
Multifaceted Roles of ALK Family Receptors and Augmentor Ligands in Health and Disease: A Comprehensive Review

Abstract
:1. Introduction
2. Biophysical and Cellular Background
3. Cancer
4. Metabolism and Body Weight Regulation
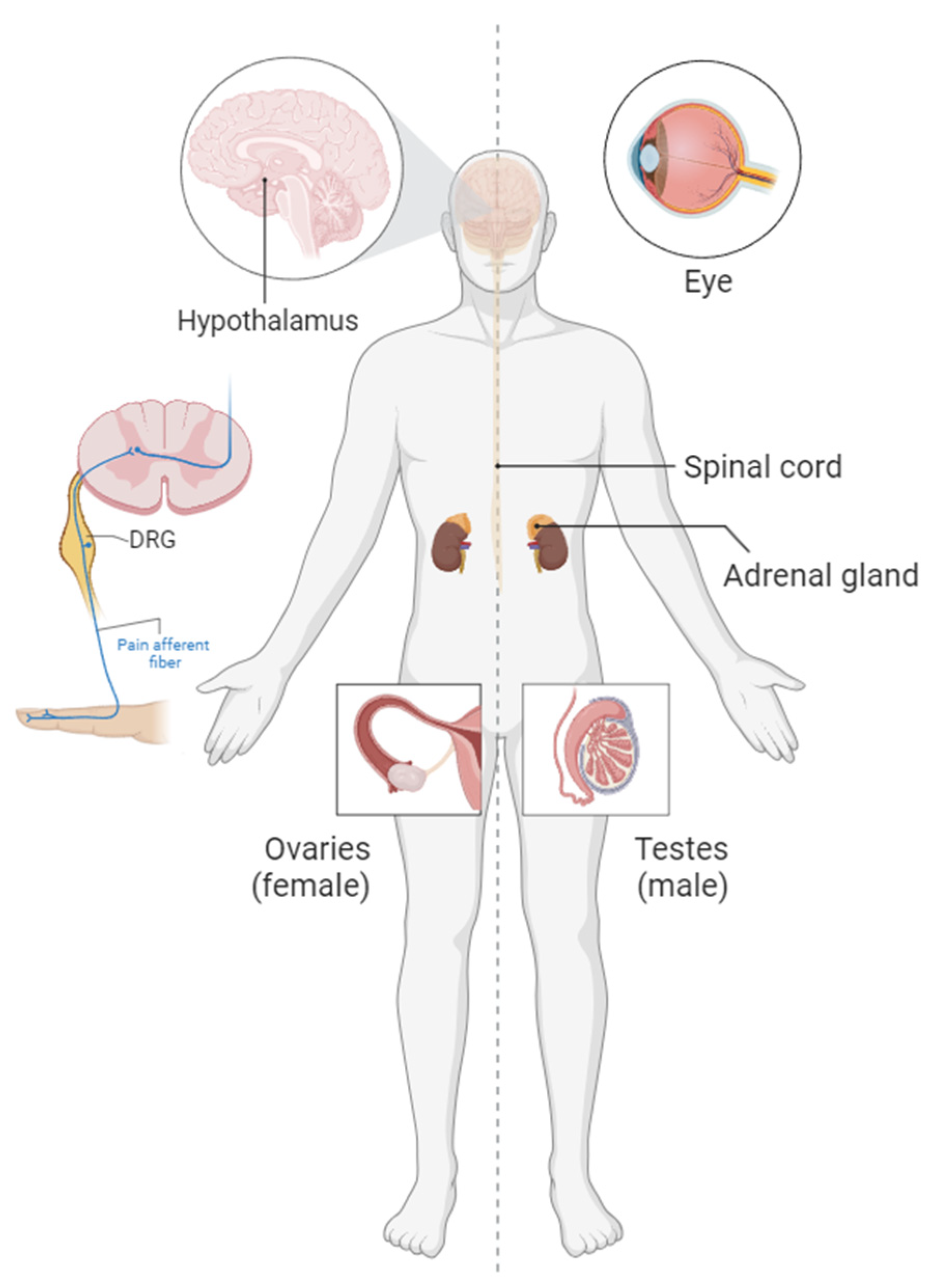
5. Pain Signaling
6. Ophthalmology and Pigmentation
7. CNS and Teratogenicity
8. Reproductive System
9. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, H.; Pao, L.I.; Zhou, A.; Brace, A.D.; Halenbeck, R.; Hsu, A.W.; Bray, T.L.; Hestir, K.; Bosch, E.; Lee, E.; et al. Deorphanization of the human leukocyte tyrosine kinase (LTK) receptor by a signaling screen of the extracellular proteome. Proc. Natl. Acad. Sci. USA 2014, 111, 15741–15745. [Google Scholar] [CrossRef]
- Palomo, V.; Nozal, V.; Rojas-Prats, E.; Gil, C.; Martinez, A. Protein kinase inhibitors for amyotrophic lateral sclerosis therapy. Br. J. Pharmacol. 2021, 178, 1316–1335. [Google Scholar] [CrossRef] [PubMed]
- Abdelsayed, M.; Kort, E.J.; Jovinge, S.; Mercola, M. Repurposing drugs to treat cardiovascular disease in the era of precision medicine. Nat. Rev. Cardiol. 2022, 19, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Duggan, B.M.; Marko, D.M.; Muzaffar, R.; Chan, D.Y.; Schertzer, J.D. Kinase inhibitors for cancer alter metabolism, blood glucose, and insulin. J. Endocrinol. 2023, 256, e220212. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef] [PubMed]
- Shepard, H.M.; Phillips, G.L.; Thanos, C.D.; Feldmann, M. Developments in therapy with monoclonal antibodies and related proteins. Clin. Med. 2017, 17, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Gharwan, H.; Groninger, H. Kinase inhibitors and monoclonal antibodies in oncology: Clinical implications. Nat. Rev. Clin. Oncol. 2016, 13, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Fauvel, B.; Yasri, A. Antibodies directed against receptor tyrosine kinases. mAbs 2014, 6, 838–851. [Google Scholar] [CrossRef]
- Maliartchouk, S.; Debeir, T.; Beglova, N.; Cuello, A.C.; Gehring, K.; Saragovi, H.U. Genuine Monovalent Ligands of TrkA Nerve Growth Factor Receptors Reveal a Novel Pharmacological Mechanism of Action. J. Biol. Chem. 2000, 275, 9946–9956. [Google Scholar] [CrossRef]
- Lampson, L.A. Monoclonal antibodies in neuro-oncology. mAbs 2011, 3, 153–160. [Google Scholar] [CrossRef]
- Moroder, L.; Musiol, H.J. Insulin—From its Discovery to the Industrial Synthesis of Modern Insulin Analogues. Angew. Chem. Int. Ed. 2017, 56, 10656–10669. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Receptor tyrosine kinases: Legacy of the first two decades. Cold Spring Harb. Perspect. Biol. 2014, 6, a008912. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Bauskin, A.R. Leukocytes express a novel gene encoding a putative transmembrane protein-kinase devoid of an extracellular domain. Nature 1988, 333, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef]
- Murray, P.B.; Lax, I.; Reshetnyak, A.; Ligon, G.F.; Lillquist, J.S.; Natoli, E.J.; Shi, X.; Folta-Stogniew, E.; Gunel, M.; Alvarado, D.; et al. Heparin is an activating ligand of the orphan receptor tyrosine kinase, ALK. Sci. Signal. 2015, 8, ra6. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, A.V.; Murray, P.B.; Shi, X.; Mo, E.S.; Mohanty, J.; Tome, F.; Bai, H.; Gunel, M.; Lax, I.; Schlessinger, J. Augmentor α and β (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: Hierarchy and specificity of ligand-receptor interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 15862–15867. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, A.V.; Rossi, P.; Myasnikov, A.G.; Sowaileh, M.; Mohanty, J.; Nourse, A.; Miller, D.J.; Lax, I.; Schlessinger, J.; Kalodimos, C.G. Mechanism for the activation of the anaplastic lymphoma kinase receptor. Nature 2021, 600, 153–157. [Google Scholar] [CrossRef]
