singer, J.M. Improved data sets and evaluation methods for the automatic prediction of DNA-binding proteins. Bioinformatics 2021, 38, 44–51. [Google Scholar] [CrossRef]
- Wang, W.; Sun, L.; Zhang, S.; Zhang, H.; Shi, J.; Xu, T.; Li, K. Analysis and prediction of single-stranded and double-stranded DNA binding proteins based on protein sequences. BMC Bioinform. 2017, 18, 300. [Google Scholar] [CrossRef]
- Sharma, R.; Kumar, S.; Tsunoda, T.; Kumarevel, T.; Sharma, A. Single-stranded and double-stranded DNA-binding protein prediction using HMM profiles. Anal. Biochem. 2021, 612, 113954. [Google Scholar] [CrossRef]
- Ali, F.; Arif, M.; Khan, Z.U.; Kabir, M.; Ahmed, S.; Yu, D.J. SDBP-Pred: Prediction of single-stranded and double-stranded DNA-binding proteins by extending consensus sequence and K-segmentation strategies into PSSM. Anal. Biochem. 2020, 589, 113494. [Google Scholar] [CrossRef]
- Tan, C.; Wang, T.; Yang, W.; Deng, L. PredPSD: A Gradient Tree Boosting Approach for Single-Stranded and Double-Stranded DNA Binding Protein Prediction. Molecules 2019, 25, 98. [Google Scholar] [CrossRef]
- Selby, C.P.; Sancar, A. A cryptochrome/photolyase class of enzymes with single-stranded DNA-specific photolyase activity. Proc. Natl. Acad. Sci. USA 2006, 103, 17696–17700. [Google Scholar] [CrossRef]
- Pokorny, R.; Klar, T.; Hennecke, U.; Carell, T.; Batschauer, A.; Essen, L.O. Recognition and repair of UV lesions in loop structures of duplex DNA by DASH-type cryptochrome. Proc. Natl. Acad. Sci. USA 2008, 105, 21023–21027. [Google Scholar] [CrossRef]
- Bakalkin, G.; Yakovleva, T.; Selivanova, G.; Magnusson, K.P.; Szekely, L.; Kiseleva, E.; Klein, G.; Terenius, L.; Wiman, K.G. p53 binds single-stranded DNA ends and catalyzes DNA renaturation and strand transfer. Proc. Natl. Acad. Sci. USA 1994, 91, 413–417. [Google Scholar] [CrossRef] [Green Version]
- Bochkareva, E.; Kaustov, L.; Ayed, A.; Yi, G.S.; Lu, Y.; Pineda-Lucena, A.; Liao, J.C.; Okorokov, A.L.; Milner, J.; Arrowsmith, C.H.; et al. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl. Acad. Sci. USA 2005, 102, 15412–15417. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Gribskov, M.; McLachlan, A.D.; Eisenberg, D. Profile analysis: Detection of distantly related proteins. Proc. Natl. Acad. Sci. USA 1987, 84, 4355–4358. [Google Scholar] [CrossRef]
- Feng, Z.P.; Zhang, C.T. Prediction of the subcellular location of prokaryotic proteins based on the hydrophobicity index of amino acids. Int. J. Biol. Macromol. 2001, 28, 255–261. [Google Scholar] [CrossRef]
- Huang, H.L.; Lin, I.C.; Liou, Y.F.; Tsai, C.T.; Hsu, K.T.; Huang, W.L.; Ho, S.J.; Ho, S.Y. Predicting and analyzing DNA-binding domains using a systematic approach to identifying a set of informative physicochemical and biochemical properties. BMC Bioinform. 2011, 12 (Suppl. S1), S47. [Google Scholar] [CrossRef]
- Wang, S.; Peng, J.; Ma, J.; Xu, J. Protein Secondary Structure Prediction Using Deep Convolutional Neural Fields. Sci. Rep. 2016, 6, 18962. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Wang, S.; Sun, S.; Xu, J. AUC-Maximized Deep Convolutional Neural Fields for Protein Sequence Labeling. In Machine Learning and Knowledge Discovery in Databases; Springer: Berlin/Heidelberg, Germany, 2016; Volume 9852, pp. 1–16. [Google Scholar]
- Hou, Q.; De Geest, P.F.G.; Vranken, W.F.; Heringa, J.; Feenstra, K.A. Seeing the trees through the forest: Sequence-based homo- and heteromeric protein-protein interaction sites prediction using random forest. Bioinformatics 2017, 33, 1479–1487. [Google Scholar] [CrossRef]
- Jo, T.; Cheng, J. Improving protein fold recognition by random forest. BMC Bioinform. 2014, 15 (Suppl. S11), S14. [Google Scholar] [CrossRef] [Green Version]
- Noble, W.S. What is a support vector machine? Nat. Biotechnol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef]
- Cheng, J.; Baldi, P. A machine learning information retrieval approach to protein fold recognition. Bioinformatics 2006, 22, 1456–1463. [Google Scholar] [CrossRef]
- Ward, J.J.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Secondary structure prediction with support vector machines. Bioinformatics 2003, 19, 1650–1655. [Google Scholar] [CrossRef]
- Ding, C.H.; Dubchak, I. Multi-class protein fold recognition using support vector machines and neural networks. Bioinformatics 2001, 17, 349–358. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| References | Predictor (If Any) | Features | Classifiers |
|---|---|---|---|
| Wang et al. [95] | NA | -OAAC -Dipeptide composition -Physicochemical properties -PSSM | SVM RF |
| Ali et al. [97] | SDBP-Pred | -PSSM -CS-PSSM -CSS-PSSM2 -CSS-PSSM3 | SVM |
| Tan et al. [98] | PredPSD | -OAAC -Dipeptide composition -Physicochemical properties -PSSM -Structural features from NetSurfP and DisEMBL | GTB |
| Sharma et al. [96] | NA | -HMM | SVM |
| -normalized profile-monogram | RF | ||
| -normalized profile-bigram | KNN |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, J.-T.; Malik, F. Single-Stranded DNA Binding Proteins and Their Identification Using Machine Learning-Based Approaches. Biomolecules 2022, 12, 1187. https://doi.org/10.3390/biom12091187
Guo J-T, Malik F. Single-Stranded DNA Binding Proteins and Their Identification Using Machine Learning-Based Approaches. Biomolecules. 2022; 12(9):1187. https://doi.org/10.3390/biom12091187
Chicago/Turabian StyleGuo, Jun-Tao, and Fareeha Malik. 2022. "Single-Stranded DNA Binding Proteins and Their Identification Using Machine Learning-Based Approaches" Biomolecules 12, no. 9: 1187. https://doi.org/10.3390/biom12091187
APA StyleGuo, J.-T., & Malik, F. (2022). Single-Stranded DNA Binding Proteins and Their Identification Using Machine Learning-Based Approaches. Biomolecules, 12(9), 1187. https://doi.org/10.3390/biom12091187