
Changes in NMDA Receptor Function in Rapid Ischemic Tolerance: A Potential Role for Tri-Heteromeric NMDA Receptors

_Maysami.png)
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Electrophysiology
2.3. Biotinylation Assay and Streptavidin-Pulldown
2.4. Immunoprecipitation
2.5. Immunoblotting
2.6. Data Analysis
3. Results
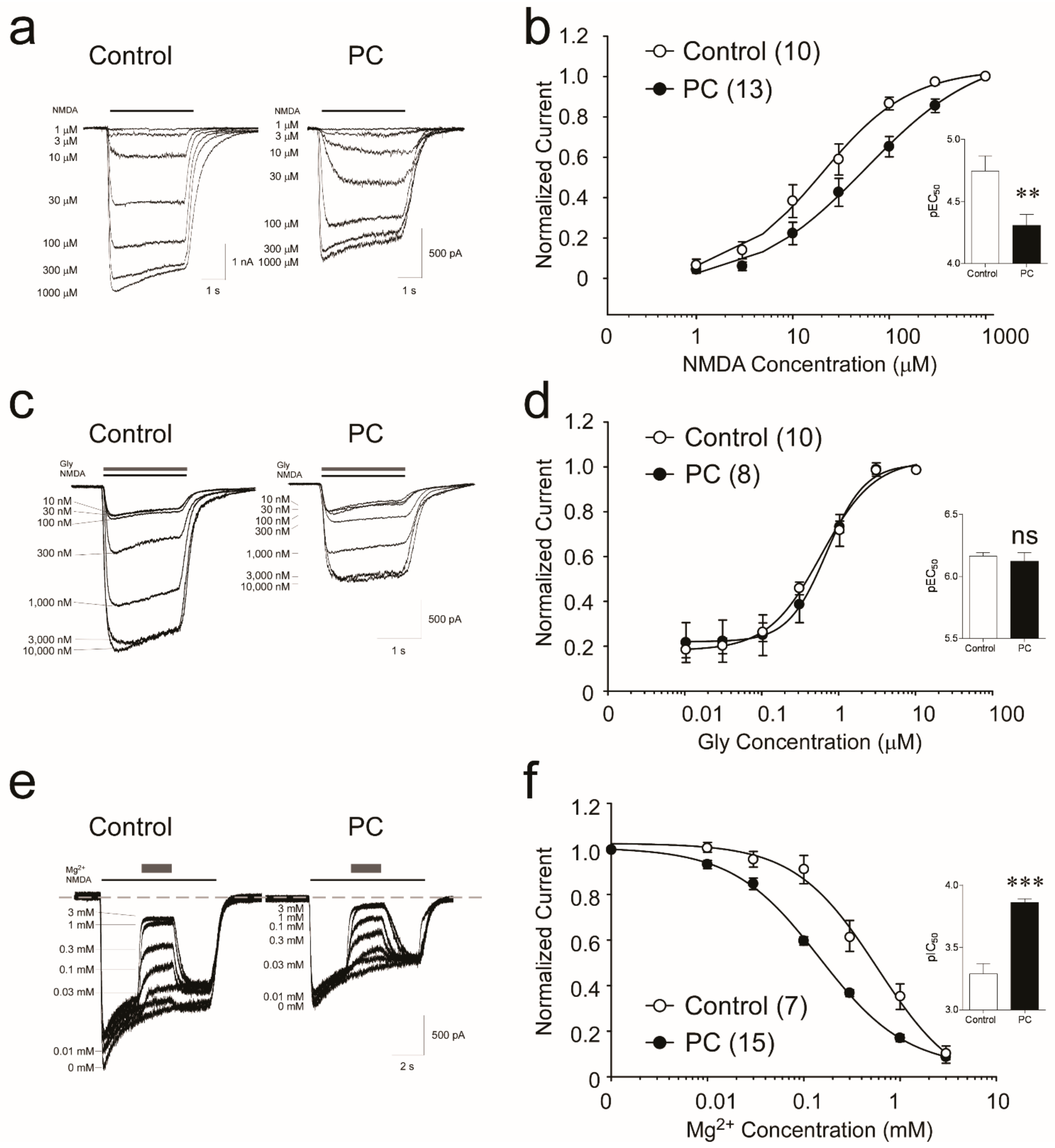
3.1. Biophysical Analysis Reveals Decreased Potency of NMDA following Preconditioning
3.2. NMDA Receptors Show Increased Sensitivity to Mg2+, but Not Glycine, following Ischemic Preconditioning
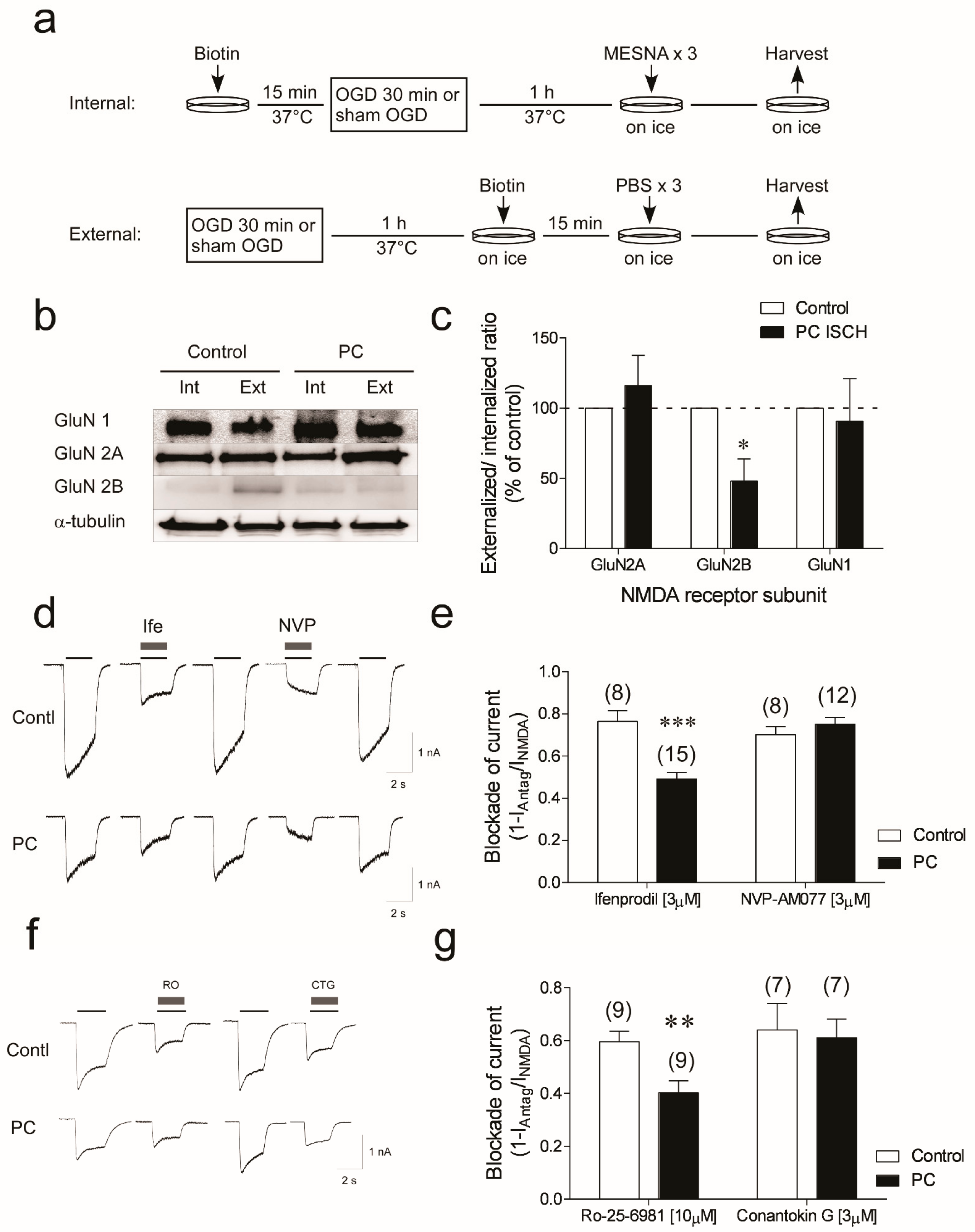
3.3. Reduction in GluN2B Membrane Localization and Functional Responses in Neurons Subjected to Ischemic Preconditioning
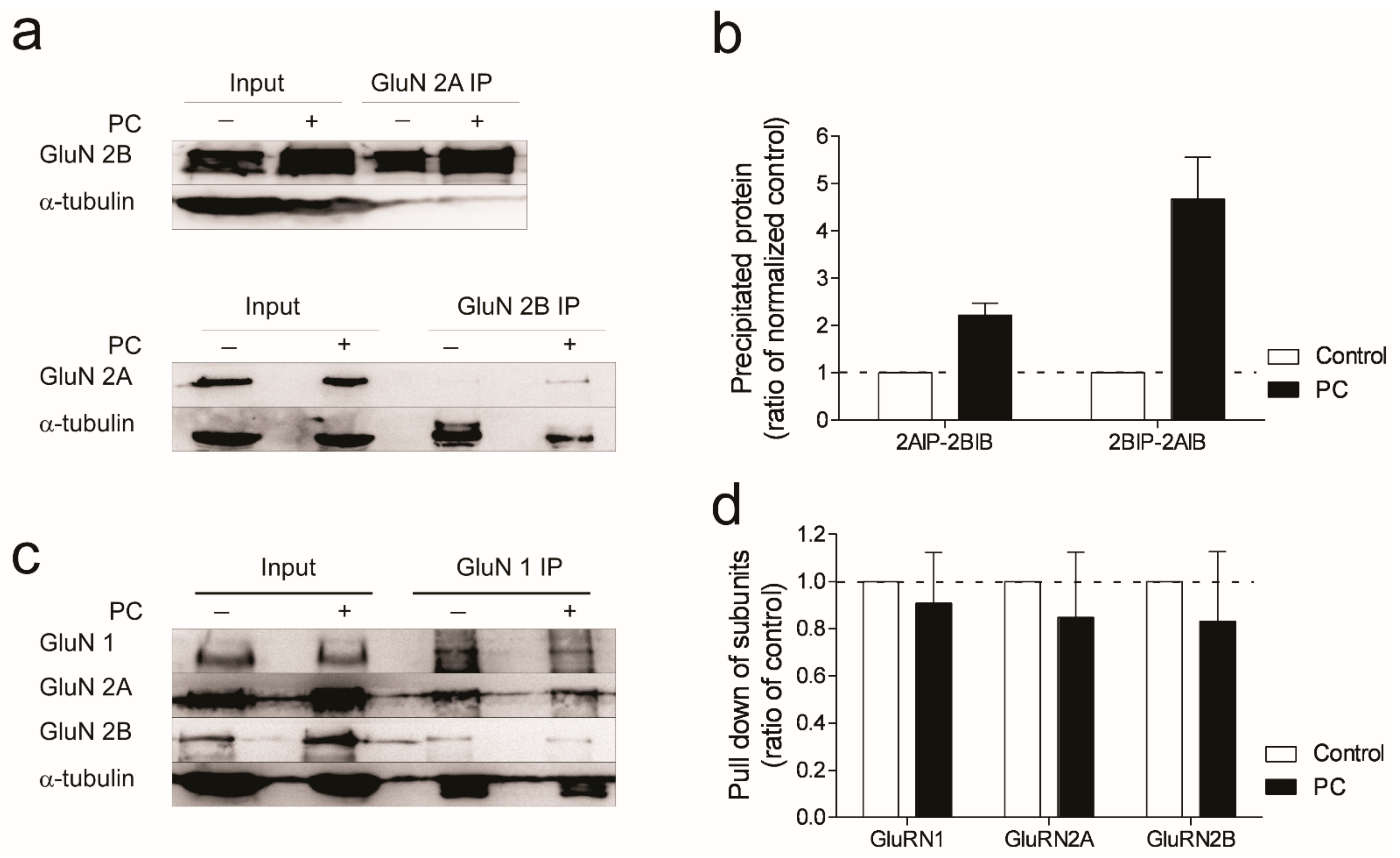
3.4. Evidence for Increased Formation of Tri-Heteromeric NMDA Receptors following Ischemic Preconditioning
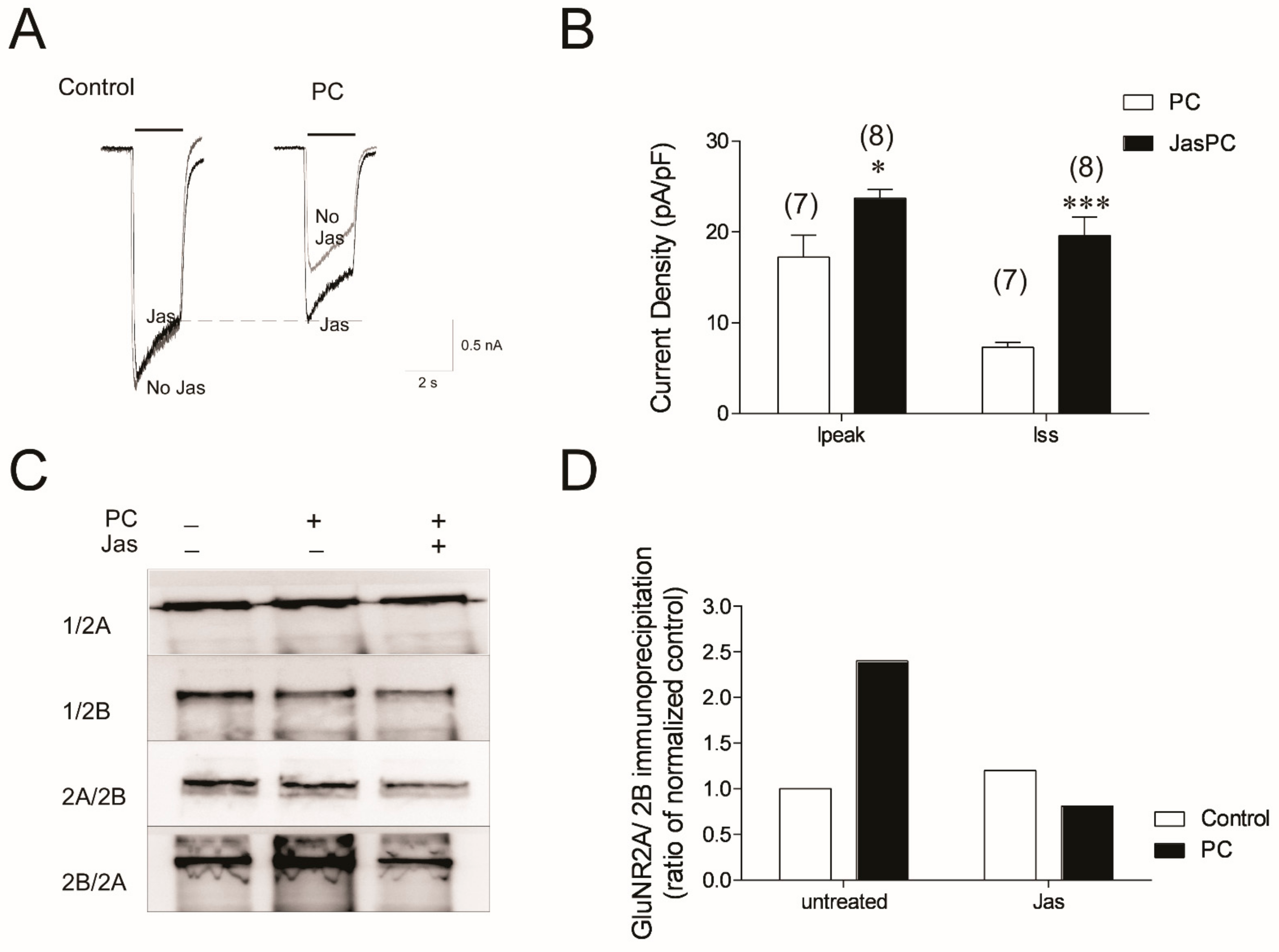
3.5. Stabilizing Actin Polymerization Partially Lessened the NMDA Current Reduction and Blocked Formation of Tri-Heteromeric NMDAR in Preconditioned Neurons
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hunt, D.L.; Castillo, P.E. Synaptic plasticity of NMDA receptors: Mechanisms and functional implications. Curr. Opin. Neurobiol. 2012, 22, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Gascon, S.; Sobrado, M.; Roda, J.M.; Rodriguez-Pena, A.; Diaz-Guerra, M. Excitotoxicity and focal cerebral ischemia induce truncation of the NR2A and NR2B subunits of the NMDA receptor and cleavage of the scaffolding protein PSD-95. Mol. Psychiatry 2007, 13, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.P.; Swan, J.H.; Griffiths, T.; Meldrum, B.S. Blockade of N-methyl-D-aspartate receptors may protect against ischemic damage in the brain. Science 1984, 226, 850–852. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.K.; Saleh, J.; DeLaPaz, R.; Kunis, D.; Zarnegar, S.R. Pretreatment with the NMDA antagonist dextrorphan reduces cerebral injury following transient focal ischemia in rabbits. Brain Res. 1989, 497, 382–386. [Google Scholar] [CrossRef]
