
The Molecular Mechanisms of Complement Receptor 1—It Is Complicated


Abstract
:1. Introduction
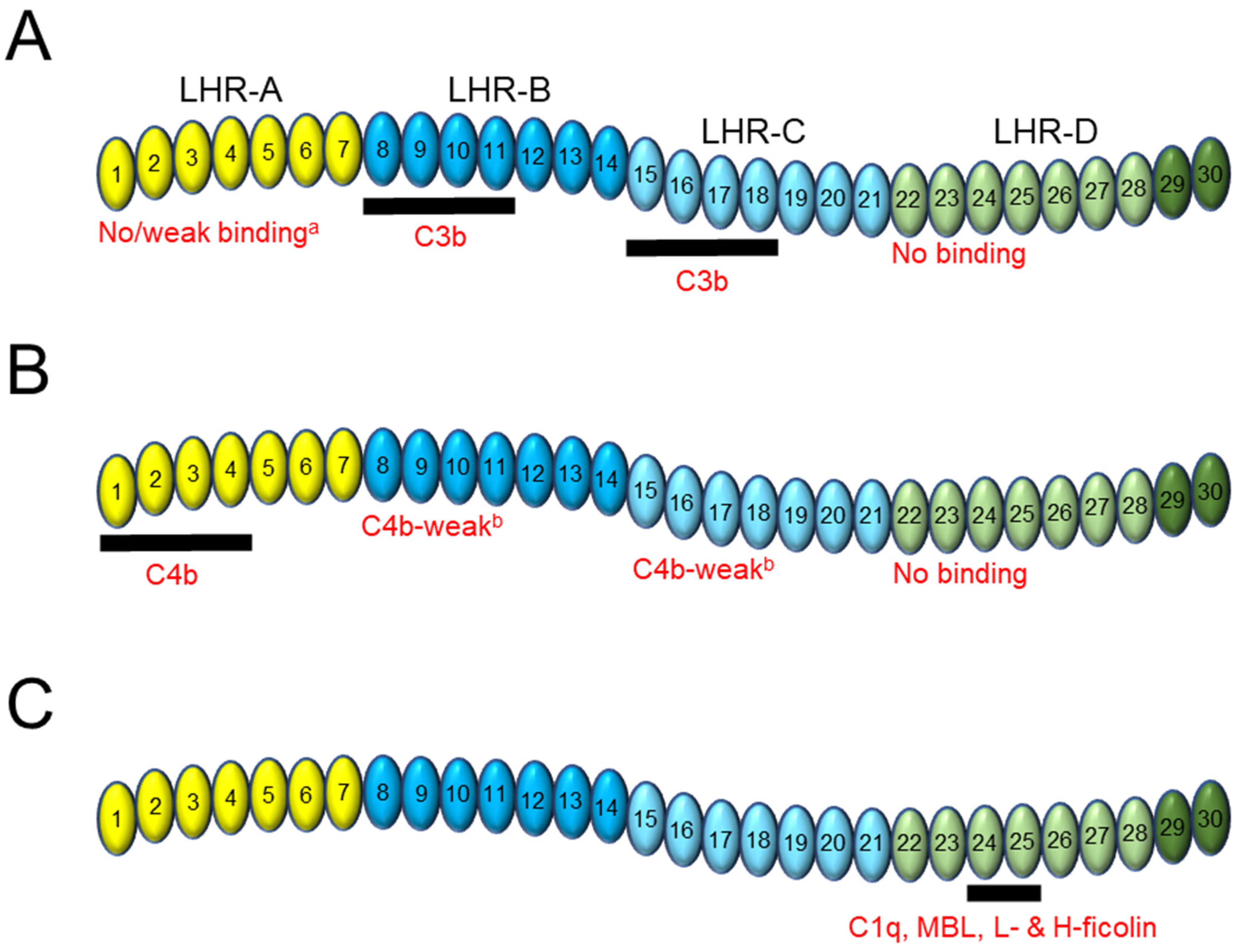
2. CR1 Binding to C3b and C4b
2.1. General and Comparative

{kind=link}
{kind=link}
| Affinity to C4b Dimer (nM) | Affinity to C4b Monomer (nM) | Method Used | CR1 Source | References |
|---|---|---|---|---|
| 12 a | >100-fold weaker b | Radioligand binding/competition | sCR1 and E-CR1 | Wiesman et al., 1990 [24] |
| 340 | >100-fold weaker b | Radioligand binding assay | E-CR1 | Reilly et al., 1994 [36] |
| 360 | >100-fold weaker b | Radioligand binding assay | Expressed on CHO cells | Reilly et al., 1994 [36] |
| 120–480 c | Not measured | Radioligand binding assay | E-CR1 | Reilly and Mold 1997 [40] |
| 330–390 d | 900 | SPR | sCR1 | Clemenza and Isenman 2004 [38] |
2.2. Domain Contribution
3. CR1 Binding to Other Ligands
4. Structural Data
5. Decay Acceleration Activity of CR1
5.1. Classical/Lectin Pathway C3 and C5 Convertases
5.2. Alternative Pathway C3 and C5 Convertases
6. Co-Factor Activity of CR1
6.1. General and Comparative
6.2. Domain Contribution
7. Domain Contribution to CR1-Mediated Complement Pathway Inhibition
8. CSL040 and Its Mechanism of Action
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Fearon, D.T. Identification of the membrane glycoprotein that is the C3b receptor of the human erythrocyte, polymorphonuclear leukocyte, B lymphocyte, and monocyte. J. Exp. Med. 1980, 152, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.D.; Lambris, J.D. Identification of a C3bi-specific membrane complement receptor that is expressed on lymphocytes, monocytes, neutrophils, and erythrocytes. J. Exp. Med. 1982, 155, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Ahearn, J.M.; Fearon, D.T. Structure and function of the complement receptors, CR1 (CD35) and CR2 (CD21). Adv. Immunol. 1989, 46, 183–219. [Google Scholar] [CrossRef] [PubMed]
- Lublin, D.M.; Griffith, R.C.; Atkinson, J.P. Influence of glycosylation on allelic and cell-specific Mr variation, receptor processing, and ligand binding of the human complement C3b/C4b receptor. J. Biol. Chem. 1986, 261, 5736–5744. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, P.J. Biological functions of the complement system. Biochem. Soc. Trans. 1990, 18, 1143–1145. [Google Scholar] [CrossRef]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Kolev, M.; Le Friec, G.; Kemper, C. Complement—Tapping into new sites and effector systems. Nat. Rev. Immunol. 2014, 14, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, P.J. The amplification loop of the complement pathways. Adv. Immunol. 2009, 104, 115–149. [Google Scholar] [CrossRef]
- Pangburn, M.K. Initiation of the alternative pathway of complement and the history of “tickover”. Immunol. Rev. 2022, 313, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Frank, M.M. Therapeutic potential of complement modulation. Nat. Rev. Drug Discov. 2010, 9, 43–56. [Google Scholar] [CrossRef]