- De Munck, S.; Provost, M.; Kurikawa, M.; Omori, I.; Mukohyama, J.; Felix, J.; Bloch, Y.; Abdel-Wahab, O.; Bazan, J.F.; Yoshimi, A.; et al. Structural basis of cytokine-mediated activation of ALK family receptors. Nature 2021, 600, 143–147. [Google Scholar] [CrossRef]
- Li, T.; Stayrook, S.E.; Tsutsui, Y.; Zhang, J.; Wang, Y.; Li, H.; Proffitt, A.; Krimmer, S.G.; Ahmed, M.; Belliveau, O.; et al. Structural basis for ligand reception by anaplastic lymphoma kinase. Nature 2021, 600, 148–152. [Google Scholar] [CrossRef]
- Lorén, C.E.; Englund, C.; Grabbe, C.; Hallberg, B.; Hunter, T.; Palmer, R.H. A crucial role for the Anaplastic lymphoma kinase receptor tyrosine kinase in gut development in Drosophila melanogaster. EMBO Rep. 2003, 4, 781–786. [Google Scholar] [CrossRef]
- Reiner, D.J.; Ailion, M.; Thomas, J.H.; Meyer, B.J.C. elegans Anaplastic Lymphoma Kinase Ortholog SCD-2 Controls Dauer Formation by Modulating TGF-β Signaling. Curr. Biol. 2008, 18, 1101–1109. [Google Scholar] [CrossRef]
- Fadeev, A.; Mendoza-Garcia, P.; Irion, U.; Guan, J.; Pfeifer, K.; Wiessner, S.; Serluca, F.; Singh, A.P.; Nüsslein-Volhard, C.; Palmer, R.H. ALKALs are in vivo ligands for ALK family receptor tyrosine kinases in the neural crest and derived cells. Proc. Natl. Acad. Sci. USA 2018, 115, E630–E638. [Google Scholar] [CrossRef]
- Degoutin, J.; Brunet-de Carvalho, N.; Cifuentes-Diaz, C.; Vigny, M. ALK (Anaplastic Lymphoma Kinase) expression in DRG neurons and its involvement in neuron-Schwann cells interaction. Eur. J. Neurosci. 2009, 29, 275–286. [Google Scholar] [CrossRef]
- Huang, H.; Gont, A.; Kee, L.; Dries, R.; Pfeifer, K.; Sharma, B.; Debruyne, D.N.; Harlow, M.; Sengupta, S.; Guan, J.; et al. Extracellular domain shedding of the ALK receptor mediates neuroblastoma cell migration. Cell Rep. 2021, 36, 109363. [Google Scholar] [CrossRef]
- Englund, C.; Lorén, C.E.; Grabbe, C.; Varshney, G.K.; Deleuil, F.; Hallberg, B.; Palmer, R.H. Jeb signals through the Alk receptor tyrosine kinase to drive visceral muscle fusion. Nature 2003, 425, 512–516. [Google Scholar] [CrossRef]
- Lee, H.H.; Norris, A.; Weiss, J.B.; Frasch, M. Jelly belly protein activates the receptor tyrosine kinase Alk to specify visceral muscle pioneers. Nature 2003, 425, 507–512. [Google Scholar] [CrossRef]
- Ishihara, T.; Iino, Y.; Mohri, A.; Mori, I.; Gengyo-Ando, K.; Mitani, S.; Katsura, I. HEN-1, a Secretory Protein with an LDL Receptor Motif, Regulates Sensory Integration and Learning in Caenorhabditis elegans. Cell 2002, 109, 639–649. [Google Scholar] [CrossRef]
- Wang, Z.; Dornburg, A.; Wang, J.; Mo, E.S.; Lopez-Giraldez, F.; Townsend, J.P. Evolutionary analyses guide selection of model systems to investigate proto-oncogene function in ALK and LTK. bioRxiv 2019. [CrossRef]
- Guan, J.; Umapathy, G.; Yamazaki, Y.; Wolfstetter, G.; Mendoza, P.; Pfeifer, K.; Mohammed, A.; Hugosson, F.; Zhang, H.; Hsu, A.W.; et al. FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. Davis R, editor. eLife 2015, 4, e09811. [Google Scholar] [CrossRef]
- Reshetnyak, A.V.; Mohanty, J.; Tomé, F.; Puleo, D.E.; Plotnikov, A.N.; Ahmed, M.; Kaur, N.; Poliakov, A.; Cinnaiyan, A.M.; Lax, I.; et al. Identification of a biologically active fragment of ALK and LTK-Ligand 2 (augmentor-α). Proc. Natl. Acad. Sci. USA 2018, 115, 8340–8345. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Duyster, J.; Bai, R.Y.; Morris, S.W. Translocations involving anaplastic lymphoma kinase (ALK). Oncogene 2001, 20, 5623–5637. [Google Scholar] [CrossRef] [PubMed]