- Bullock, R.; Graham, D.I.; Chen, M.H.; Lowe, D.; McCulloch, J. Focal cerebral ischemia in the cat: Pretreatment with a competitive NMDA receptor antagonist, D-CPP-ene. J. Cereb. Blood Flow. Metab. 1990, 10, 668–674. [Google Scholar] [CrossRef]
- Boireau, A.; Malgouris, C.; Burgevin, M.C.; Peny, C.; Durand, G.; Bordier, F.; Meunier, M.; Miquet, J.M.; Daniel, M.; Chevet, T.; et al. Neuroprotective effects of RPR 104632, a novel antagonist at the glycine site of the NMDA receptor, in vitro. Eur. J. Pharmacol. 1996, 300, 237–246. [Google Scholar] [CrossRef]
- Ripley, T.L.; Little, H.J. Ethanol withdrawal hyperexcitability in vitro is selectively decreased by a competitive NMDA receptor antagonist. Brain Res. 1995, 699, 1–11. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Turski, L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet. Neurol. 2002, 1, 383–386. [Google Scholar] [CrossRef]
- Lai, T.W.; Shyu, W.C.; Wang, Y.T. Stroke intervention pathways: NMDA receptors and beyond. Trends Mol. Med. 2011, 17, 266–275. [Google Scholar] [CrossRef]
- Biegon, A.; Fry, P.A.; Paden, C.M.; Alexandrovich, A.; Tsenter, J.; Shohami, E. Dynamic changes in N-methyl-D-aspartate receptors after closed head injury in mice: Implications for treatment of neurological and cognitive deficits. Proc. Nat. Acad. Sci. USA 2004, 101, 5117–5122. [Google Scholar] [CrossRef] [Green Version]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wong, T.P.; Aarts, M.; Rooyakkers, A.; Liu, L.; Lai, T.W.; Wu, D.C.; Lu, J.; Tymianski, M.; Craig, A.M.; et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. 2007, 27, 2846–2857. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Papadia, S.; Soriano, F.X.; Leveille, F.; Martel, M.A.; Dakin, K.A.; Hansen, H.H.; Kaindl, A.; Sifringer, M.; Fowler, J.; Stefovska, V.; et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat. Neurosci. 2008, 11, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, Y.; Yasuda, R.P.; Dunah, A.W.; Wolfe, B.B. The majority of N-methyl-D-aspartate receptor complexes in adult rat cerebral cortex contain at least three different subunits (NR1/NR2A/NR2B). Mol. Pharmacol. 1997, 51, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Tovar, K.R.; McGinley, M.J.; Westbrook, G.L. Triheteromeric NMDA receptors at hippocampal synapses. J. Neurosci. 2013, 33, 9150–9160. [Google Scholar] [CrossRef]
- Rauner, C.; Kohr, G. Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-D-aspartate receptor population in adult hippocampal synapses. J. Biol. Chem. 2011, 286, 7558–7566. [Google Scholar] [CrossRef]
- Meller, R.; Thompson, S.J.; Lusardi, T.A.; Ordonez, A.N.; Ashley, M.D.; Jessick, V.; Wang, W.; Torrey, D.J.; Henshall, D.C.; Gafken, P.R.; et al. Ubiquitin proteasome-mediated synaptic reorganization: A novel mechanism underlying rapid ischemic tolerance. J. Neurosci. 2008, 28, 50–59. [Google Scholar] [CrossRef]
- Jessick, V.J.; Xie, M.; Pearson, A.N.; Torrey, D.J.; Ashley, M.D.; Thompson, S.; Meller, R. Investigating the role of the actin regulating complex ARP2/3 in rapid ischemic tolerance induced neuro-protection. Int. J. Physiol. Pathophysiol. Pharmacol. 2013, 5, 216–227. [Google Scholar]
- Kleckner, N.W.; Dingledine, R. Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science 1988, 241, 835–837. [Google Scholar] [CrossRef] [PubMed]