- Krych-Goldberg, M.; Atkinson, J.P. Structure-function relationships of complement receptor type 1. Immunol. Rev. 2001, 180, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Furtado, P.B.; Huang, C.Y.; Ihyembe, D.; Hammond, R.A.; Marsh, H.C.; Perkins, S.J. The partly folded back solution structure arrangement of the 30 SCR domains in human complement receptor type 1 (CR1) permits access to its C3b and C4b ligands. J. Mol. Biol. 2008, 375, 102–118. [Google Scholar] [CrossRef] [PubMed]
- Weis, J.H.; Morton, C.C.; Bruns, G.A.; Weis, J.J.; Klickstein, L.B.; Wong, W.W.; Fearon, D.T. A complement receptor locus: Genes encoding C3b/C4b receptor and C3d/Epstein-Barr virus receptor map to 1q32. J. Immunol. 1987, 138, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Klickstein, L.B.; Wong, W.W.; Smith, J.A.; Weis, J.H.; Wilson, J.G.; Fearon, D.T. Human C3b/C4b receptor (CR1). Demonstration of long homologous repeating domains that are composed of the short consensus repeats characteristics of C3/C4 binding proteins. J. Exp. Med. 1987, 165, 1095–1112. [Google Scholar] [CrossRef]
- Liu, D.; Niu, Z.X. The structure, genetic polymorphisms, expression and biological functions of complement receptor type 1 (CR1/CD35). Immunopharmacol. Immunotoxicol. 2009, 31, 524–535. [Google Scholar] [CrossRef]
- Kalli, K.R.; Hsu, P.H.; Bartow, T.J.; Ahearn, J.M.; Matsumoto, A.K.; Klickstein, L.B.; Fearon, D.T. Mapping of the C3b-binding site of CR1 and construction of a (CR1)2-F(ab’)2 chimeric complement inhibitor. J. Exp. Med. 1991, 174, 1451–1460. [Google Scholar] [CrossRef]
- Mqadmi, A.; Abdullah, Y.; Yazdanbakhsh, K. Characterization of complement receptor 1 domains for prevention of complement-mediated red cell destruction. Transfusion 2005, 45, 234–244. [Google Scholar] [CrossRef]
- Wymann, S.; Dai, Y.; Nair, A.G.; Cao, H.; Powers, G.A.; Schnell, A.; Martin-Roussety, G.; Leong, D.; Simmonds, J.; Lieu, K.G.; et al. A novel soluble complement receptor 1 fragment with enhanced therapeutic potential. J. Biol. Chem. 2021, 296, 100200. [Google Scholar] [CrossRef]
- Hardy, M.P.; Rowe, T.; Wymann, S. Soluble Complement Receptor 1 Therapeutics. J. Immunol. Sci. 2022, 6, 1–17. [Google Scholar] [CrossRef]
- Fearon, D.T. Regulation of the amplification C3 convertase of human complement by an inhibitory protein isolated from human erythrocyte membrane. Proc. Natl. Acad. Sci. USA 1979, 76, 5867–5871. [Google Scholar] [CrossRef]
- Karthikeyan, G.; Baalasubramanian, S.; Seth, S.; Das, N. Low levels of plasma soluble complement receptor type 1 in patients receiving thrombolytic therapy for acute myocardial infarction. J. Thromb. Thrombolysis 2007, 23, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Arnaout, M.A.; Melamed, J.; Tack, B.F.; Colten, H.R. Characterization of the human complement (c3b) receptor with a fluid phase C3b dimer. J. Immunol. 1981, 127, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Arnaout, M.A.; Dana, N.; Melamed, J.; Medicus, R.; Colten, H.R. Low ionic strength or chemical cross-linking of monomeric C3b increases its binding affinity to the human complement C3b receptor. Immunology 1983, 48, 229–237. [Google Scholar] [PubMed]