- Mano, H. Non-solid oncogenes in solid tumors: EML4–ALK fusion genes in lung cancer. Cancer Sci. 2008, 99, 2349–2355. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. The role of the ALK receptor in cancer biology. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27 (Suppl. S3), iii4–iii15. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.M.; Heuckmann, J.M.; de Stanchina, E.; Chen, H.; Thomas, R.K.; Liang, C.; Pao, W. Insights into ALK-Driven Cancers Revealed through Development of Novel ALK Tyrosine Kinase Inhibitors. Cancer Res. 2011, 71, 4920–4931. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Riely, G.J.; Shaw, A.T. Targeting ALK: Precision Medicine Takes on Drug Resistance. Cancer Discov. 2017, 7, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Xing, M. Anaplastic thyroid cancers harbor novel oncogenic mutations of the ALK gene. Cancer Res. 2011, 71, 4403–4411. [Google Scholar] [CrossRef]
- Guan, J.; Wolfstetter, G.; Siaw, J.; Chand, D.; Hugosson, F.; Palmer, R.H.; Hallberg, B. Anaplastic lymphoma kinase L1198F and G1201E mutations identified in anaplastic thyroid cancer patients are not ligand-independent. Oncotarget 2016, 8, 11566–11578. [Google Scholar] [CrossRef]
- Ono, S.; Saito, T.; Terui, K.; Yoshida, H.; Enomoto, H. Generation of conditional ALK F1174L mutant mouse models for the study of neuroblastoma pathogenesis. Genesis 2019, 57, e23323. [Google Scholar] [CrossRef]
- Cazes, A.; Lopez-Delisle, L.; Tsarovina, K.; Pierre-Eugène, C.; De Preter, K.; Peuchmaur, M.; Nicolas, A.; Provost, C.; Louis-Brennetot, C.; Daveau, R.; et al. Activated Alk triggers prolonged neurogenesis and Ret upregulation providing a therapeutic target in ALK-mutated neuroblastoma. Oncotarget 2014, 5, 2688–2702. [Google Scholar] [CrossRef] [PubMed]
- Berry, T.; Luther, W.; Bhatnagar, N.; Jamin, Y.; Poon, E.; Sanda, T.; Pei, D.; Sharma, B.; Vetharoy, W.R.; Hallsworth, A.; et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell 2012, 22, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Heukamp, L.C.; Thor, T.; Schramm, A.; De Preter, K.; Kumps, C.; De Wilde, B.; Odersky, A.; Peifer, M.; Lindner, S.; Spruessel, A.; et al. Targeted expression of mutated ALK induces neuroblastoma in transgenic mice. Sci. Transl. Med. 2012, 4, 141ra91. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Lee, J.S.; Guo, F.; Shin, J.; Perez-Atayde, A.R.; Kutok, J.L.; Rodig, S.J.; Neuberg, D.S.; Helman, D.; Feng, H.; et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell 2012, 21, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Schönherr, C.; Ruuth, K.; Kamaraj, S.; Wang, C.L.; Yang, H.L.; Combaret, V.; Djos, A.; Martinsson, T.; Christensen, J.G.; Palmer, R.H.; et al. Anaplastic Lymphoma Kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene 2012, 31, 5193–5200. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.K.; Nafady, A.; Takatori, A.; Kishida, S.; Ohira, M.; Suenaga, Y.; Hossain, S.; Akter, J.; Ogura, A.; Nakamura, Y.; et al. ALK is a MYCN target gene and regulates cell migration and invasion in neuroblastoma. Sci. Rep. 2013, 3, 3450. [Google Scholar] [CrossRef] [PubMed]
- Borenäs, M.; Umapathy, G.; Lai, W.Y.; Lind, D.E.; Witek, B.; Guan, J.; Mendoza-Garcia, P.; Masudi, T.; Claeys, A.; Chuang, T.-P.; et al. ALK ligand ALKAL2 potentiates MYCN-driven neuroblastoma in the absence of ALK mutation. EMBO J. 2021, 40, e105784. [Google Scholar] [CrossRef]
- Mossé, Y.P.; Lim, M.S.; Voss, S.D.; Wilner, K.; Ruffner, K.; Laliberte, J.; Rolland, D.; Balis, F.M.; Maris, J.M.; Weigel, B.J.; et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: A Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013, 14, 472–480. [Google Scholar] [CrossRef]
- Javanmardi, N.; Fransson, S.; Djos, A.; Umapathy, G.; Östensson, M.; Milosevic, J.; Borenäs, M.; Hallberg, B.; Kogner, P.; Martinsson, T.; et al. Analysis of ALK, MYCN, and the ALK ligand ALKAL2 (FAM150B/AUGα) in neuroblastoma patient samples with chromosome arm 2p rearrangements. Genes Chromosomes Cancer 2020, 59, 50–57. [Google Scholar] [CrossRef]