- Vyklicky, L., Jr.; Benveniste, M.; Mayer, M.L. Modulation of N-methyl-D-aspartic acid receptor desensitization by glycine in mouse cultured hippocampal neurones. J. Physiol. 1990, 428, 313–331. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, M.; Clements, J.; Vyklicky, L., Jr.; Mayer, M.L. A kinetic analysis of the modulation of N-methyl-D-aspartic acid receptors by glycine in mouse cultured hippocampal neurones. J. Physiol. 1990, 428, 333–357. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.A.; Lysko, P.G.; Henneberry, R.C. Excitatory amino acid neurotoxicity at the N-methyl-D-aspartate receptor in cultured neurons: Role of the voltage-dependent magnesium block. Brain Res. 1989, 499, 267–272. [Google Scholar] [CrossRef]
- Chai, S.; Li, M.; Branigan, D.; Xiong, Z.G.; Simon, R.P. Activation of acid-sensing ion channel 1a (ASIC1a) by surface trafficking. J. Biol. Chem. 2010, 285, 13002–13011. [Google Scholar] [CrossRef]
- Williams, K. Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: Selectivity and mechanisms at recombinant heteromeric receptors. Mol. Pharmacol. 1993, 44, 851–859. [Google Scholar]
- Sheng, Z.; Dai, Q.; Prorok, M.; Castellino, F.J. Subtype-selective antagonism of N-methyl-D-aspartate receptor ion channels by synthetic conantokin peptides. Neuropharmacology 2007, 53, 145–156. [Google Scholar] [CrossRef]
- Sheng, Z.; Liang, Z.; Geiger, J.H.; Prorok, M.; Castellino, F.J. The selectivity of conantokin-G for ion channel inhibition of NR2B subunit-containing NMDA receptors is regulated by amino acid residues in the S2 region of NR2B. Neuropharmacology 2009, 57, 127–136. [Google Scholar] [CrossRef]
- Lu, C.; Fu, Z.; Karavanov, I.; Yasuda, R.P.; Wolfe, B.B.; Buonanno, A.; Vicini, S. NMDA receptor subtypes at autaptic synapses of cerebellar granule neurons. J. Neurophysiol. 2006, 96, 2282–2294. [Google Scholar] [CrossRef]
- Barton, M.E.; White, H.S.; Wilcox, K.S. The effect of CGX-1007 and CI-1041, novel NMDA receptor antagonists, on NMDA receptor-mediated EPSCs. Epilepsy Res. 2004, 59, 13–24. [Google Scholar] [CrossRef]