- Weisman, H.F.; Bartow, T.; Leppo, M.K.; Marsh, H.C., Jr.; Carson, G.R.; Concino, M.F.; Boyle, M.P.; Roux, K.H.; Weisfeldt, M.L.; Fearon, D.T. Soluble human complement receptor type 1: In vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science 1990, 249, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.W.; Farrell, S.A. Proposed structure of the F’ allotype of human CR1. Loss of a C3b binding site may be associated with altered function. J. Immunol. 1991, 146, 656–662. [Google Scholar] [CrossRef]
- Makrides, S.C.; Scesney, S.M.; Ford, P.J.; Evans, K.S.; Carson, G.R.; Marsh, H.C., Jr. Cell surface expression of the C3b/C4b receptor (CR1) protects Chinese hamster ovary cells from lysis by human complement. J. Biol. Chem. 1992, 267, 24754–24761. [Google Scholar] [CrossRef]
- Scesney, S.M.; Makrides, S.C.; Gosselin, M.L.; Ford, P.J.; Andrews, B.M.; Hayman, E.G.; Marsh, H.C., Jr. A soluble deletion mutant of the human complement receptor type 1, which lacks the C4b binding site, is a selective inhibitor of the alternative complement pathway. Eur. J. Immunol. 1996, 26, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Klickstein, L.B.; Barbashov, S.F.; Liu, T.; Jack, R.M.; Nicholson-Weller, A. Complement receptor type 1 (CR1, CD35) is a receptor for C1q. Immunity 1997, 7, 345–355. [Google Scholar] [CrossRef]
- Alcorlo, M.; Martinez-Barricarte, R.; Fernandez, F.J.; Rodriguez-Gallego, C.; Round, A.; Vega, M.C.; Harris, C.L.; de Cordoba, S.R.; Llorca, O. Unique structure of iC3b resolved at a resolution of 24 A by 3D-electron microscopy. Proc. Natl. Acad. Sci. USA 2011, 108, 13236–13240. [Google Scholar] [CrossRef]
- Schramm, E.C.; Roumenina, L.T.; Rybkine, T.; Chauvet, S.; Vieira-Martins, P.; Hue, C.; Maga, T.; Valoti, E.; Wilson, V.; Jokiranta, S.; et al. Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood 2015, 125, 2359–2369. [Google Scholar] [CrossRef]
- Dobson, N.J.; Lambris, J.D.; Ross, G.D. Characteristics of isolated erythrocyte complement receptor type one (CR1, C4b-C3b receptor) and CR1-specific antibodies. J. Immunol. 1981, 126, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.D.; Lambris, J.D.; Cain, J.A.; Newman, S.L. Generation of three different fragments of bound C3 with purified factor I or serum. I. Requirements for factor H vs. CR1 cofactor activity. J. Immunol. 1982, 129, 2051–2060. [Google Scholar] [CrossRef]
- Ross, G.D. Analysis of the different types of leukocyte membrane complement receptors and their interaction with the complement system. J. Immunol. Methods 1980, 37, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Klickstein, L.B.; Bartow, T.J.; Miletic, V.; Rabson, L.D.; Smith, J.A.; Fearon, D.T. Identification of distinct C3b and C4b recognition sites in the human C3b/C4b receptor (CR1, CD35) by deletion mutagenesis. J. Exp. Med. 1988, 168, 1699–1717. [Google Scholar] [CrossRef] [PubMed]