- Guan, J.; Hallberg, B.; Palmer, R.H. Chromosome Imbalances in Neuroblastoma-Recent Molecular Insight into Chromosome 1p-deletion, 2p-gain, and 11q-deletion Identifies New Friends and Foes for the Future. Cancers 2021, 13, 5897. [Google Scholar] [CrossRef]
- Schleiermacher, G.; Javanmardi, N.; Bernard, V.; Leroy, Q.; Cappo, J.; Rio Frio, T.; Pierron, G.; Lapouble, E.; Combaret, V.; Speleman, F.; et al. Emergence of new ALK mutations at relapse of neuroblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 2727–2734. [Google Scholar] [CrossRef]
- Treis, D.; Umapathy, G.; Fransson, S.; Guan, J.; Mendoza-García, P.; Siaw, J.T.; Wessman, S.; Gordon Murkes, L.; Stenman, J.J.E.; Djos, A.; et al. Sustained Response to Entrectinib in an Infant With a Germline ALKAL2 Variant and Refractory Metastatic Neuroblastoma With Chromosomal 2p Gain and Anaplastic Lymphoma Kinase and Tropomyosin Receptor Kinase Activation. JCO Precis. Oncol. 2022, 6, e2100271. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M. Spontaneous regression of neuroblastoma. Cell Tissue Res. 2018, 372, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.S.; Pearson, J.; Willatt, L.; Andrews, T.; Beach, R.; Green, A. Germline duplication of chromosome 2p and neuroblastoma. J. Med. Genet. 1997, 34, 949–951. [Google Scholar] [CrossRef] [PubMed]
- Dowa, Y.; Yamamoto, T.; Abe, Y.; Kobayashi, M.; Hoshino, R.; Tanaka, K.; Aida, N.; Take, H.; Kato, K.; Tanaka, Y.; et al. Congenital Neuroblastoma in a Patient With Partial Trisomy of 2p. J. Pediatr. Hematol. Oncol. 2006, 28, 379. [Google Scholar] [CrossRef] [PubMed]
- Nagano, H.; Kano, Y.; Kobuchi, S.; Kajitani, T. A case of partial 2p trisomy with neuroblastoma. Jpn. J. Hum. Genet. 1980, 25, 39–45. [Google Scholar] [CrossRef]
- Lopez, G.; Conkrite, K.L.; Doepner, M.; Rathi, K.S.; Modi, A.; Vaksman, Z.; Farra, L.M.; Hyson, E.; Noureddine, M.; Wei, J.S.; et al. Somatic structural variation targets neurodevelopmental genes and identifies SHANK2 as a tumor suppressor in neuroblastoma. Genome Res. 2020, 30, 1228–1242. [Google Scholar] [CrossRef]
- Siaw, J.T.; Javanmardi, N.; Van den Eynden, J.; Lind, D.E.; Fransson, S.; Martinez-Monleon, A.; Djos, A.; Sjöberg, R.-M.; Östensson, M.; Carén, H.; et al. 11q Deletion or ALK Activity Curbs DLG2 Expression to Maintain an Undifferentiated State in Neuroblastoma. Cell Rep. 2020, 32, 108171. [Google Scholar] [CrossRef]
- van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef]
- Peterson, S.; Bogenmann, E. The RET and TRKA pathways collaborate to regulate neuroblastoma differentiation. Oncogene 2004, 23, 213–225. [Google Scholar] [CrossRef]
- Siaw, J.T.; Gabre, J.L.; Uçkun, E.; Vigny, M.; Zhang, W.; Van den Eynden, J.; Hallberg, B.; Palmer, R.H.; Guan, J. Loss of RET Promotes Mesenchymal Identity in Neuroblastoma Cells. Cancers 2021, 13, 1909. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Delisle, L.; Pierre-Eugène, C.; Louis-Brennetot, C.; Surdez, D.; Raynal, V.; Baulande, S.; Boeva, V.; Grossetête-Lalami, S.; Combaret, V.; Peuchmaur, M.; et al. Activated ALK signals through the ERK-ETV5-RET pathway to drive neuroblastoma oncogenesis. Oncogene 2018, 37, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Lambertz, I.; Kumps, C.; Claeys, S.; Lindner, S.; Beckers, A.; Janssens, E.; Carter, D.R.; Cazes, A.; Cheung, B.B.; De Mariano, M.; et al. Upregulation of MAPK Negative Feedback Regulators and RET in Mutant ALK Neuroblastoma: Implications for Targeted Treatment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 3327–3339. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, B.; Fu, X.; Liang, Y.; Chai, X.; Ye, Z.; Li, R.; He, Y.; Kong, G.; Lian, J.; et al. ALKAL1 gene silencing prevents colorectal cancer progression via suppressing Sonic Hedgehog (SHH) signaling pathway. J. Cancer 2021, 12, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Mazzeschi, M.; Sgarzi, M.; Romaniello, D.; Gelfo, V.; Cavallo, C.; Ambrosi, F.; Morselli, A.; Miano, C.; Laprovitera, N.; Girone, C.; et al. The autocrine loop of ALK receptor and ALKAL2 ligand is an actionable target in consensus molecular subtype 1 colon cancer. J. Exp. Clin. Cancer Res. 2022, 41, 113. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Bilsland, J.G.; Wheeldon, A.; Mead, A.; Znamenskiy, P.; Almond, S.; Waters, K.A.; Thakur, M.; Beaumont, V.; Bonnert, T.P.; Heavens, R.; et al. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2008, 33, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Orthofer, M.; Valsesia, A.; Mägi, R.; Wang, Q.P.; Kaczanowska, J.; Kozieradzki, I.; Leopoldi, A.; Cikes, D.; Zopf, L.M.; Tretiakov, E.O.; et al. Identification of ALK in Thinness. Cell 2020, 181, 1246–1262.e22. [Google Scholar] [CrossRef]
- Ahmed, M.; Kaur, N.; Cheng, Q.; Shanabrough, M.; Tretiakov, E.O.; Harkany, T.; Horvath, T.L.; Schlessinger, J. A hypothalamic pathway for Augmentor α-controlled body weight regulation. Proc. Natl. Acad. Sci. USA 2022, 119, e2200476119. [Google Scholar] [CrossRef]
- Berthoud, H.R.; Neuhuber, W.L. Functional and chemical anatomy of the afferent vagal system. Auton. Neurosci. 2000, 85, 1–17. [Google Scholar] [CrossRef]
- Affleck, V.S.; Coote, J.H.; Pyner, S. The projection and synaptic organisation of NTS afferent connections with presympathetic neurons, GABA and nNOS neurons in the paraventricular nucleus of the hypothalamus. Neuroscience 2012, 219, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Defaye, M.; Iftinca, M.C.; Gadotti, V.M.; Basso, L.; Abdullah, N.S.; Cuménal, M.; Agosti, F.; Hassan, A.; Flynn, R.; Martin, J.; et al. The neuronal tyrosine kinase receptor ligand ALKAL2 mediates persistent pain. J. Clin. Investig. 2022, 132, e154317. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.; Rosales, A.L.S.; Chioda, M.D.; Parker, L.; Devgan, G.; Kettle, J. Consensus Recommendations for Management and Counseling of Adverse Events Associated with Lorlatinib: A Guide for Healthcare Practitioners. Adv. Ther. 2020, 37, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, K.C.; Park, J.R.; Kayser, K.; Malvar, J.; Chi, Y.Y.; Groshen, S.G.; Villablanca, J.G.; Krytska, K.; Lai, L.M.; Acharya, P.T.; et al. Lorlatinib with or without chemotherapy in ALK-driven refractory/relapsed neuroblastoma: Phase 1 trial results. Nat. Med. 2023, 29, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Wu, H.; Ding, J.-M.; Wang, Z.-X.; Ullah, T.; Dong, S.-S.; Chen, H.; Guo, Y. Transcriptome-wide association study identifies multiple genes associated with childhood body mass index. Int. J. Obes. 2021, 45, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Madrid, D.; Szydzik, J.; Lind, D.E.; Borenäs, M.; Bemark, M.; Cui, J.; Palmer, R.H.; Hallberg, B. Repotrectinib (TPX-0005), effectively reduces growth of ALK driven neuroblastoma cells. Sci. Rep. 2019, 9, 19353. [Google Scholar] [CrossRef] [PubMed]
- Marchand, S. Mechanisms Challenges of the Pain Phenomenon. Front. Pain Res. 2021, 1, 574370. [Google Scholar] [CrossRef]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and Molecular Mechanisms of Pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef]
- Ji, R.R.; Strichartz, G. Cell Signaling and the Genesis of Neuropathic Pain. Sci. STKE 2004, 2004, re14. [Google Scholar] [CrossRef]
- Woolf, C.J.; Max, M.B. Mechanism-based Pain Diagnosis: Issues for Analgesic Drug Development. Anesthesiology 2001, 95, 241–249. [Google Scholar] [CrossRef]