- Frizelle, P.A.; Chen, P.E.; Wyllie, D.J. Equilibrium constants for (R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5 -yl)-methyl]-phosphonic acid (NVP-AAM077) acting at recombinant NR1/NR2A and NR1/NR2B N-methyl-D-aspartate receptors: Implications for studies of synaptic transmission. Mol. Pharmacol. 2006, 70, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Halpain, S.; Hipolito, A.; Saffer, L. Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J. Neurosci. 1998, 18, 9835–9844. [Google Scholar] [CrossRef] [PubMed]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar] [PubMed]
- Donevan, S.D.; McCabe, R.T. Conantokin G is an NR2B-selective competitive antagonist of N-methyl-D-aspartate receptors. Mol. Pharmacol. 2000, 58, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.C.; Prorok, M.; Galdzicki, Z.; Castellino, F.J. The amino acid residue at sequence position 5 in the conantokin peptides partially governs subunit-selective antagonism of recombinant N-methyl-D-aspartate receptors. J. Biol. Chem. 2001, 276, 26860–26867. [Google Scholar] [CrossRef]
- Wittekindt, B.; Malany, S.; Schemm, R.; Otvos, L.; Maccecchini, M.L.; Laube, B.; Betz, H. Point mutations identify the glutamate binding pocket of the N-methyl-D-aspartate receptor as major site of conantokin-G inhibition. Neuropharmacology 2001, 41, 753–761. [Google Scholar] [CrossRef]
- Cheriyan, J.; Balsara, R.D.; Hansen, K.B.; Castellino, F.J. Pharmacology of triheteromeric N-Methyl-D-Aspartate Receptors. Neurosci. Lett. 2016, 617, 240–246. [Google Scholar] [CrossRef]
- Hansen, K.B.; Ogden, K.K.; Yuan, H.; Traynelis, S.F. Distinct functional and pharmacological properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron 2014, 81, 1084–1096. [Google Scholar] [CrossRef]