- Pangburn, M.K. Differences between the binding sites of the complement regulatory proteins DAF, CR1, and factor H on C3 convertases. J. Immunol. 1986, 136, 2216–2221. [Google Scholar] [CrossRef] [PubMed]
- Reilly, B.D.; Makrides, S.C.; Ford, P.J.; Marsh, H.C., Jr.; Mold, C. Quantitative analysis of C4b dimer binding to distinct sites on the C3b/C4b receptor (CR1). J. Biol. Chem. 1994, 269, 7696–7701. [Google Scholar] [CrossRef] [PubMed]
- Belt, K.T.; Carroll, M.C.; Porter, R.R. The structural basis of the multiple forms of human complement component C4. Cell 1984, 36, 907–914. [Google Scholar] [CrossRef]
- Clemenza, L.; Isenman, D.E. The C4A and C4B isotypic forms of human complement fragment C4b have the same intrinsic affinity for complement receptor 1 (CR1/CD35). J. Immunol. 2004, 172, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, P.A.; Barbosa, J.E.; Lachmann, P.J. Differences between C4A and C4B in the handling of immune complexes: The enhancement of CR1 binding is more important than the inhibition of immunoprecipitation. Clin. Exp. Immunol. 1990, 79, 158–163. [Google Scholar] [CrossRef]
- Reilly, B.D.; Mold, C. Quantitative analysis of C4Ab and C4Bb binding to the C3b/C4b receptor (CR1, CD35). Clin. Exp. Immunol. 1997, 110, 310–316. [Google Scholar] [CrossRef]
- Prohaska, R.; Adolf, G.R. Characterization of the human erythrocyte complement receptor CR1 (C3b receptor) by epitope mapping. Immunobiology 1987, 174, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Krych, M.; Clemenza, L.; Howdeshell, D.; Hauhart, R.; Hourcade, D.; Atkinson, J.P. Analysis of the functional domains of complement receptor type 1 (C3b/C4b receptor; CD35) by substitution mutagenesis. J. Biol. Chem. 1994, 269, 13273–13278. [Google Scholar] [CrossRef] [PubMed]
- Krych, M.; Hauhart, R.; Atkinson, J.P. Structure-function analysis of the active sites of complement receptor type 1. J. Biol. Chem. 1998, 273, 8623–8629. [Google Scholar] [CrossRef] [PubMed]
- Krych, M.; Hourcade, D.; Atkinson, J.P. Sites within the complement C3b/C4b receptor important for the specificity of ligand binding. Proc. Natl. Acad. Sci. USA 1991, 88, 4353–4357. [Google Scholar] [CrossRef] [PubMed]
- Yazdanbakhsh, K.; Kang, S.; Tamasauskas, D.; Sung, D.; Scaradavou, A. Complement receptor 1 inhibitors for prevention of immune-mediated red cell destruction: Potential use in transfusion therapy. Blood 2003, 101, 5046–5052. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.O.; Mallin, R.L.; Krych-Goldberg, M.; Wang, X.; Hauhart, R.E.; Bromek, K.; Uhrin, D.; Atkinson, J.P.; Barlow, P.N. Structure of the C3b binding site of CR1 (CD35), the immune adherence receptor. Cell 2002, 108, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Korb, L.C.; Ahearn, J.M. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: Complement deficiency and systemic lupus erythematosus revisited. J. Immunol. 1997, 158, 4525–4528. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer, R.D.; Racila, E.; Racila, D.M. C1q: Its functions within the innate and adaptive immune responses and its role in lupus autoimmunity. J. Investig. Dermatol. 2005, 125, 14–23. [Google Scholar] [CrossRef]