- Modi, A.D.; Parekh, A.; Pancholi, Y.N. Evaluating pain behaviours: Widely used mechanical and thermal methods in rodents. Behav. Brain Res. 2023, 446, 114417. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Gereau, R.W.; Malcangio, M.; Strichartz, G.R. MAP kinase and pain. Brain Res. Rev. 2009, 60, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Stamboulian, S.; Choi, J.S.; Ahn, H.S.; Chang, Y.W.; Tyrrell, L.; Black, J.A.; Waxman, S.G.; Dib-Hajj, S.D. ERK1/2 Mitogen-Activated Protein Kinase Phosphorylates Sodium Channel Nav1.7 and Alters Its Gating Properties. 7 and Alters Its Gating Properties. J. Neurosci. 2010, 30, 1637–1647. [Google Scholar] [CrossRef]
- Devor, M. Unexplained peculiarities of the dorsal root ganglion. Pain 1999, 82, S27–S35. [Google Scholar] [CrossRef]
- Pleasure, S.J.; Page, C.; Lee, V.M. Pure, postmitotic, polarized human neurons derived from NTera 2 cells provide a system for expressing exogenous proteins in terminally differentiated neurons. J. Neurosci. 1992, 12, 1802–1815. [Google Scholar] [CrossRef] [PubMed]
- Kersten, C.; Cameron, M.G. Cetuximab alleviates neuropathic pain despite tumour progression. BMJ Case Rep. 2012, 2012, bcr1220115374. [Google Scholar] [CrossRef] [PubMed]
- Kersten, C.; Cameron, M.G.; Laird, B.; Mjåland, S. Epidermal growth factor receptor-inhibition (EGFR-I) in the treatment of neuropathic pain. Br. J. Anaesth. 2015, 115, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Lin, J.; Liao, G.; Tian, Y.; Liang, Y.; Li, R.; Liu, M.; Yuan, Y. ALK Inhibitors in the Treatment of ALK Positive, NSCLC. Front. Oncol. 2019, 8, 557. [Google Scholar] [CrossRef]
- Hochberg, M.C. Serious joint-related adverse events in randomized controlled trials of anti-nerve growth factor monoclonal antibodies. Osteoarthr. Cartil. 2015, 23, S18–S21. [Google Scholar] [CrossRef]
- Mahato, A.K.; Sidorova, Y.A. Glial cell line-derived neurotrophic factors (GFLs) and small molecules targeting RET receptor for the treatment of pain and Parkinson’s disease. Cell Tissue Res. 2020, 382, 147–160. [Google Scholar] [CrossRef]
- Andres, C.; Hasenauer, J.; Ahn, H.S.; Joseph, E.K.; Isensee, J.; Theis, F.J.; Allgöwer, F.; Levine, J.D.; Dib-Hajj, S.D.; Waxman, S.G.; et al. Wound-healing growth factor, basic FGF, induces Erk1/2-dependent mechanical hyperalgesia. Pain 2013, 154, 2216–2226. [Google Scholar] [CrossRef] [PubMed]
- Lázár, B.A.; Jancsó, G.; Sántha, P. Modulation of Sensory Nerve Function by Insulin: Possible Relevance to Pain, Inflammation and Axon Growth. Int. J. Mol. Sci. 2020, 21, 2507. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Barker, K.; Shi, S.; Diaz, M.; Mo, B.; Gutstein, H.B. Blockade of PDGFR-β activation eliminates morphine analgesic tolerance. Nat. Med. 2012, 18, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.G.; Gracias, N.G.; Drobish, J.K.; Vasko, M.R.; Gereau, R.W.; Chen, Z.F. The c-kit signaling pathway is involved in the development of persistent pain. Pain 2009, 144, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Stover, J.D.; Farhang, N.; Berrett, K.C.; Gertz, J.; Lawrence, B.; Bowles, R.D. CRISPR Epigenome Editing of AKAP150 in DRG Neurons Abolishes Degenerative IVD-Induced Neuronal Activation. Mol. Ther. 2017, 25, 2014–2027. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.B.; Xue, C.; Benice, T.; Xue, L.; Morris, S.W.; Raber, J. Anaplastic lymphoma kinase and leukocyte tyrosine kinase: Functions and genetic interactions in learning, memory and adult neurogenesis. Pharmacol. Biochem. Behav. 2012, 100, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Chelala, E.; Hoyek, S.; Arej, N.; Kattan, J.; Kourie, H.R.; Baakliny, J.; Antoun, J. Ocular and orbital side effects of ALK inhibitors: A review article. Future Oncol. 2019, 15, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Vernersson, E.; Khoo, N.K.S.; Henriksson, M.L.; Roos, G.; Palmer, R.H.; Hallberg, B. Characterization of the expression of the ALK receptor tyrosine kinase in mice. Gene Expr. Patterns 2006, 6, 448–461. [Google Scholar] [CrossRef]