- Stroebel, D.; Carvalho, S.; Grand, T.; Zhu, S.; Paoletti, P. Controlling NMDA receptor subunit composition using ectopic retention signals. J. Neurosci. 2014, 34, 16630–16636. [Google Scholar] [CrossRef]
- Auberson, Y.P.; Allgeier, H.; Bischoff, S.; Lingenhoehl, K.; Moretti, R.; Schmutz, M. 5-Phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg. Med. Chem. Lett. 2002, 12, 1099–1102. [Google Scholar] [CrossRef]
- Erreger, K.; Geballe, M.T.; Kristensen, A.; Chen, P.E.; Hansen, K.B.; Lee, C.J.; Yuan, H.; Le, P.; Lyuboslavsky, P.N.; Micale, N.; et al. Subunit-specific agonist activity at NR2A-, NR2B-, NR2C-, and NR2D-containing N-methyl-D-aspartate glutamate receptors. Mol. Pharmacol. 2007, 72, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tsien, J.Z. Memory and the NMDA receptors. N. Eng. J. Med. 2009, 361, 302–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yashiro, K.; Philpot, B.D. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 2008, 55, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- von Engelhardt, J.; Coserea, I.; Pawlak, V.; Fuchs, E.C.; Kohr, G.; Seeburg, P.H.; Monyer, H. Excitotoxicity in vitro by NR2A- and NR2B-containing NMDA receptors. Neuropharmacology 2007, 53, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Lu, T.J.; Chen, X.J.; Zhou, Y.; Chen, Q.; Feng, X.Y.; Xu, L.; Duan, W.H.; Xiong, Z.Q. Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke 2008, 39, 3042–3048. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Sasaki, Y.F.; Rothe, T.; Premkumar, L.S.; Takasu, M.; Crandall, J.E.; Dikkes, P.; Conner, D.A.; Rayudu, P.V.; Cheung, W.; et al. Increased NMDA current and spine density in mice lacking the NMDA receptor subunit NR3A. Nature 1998, 393, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, N.; Tu, S.; Shin, Y.; Cui, J.; Kurokawa, T.; Zhang, D.; Chen, H.S.; Tong, G.; Lipton, S.A. Neuroprotection by the NR3A subunit of the NMDA receptor. J. Neurosci. 2009, 29, 5260–5265. [Google Scholar] [CrossRef]