- Tas, S.W.; Klickstein, L.B.; Barbashov, S.F.; Nicholson-Weller, A. C1q and C4b bind simultaneously to CR1 and additively support erythrocyte adhesion. J. Immunol. 1999, 163, 5056–5063. [Google Scholar] [CrossRef]
- Jacquet, M.; Cioci, G.; Fouet, G.; Bally, I.; Thielens, N.M.; Gaboriaud, C.; Rossi, V. C1q and Mannose-Binding Lectin Interact with CR1 in the Same Region on CCP24-25 Modules. Front. Immunol. 2018, 9, 453. [Google Scholar] [CrossRef]
- Ghiran, I.; Barbashov, S.F.; Klickstein, L.B.; Tas, S.W.; Jensenius, J.C.; Nicholson-Weller, A. Complement receptor 1/CD35 is a receptor for mannan-binding lectin. J. Exp. Med. 2000, 192, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, M.; Lacroix, M.; Ancelet, S.; Gout, E.; Gaboriaud, C.; Thielens, N.M.; Rossi, V. Deciphering complement receptor type 1 interactions with recognition proteins of the lectin complement pathway. J. Immunol. 2013, 190, 3721–3731. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, D.C.; Chalmers, J.D. Human L-ficolin (ficolin-2) and its clinical significance. J. Biomed. Biotechnol. 2012, 2012, 138797. [Google Scholar] [CrossRef] [PubMed]
- Emlen, W.; Burdick, G.; Carl, V.; Lachmann, P.J. Binding of model immune complexes to erythrocyte CR1 facilitates immune complex uptake by U937 cells. J. Immunol. 1989, 142, 4366–4371. [Google Scholar] [CrossRef] [PubMed]
- Nardin, A.; Lindorfer, M.A.; Taylor, R.P. How are immune complexes bound to the primate erythrocyte complement receptor transferred to acceptor phagocytic cells? Mol. Immunol. 1999, 36, 827–835. [Google Scholar] [CrossRef]
- Janssen, B.J.; Christodoulidou, A.; McCarthy, A.; Lambris, J.D.; Gros, P. Structure of C3b reveals conformational changes that underlie complement activity. Nature 2006, 444, 213–216. [Google Scholar] [CrossRef]
- Park, H.J.; Guariento, M.; Maciejewski, M.; Hauhart, R.; Tham, W.H.; Cowman, A.F.; Schmidt, C.Q.; Mertens, H.D.; Liszewski, M.K.; Hourcade, D.E.; et al. Using mutagenesis and structural biology to map the binding site for the Plasmodium falciparum merozoite protein PfRh4 on the human immune adherence receptor. J. Biol. Chem. 2014, 289, 450–463. [Google Scholar] [CrossRef]
- Forneris, F.; Wu, J.; Xue, X.; Ricklin, D.; Lin, Z.; Sfyroera, G.; Tzekou, A.; Volokhina, E.; Granneman, J.C.; Hauhart, R.; et al. Regulators of complement activity mediate inhibitory mechanisms through a common C3b-binding mode. EMBO J. 2016, 35, 1133–1149. [Google Scholar] [CrossRef]
- Iida, K.; Nussenzweig, V. Complement receptor is an inhibitor of the complement cascade. J. Exp. Med. 1981, 153, 1138–1150. [Google Scholar] [CrossRef]