- Belmonte, C.; Acosta, M.C.; Merayo-Lloves, J.; Gallar, J. What Causes Eye Pain? Curr. Ophthalmol. Rep. 2015, 3, 111–121. [Google Scholar] [CrossRef]
- Bazigou, E.; Apitz, H.; Johansson, J.; Lorén, C.E.; Hirst, E.M.A.; Chen, P.-L.; Palmer, R.H.; Salecker, I. Anterograde Jelly belly and Alk receptor tyrosine kinase signaling mediates retinal axon targeting in Drosophila. Cell 2007, 128, 961–975. [Google Scholar] [CrossRef]
- Martinsson, T.; Eriksson, T.; Abrahamsson, J.; Caren, H.; Hansson, M.; Kogner, P.; Kamaraj, S.; Schönherr, C.; Weinmar, J.; Ruuth, K.; et al. Appearance of the Novel Activating F1174S ALK Mutation in Neuroblastoma Correlates with Aggressive Tumor Progression and Unresponsiveness to Therapy. Cancer Res. 2011, 71, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Mo, E.S.; Cheng, Q.; Reshetnyak, A.V.; Schlessinger, J.; Nicoli, S. Alk and Ltk ligands are essential for iridophore development in zebrafish mediated by the receptor tyrosine kinase Ltk. Proc. Natl. Acad. Sci. USA 2017, 114, 12027–12032. [Google Scholar] [CrossRef]
- Fadeev, A.; Krauss, J.; Singh, A.P.; Nüsslein-Volhard, C. Zebrafish Leucocyte tyrosine kinase controls iridophore establishment, proliferation and survival. Pigment Cell Melanoma Res. 2016, 29, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Kelsh, R.N.; Brand, M.; Jiang, Y.J.; Heisenberg, C.P.; Lin, S.; Haffter, P.; Odenthal, J.; Mullins, M.C.; van Eeden, F.J.; Furutani-Seiki, M.; et al. Zebrafish pigmentation mutations and the processes of neural crest development. Development 1996, 123, 369–389. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Schach, U.; Nüsslein-Volhard, C. Proliferation, dispersal and patterned aggregation of iridophores in the skin prefigure striped colouration of zebrafish. Nat. Cell Biol. 2014, 16, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Selleck, M.A.J.; Bronner-Fraser, M. Origins of the avian neural crest: The role of neural plate-epidermal interactions. Development 1995, 121, 525–538. [Google Scholar] [CrossRef]
- Kabangu, M.; Cecil, R.; Strohl, L.; Timoshevskaya, N.; Smith, J.J.; Voss, S.R. Leukocyte Tyrosine Kinase (Ltk) Is the Mendelian Determinant of the Axolotl Melanoid Color Variant. Genes 2023, 14, 904. [Google Scholar] [CrossRef]
- Coblentz, J.; Logan, P.T.; Dias, A.B.T.; Esposito, E.; Mansure, J.J.; Burnier, M.N. Expression of anaplastic lymphoma kinase in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3955. [Google Scholar] [CrossRef]
- Surriga, O.; Rajasekhar, V.K.; Ambrosini, G.; Dogan, Y.; Huang, R.; Schwartz, G.K. Crizotinib, a c-Met Inhibitor, Prevents Metastasis in a Metastatic Uveal Melanoma Model. Mol. Cancer Ther. 2013, 12, 2817–2826. [Google Scholar] [CrossRef]
- Iwahara, T.; Fujimoto, J.; Wen, D.; Cupples, R.; Bucay, N.; Arakawa, T.; Mori, S.; Ratzkin, B.; Yamamoto, T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997, 14, 439–449. [Google Scholar] [CrossRef]
- Rohrbough, J.; Kent, K.S.; Broadie, K.; Weiss, J.B. Jelly Belly Trans-Synaptic Signaling to Anaplastic Lymphoma Kinase Regulates Neurotransmission Strength and Synapse Architecture. Dev. Neurobiol. 2013, 73, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Lasek, A.W.; Gesch, J.; Giorgetti, F.; Kharazia, V.; Heberlein, U. Alk is a transcriptional target of LMO4 and ERα that promotes cocaine sensitization and reward. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 14134–14141. [Google Scholar] [CrossRef] [PubMed]