- Chatterton, J.E.; Awobuluyi, M.; Premkumar, L.S.; Takahashi, H.; Talantova, M.; Shin, Y.; Cui, J.; Tu, S.; Sevarino, K.A.; Nakanishi, N.; et al. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature 2002, 415, 793–798. [Google Scholar] [CrossRef]
- Chen, L.; Huang, L.Y. Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature 1992, 356, 521–523. [Google Scholar] [CrossRef]
- Kawajiri, S.; Dingledine, R. Multiple structural determinants of voltage-dependent magnesium block in recombinant NMDA receptors. Neuropharmacology 1993, 32, 1203–1211. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, M.; Leng, T.; Maysami, S.; Pearson, A.; Simon, R.; Xiong, Z.-G.; Meller, R. Changes in NMDA Receptor Function in Rapid Ischemic Tolerance: A Potential Role for Tri-Heteromeric NMDA Receptors. Biomolecules 2022, 12, 1214. https://doi.org/10.3390/biom12091214
Xie M, Leng T, Maysami S, Pearson A, Simon R, Xiong Z-G, Meller R. Changes in NMDA Receptor Function in Rapid Ischemic Tolerance: A Potential Role for Tri-Heteromeric NMDA Receptors. Biomolecules. 2022; 12(9):1214. https://doi.org/10.3390/biom12091214
Chicago/Turabian StyleXie, Mian, Tiandong Leng, Samaneh Maysami, Andrea Pearson, Roger Simon, Zhi-Gang Xiong, and Robert Meller. 2022. "Changes in NMDA Receptor Function in Rapid Ischemic Tolerance: A Potential Role for Tri-Heteromeric NMDA Receptors" Biomolecules 12, no. 9: 1214. https://doi.org/10.3390/biom12091214
APA StyleXie, M., Leng, T., Maysami, S., Pearson, A., Simon, R., Xiong, Z.-G., & Meller, R. (2022). Changes in NMDA Receptor Function in Rapid Ischemic Tolerance: A Potential Role for Tri-Heteromeric NMDA Receptors. Biomolecules, 12(9), 1214. https://doi.org/10.3390/biom12091214