- Takata, Y.; Kinoshita, T.; Kozono, H.; Takeda, J.; Tanaka, E.; Hong, K.; Inoue, K. Covalent association of C3b with C4b within C5 convertase of the classical complement pathway. J. Exp. Med. 1987, 165, 1494–1507. [Google Scholar] [CrossRef]
- Krych-Goldberg, M.; Hauhart, R.E.; Subramanian, V.B.; Yurcisin, B.M., 2nd; Crimmins, D.L.; Hourcade, D.E.; Atkinson, J.P. Decay accelerating activity of complement receptor type 1 (CD35). Two active sites are required for dissociating C5 convertases. J. Biol. Chem. 1999, 274, 31160–31168. [Google Scholar] [CrossRef] [PubMed]
- Krych-Goldberg, M.; Hauhart, R.E.; Porzukowiak, T.; Atkinson, J.P. Synergy between two active sites of human complement receptor type 1 (CD35) in complement regulation: Implications for the structure of the classical pathway C3 convertase and generation of more potent inhibitors. J. Immunol. 2005, 175, 4528–4535. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Takata, Y.; Kozono, H.; Takeda, J.; Hong, K.S.; Inoue, K. C5 convertase of the alternative complement pathway: Covalent linkage between two C3b molecules within the trimolecular complex enzyme. J. Immunol. 1988, 141, 3895–3901. [Google Scholar] [CrossRef] [PubMed]
- Medof, M.E.; Iida, K.; Mold, C.; Nussenzweig, V. Unique role of the complement receptor CR1 in the degradation of C3b associated with immune complexes. J. Exp. Med. 1982, 156, 1739–1754. [Google Scholar] [CrossRef] [PubMed]
- Medof, M.E.; Nussenzweig, V. Control of the function of substrate-bound C4b-C3b by the complement receptor Cr1. J. Exp. Med. 1984, 159, 1669–1685. [Google Scholar] [CrossRef] [PubMed]
- Mossakowska, D.; Dodd, I.; Pindar, W.; Smith, R.A. Structure-activity relationships within the N-terminal short consensus repeats (SCR) of human CR1 (C3b/C4b receptor, CD35): SCR 3 plays a critical role in inhibition of the classical and alternative pathways of complement activation. Eur. J. Immunol. 1999, 29, 1955–1965. [Google Scholar] [CrossRef]
- Smith, R.A. Targeting anticomplement agents. Biochem. Soc. Trans. 2002, 30, 1037–1041. [Google Scholar] [CrossRef]
- Bongoni, A.K.; Vikstrom, I.B.; McRae, J.L.; Salvaris, E.J.; Fisicaro, N.; Pearse, M.J.; Wymann, S.; Rowe, T.; Morelli, A.B.; Hardy, M.P.; et al. A potent truncated form of human soluble CR1 is protective in a mouse model of renal ischemia-reperfusion injury. Sci. Rep. 2021, 11, 21873. [Google Scholar] [CrossRef]
- Wymann, S.; Mischnik, M.; Leong, D.; Ghosh, S.; Tan, X.; Cao, H.; Kuehnemuth, B.; Powers, G.A.; Halder, P.; de Souza, M.J.; et al. Sialylation-dependent pharmacokinetics and differential complement pathway inhibition are hallmarks of CR1 activity in vivo. Biochem. J. 2022, 479, 1007–1030. [Google Scholar] [CrossRef]

| Affinity to C3b Dimer (nM) | Affinity to C3b Monomer (nM) | Method Used | CR1 Source | References |
|---|---|---|---|---|
| 9–16 | No binding | Radioligand binding assay | E-CR1 | Arnaout et al., 1981 [22] |