- Mangieri, R.A.; Maier, E.Y.; Buske, T.R.; Lasek, A.W.; Morrisett, R.A. Anaplastic Lymphoma Kinase Is a Regulator of Alcohol Consumption and Excitatory Synaptic Plasticity in the Nucleus Accumbens Shell. Front. Pharmacol. 2017, 8, 533. [Google Scholar] [CrossRef] [PubMed]
- De Smedt, F.; Dessy, F.; Carestia, L.; Baldin, P.; Nana, F.A.; Clapuyt, P.; Boon, V.; Amant, F.; Gziri, M.M. A pregnant patient with ALK-positive non-small cell lung cancer treated with alectinib: A case report and review of the literature. Oncol. Lett. 2023, 25, 54. [Google Scholar] [CrossRef]
- Scarfone, G.; Fumagalli, M.; Imbimbo, M.; Ceruti, T.; Cribiù, F.M.; Di Loreto, E.; D’Incalci, M.; Facchin, F.; Fontana, C.; Garassino, M.C.; et al. First Case Report of Pregnancy on Alectinib in a Woman With Metastatic ALK-Rearranged Lung Cancer: A Case Report. J. Thorac. Oncol. 2021, 16, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Gouzi, J.Y.; Bouraimi, M.; Roussou, I.G.; Moressis, A.; Skoulakis, E.M.C. The Drosophila Receptor Tyrosine Kinase Alk Constrains Long-Term Memory Formation. J. Neurosci. 2018, 38, 7701–7712. [Google Scholar] [CrossRef] [PubMed]
- Kunugi, H.; Hashimoto, R.; Okada, T.; Hori, H.; Nakabayashi, T.; Baba, A.; Kudo, K.; Omori, M.; Takahashi, S.; Tsukue, R.; et al. Possible association between nonsynonymous polymorphisms of the anaplastic lymphoma kinase (ALK) gene and schizophrenia in a Japanese population. J. Neural Transm. 2006, 113, 1569–1573. [Google Scholar] [CrossRef] [PubMed]
- Weickhardt, A.J.; Rothman, M.S.; Salian-Mehta, S.; Kiseljak-Vassiliades, K.; Oton, A.B.; Doebele, R.C.; Wierman, M.E.; Camidge, D.R. Rapid-onset hypogonadism secondary to crizotinib use in men with metastatic nonsmall cell lung cancer. Cancer 2012, 118, 5302–5309. [Google Scholar] [CrossRef]
- Witek, B.; El Wakil, A.; Nord, C.; Ahlgren, U.; Eriksson, M.; Vernersson-Lindahl, E.; Helland, Å.; Alexeyev, O.A.; Hallberg, B.; Palmer, R.H. Targeted Disruption of ALK Reveals a Potential Role in Hypogonadotropic Hypogonadism. PLoS ONE 2015, 10, e0123542. [Google Scholar] [CrossRef]
- Fuqua, J.S. Treatment and Outcomes of Precocious Puberty: An Update. J. Clin. Endocrinol. Metab. 2013, 98, 2198–2207. [Google Scholar] [CrossRef]
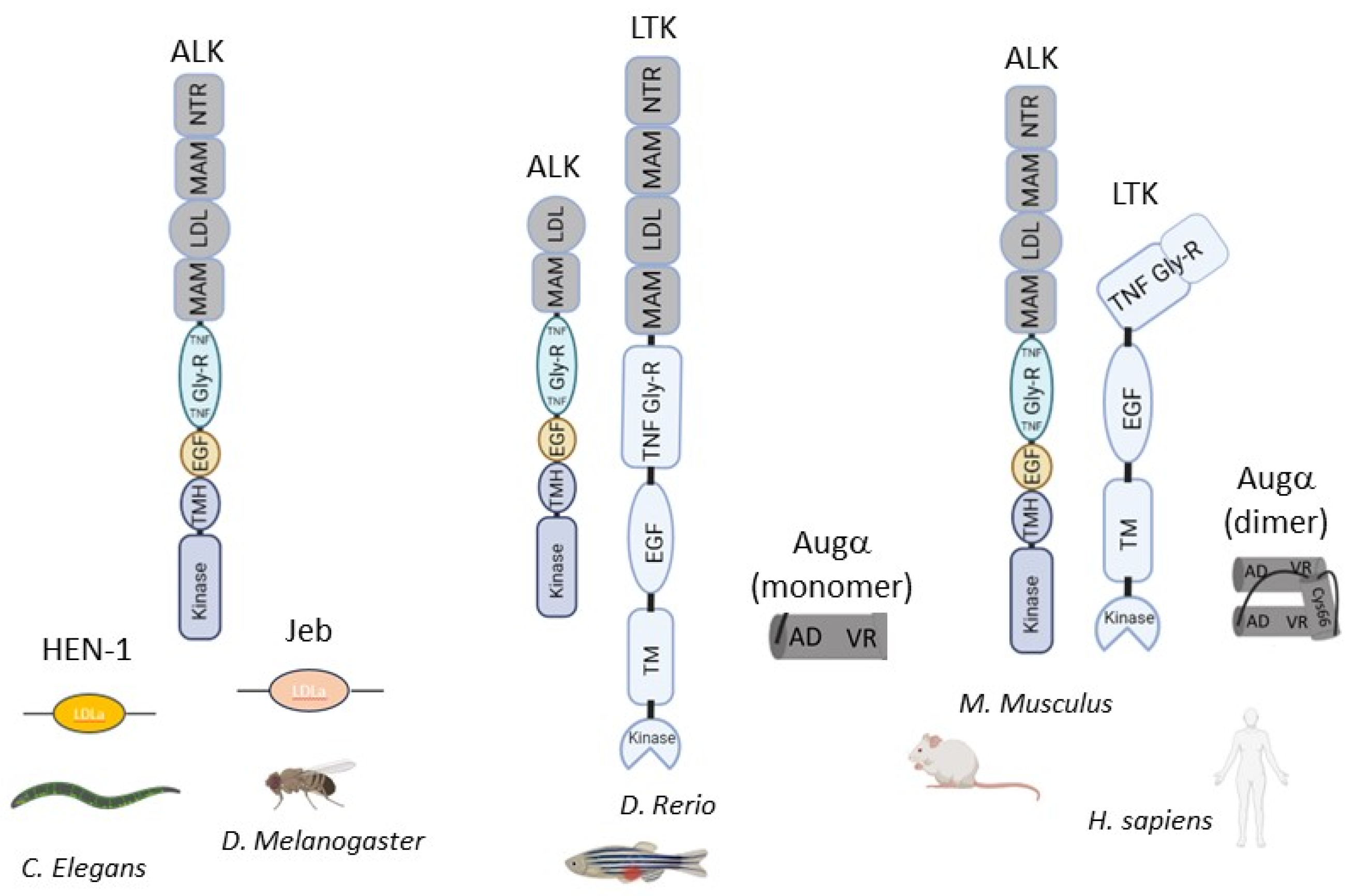
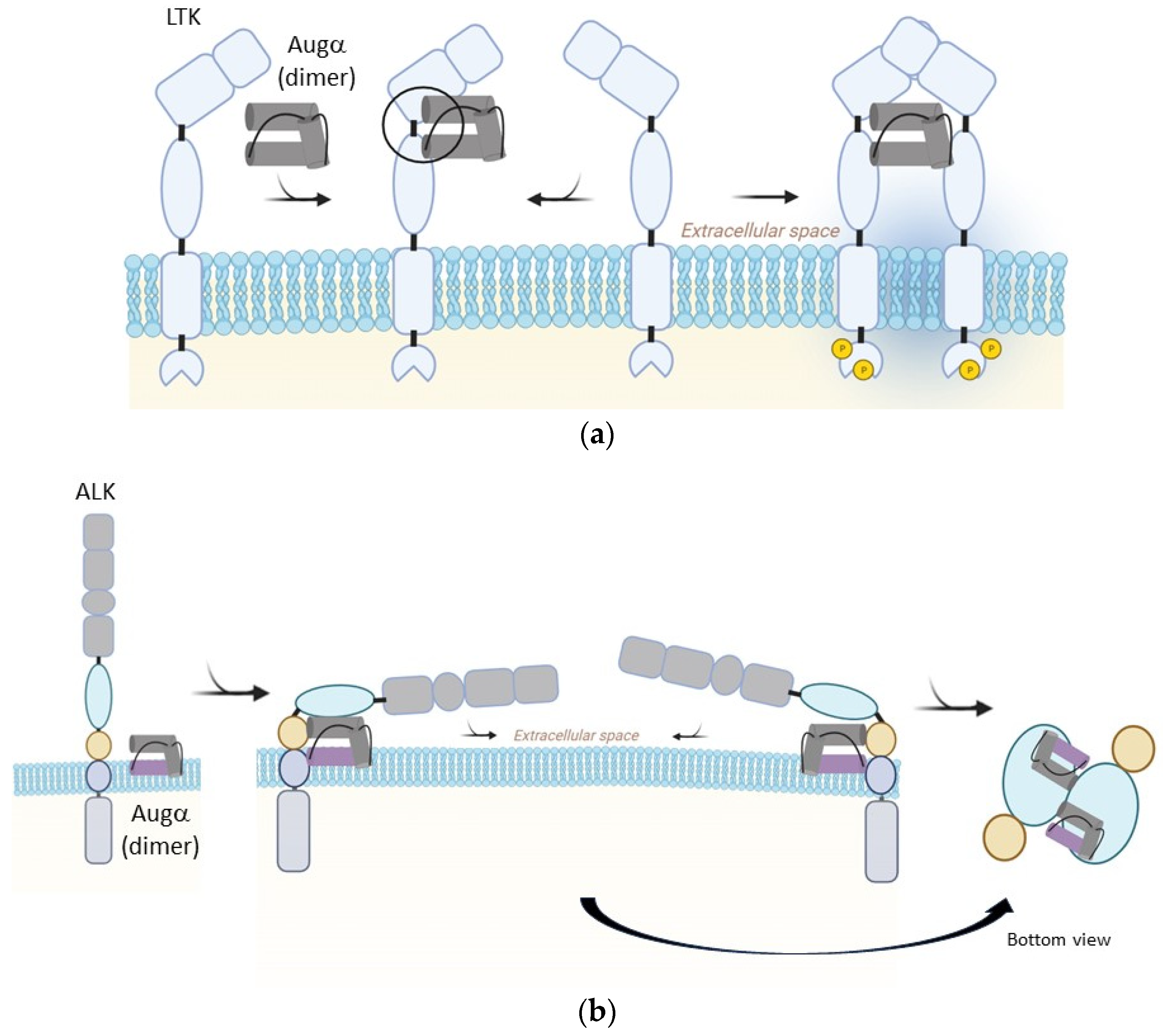

{kind=link}
{kind=link}
{kind=link}
| Research Group | Truncations | EGF-like Domain Importance | Size Exclusion Chromatography (Receptor:Ligand) | Method Used | Structural Complex (Receptor:Ligand) | |
|---|---|---|---|---|---|---|
| ALK | Augα | |||||
| Savvides | TG-EGFL (648–1038) | AD (78–152) | Yes | No shift (1:1) | X-ray crystallography | 1:2 |
| Klein, Schlessinger | TG-EGFL (678–1030) | AD (71–152) | Yes | (fusion protein) | X-ray crystallography | 2:2 |
| Kalodimos, Schlessinger | TG-EGFL (673–1025) | Full size + C66Y | Yes | Shift (2:2) | cryoEM NMR | 2:2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katic, L.; Priscan, A. Multifaceted Roles of ALK Family Receptors and Augmentor Ligands in Health and Disease: A Comprehensive Review. Biomolecules 2023, 13, 1490. https://doi.org/10.3390/biom13101490
Katic L, Priscan A. Multifaceted Roles of ALK Family Receptors and Augmentor Ligands in Health and Disease: A Comprehensive Review. Biomolecules. 2023; 13(10):1490. https://doi.org/10.3390/biom13101490
Chicago/Turabian StyleKatic, Luka, and Anamarija Priscan. 2023. "Multifaceted Roles of ALK Family Receptors and Augmentor Ligands in Health and Disease: A Comprehensive Review" Biomolecules 13, no. 10: 1490. https://doi.org/10.3390/biom13101490
APA StyleKatic, L., & Priscan, A. (2023). Multifaceted Roles of ALK Family Receptors and Augmentor Ligands in Health and Disease: A Comprehensive Review. Biomolecules, 13(10), 1490. https://doi.org/10.3390/biom13101490