| 4.62 | No binding a | Radioligand binding assay | E-CR1 | Arnaout et al., 1983 [23] |
| 1.3 b | >100-fold weaker c | Radioligand binding/competition | sCR1 and E-CR1 | Wiesman et al., 1990 [24] |
| 10 b | 1000 b | Radioligand binding/competition | sCR1 and E-CR1 | Wong and Farrell 1991 [25] |
| 1.0 | Not measured | Radioligand binding assay | Expressed on K562 cells | Kalli et al., 1991 [16] |
| 19 | 200 | Radioligand binding assay | E-CR1 | Makrides et al., 1992 [26] |
| 12–19 | 800 | Radioligand binding assay | Expressed on CHO cells | Makrides et al., 1992 [26] |
| 30 b | 600 b | Radioligand binding/competition | sCR1 and E-CR1 | Scesney et al., 1996 [27] |
| 17.9–40.6 | Not measured | SPR | sCR1 | Klickstein et al., 1997 [28] |
| 140 d | Not measured | SPR | sCR1 | Alcorlo et al., 2011 [29] |
| 69 e | SPR | sCR1 | Schramm et al., 2015 [30] | |
| 385.7 e | SPR | sCR1 | Wymann et al., 2021 [18] | |
| LHR-A | LHR-B | LHR-C | LHR-D | CP C3 DAA Activity Relative to LHR-ABCD |
|---|---|---|---|---|
| 60%, 43% a | ||||
| 12% | ||||
| 8%, 10% b | ||||
| 0% | ||||
| 100% | ||||
| 100% | ||||
| 8% | ||||
| Required | Either site required | Not Required | ||
| LHR-A | LHR-B | LHR-C | LHR-D | CP C5 DAA Activity Relative to LHR-ABCD |
|---|---|---|---|---|
| 0.5%, 1% a | ||||
| 3% | ||||
| 95% b | ||||
| 100% | ||||
| Required | Either site required | Not Required | ||
| LHR-A | LHR-B | LHR-C | LHR-D | AP C5 DAA Activity Relative to LHR-ABCD |
|---|---|---|---|---|
| 0.5%, 10% a | ||||
| 0% | ||||
| 0% | ||||
| 0% | ||||
| 50% | ||||
| 30-fold weaker | ||||
| 0.6% | ||||
| Required | Required | Required | Not Required |
| CR1 Domain(s) | Number Of C3b/C4b Binding Sites | Classical IC50 (nM) ± S.D. | Alternative IC50 (nM) ± S.D. |
|---|---|---|---|
| LHR-ABC/CSL040 | 3 | 0.42 ± 0.10 | 0.90 ± 0.54 |
| LHR-AB | 2 | 4.61 ± 1.85 | 13.15 ± 2.18 |
| LHR-BC | 2 | 10.08 ± 5.52 | 12.48 ± 4.30 |
| LHR-A | 1 | 68.38 ± 9.52 | 49.87 ± 24.76 |
| LHR-B | 1 | 959.37 ± 243.92 | 63.38 ± 31.03 |
| LHR-C | 1 | 733.10 ± 203.47 | 66.36 ± 18.10 |
| LHR-D | 0 | No Activity | No Activity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hardy, M.P.; Mansour, M.; Rowe, T.; Wymann, S. The Molecular Mechanisms of Complement Receptor 1—It Is Complicated. Biomolecules 2023, 13, 1522. https://doi.org/10.3390/biom13101522
Hardy MP, Mansour M, Rowe T, Wymann S. The Molecular Mechanisms of Complement Receptor 1—It Is Complicated. Biomolecules. 2023; 13(10):1522. https://doi.org/10.3390/biom13101522
Chicago/Turabian StyleHardy, Matthew P., Mariam Mansour, Tony Rowe, and Sandra Wymann. 2023. "The Molecular Mechanisms of Complement Receptor 1—It Is Complicated" Biomolecules 13, no. 10: 1522. https://doi.org/10.3390/biom13101522
APA StyleHardy, M. P., Mansour, M., Rowe, T., & Wymann, S. (2023). The Molecular Mechanisms of Complement Receptor 1—It Is Complicated. Biomolecules, 13(10), 1522. https://doi.org/10.3390/biom13101522