
High Fructose Corn Syrup Accelerates Kidney Disease and Mortality in Obese Mice with Metabolic Syndrome
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Approval
2.2. Animal Experiments
2.3. Oral Fructose Tolerance Test
2.4. Intestinal Permeability Assay
2.5. Tissue KHK Activity
2.6. Biochemical Analysis
2.7. Histopathology
2.8. Western Blot Analysis
2.9. DNA Isolation and Quantitative Real-Time PCR
2.10. Statistical Analysis
3. Results
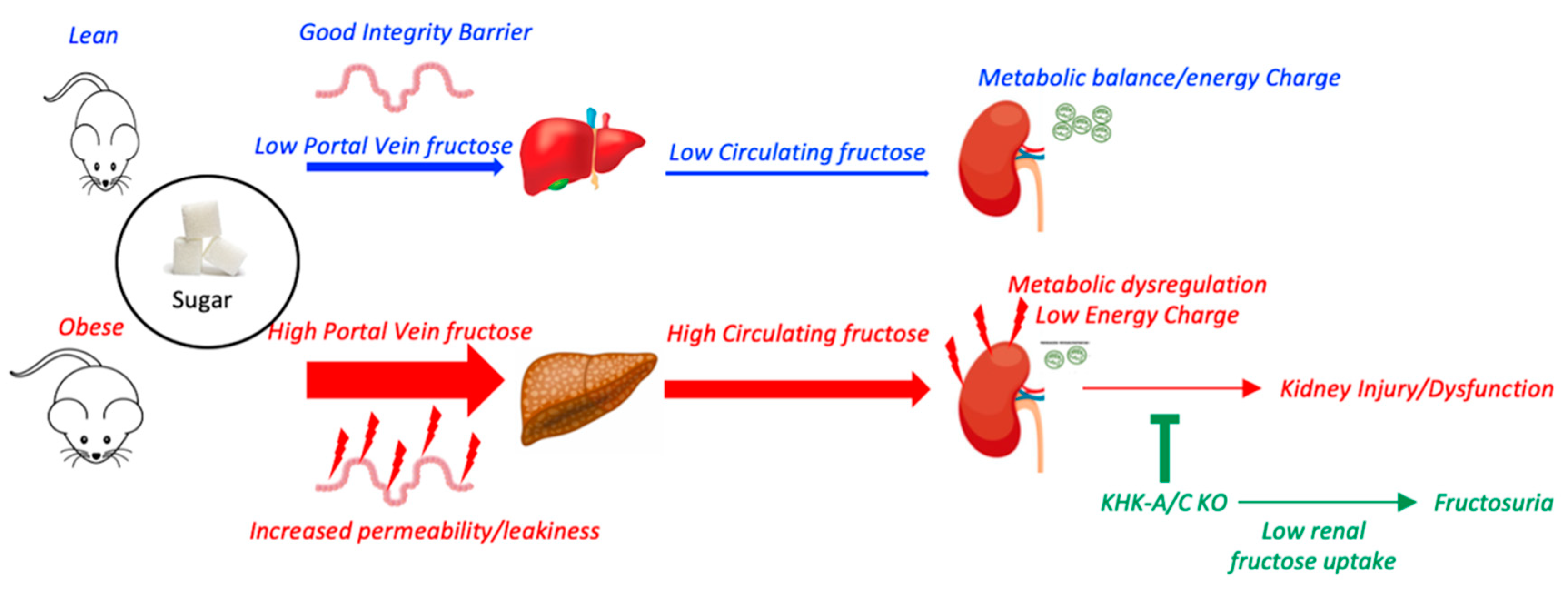
3.1. Metabolic Syndrome Is Associated with Leaky Gut and Enhanced Fructose Absorption
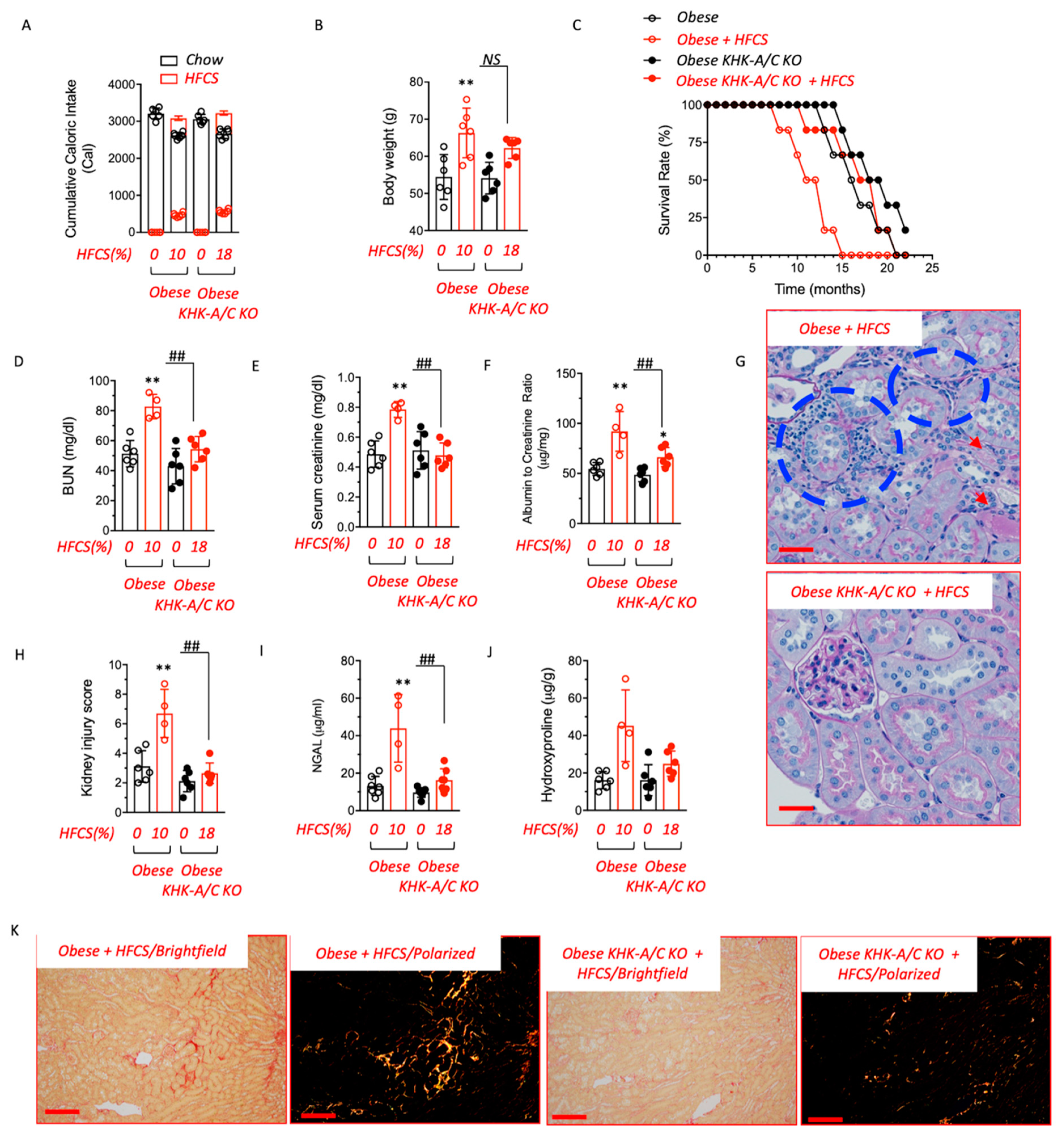
3.2. Chronic Intake of a HFCS-like Beverage Accelerates Development of CKD in Obese Mice
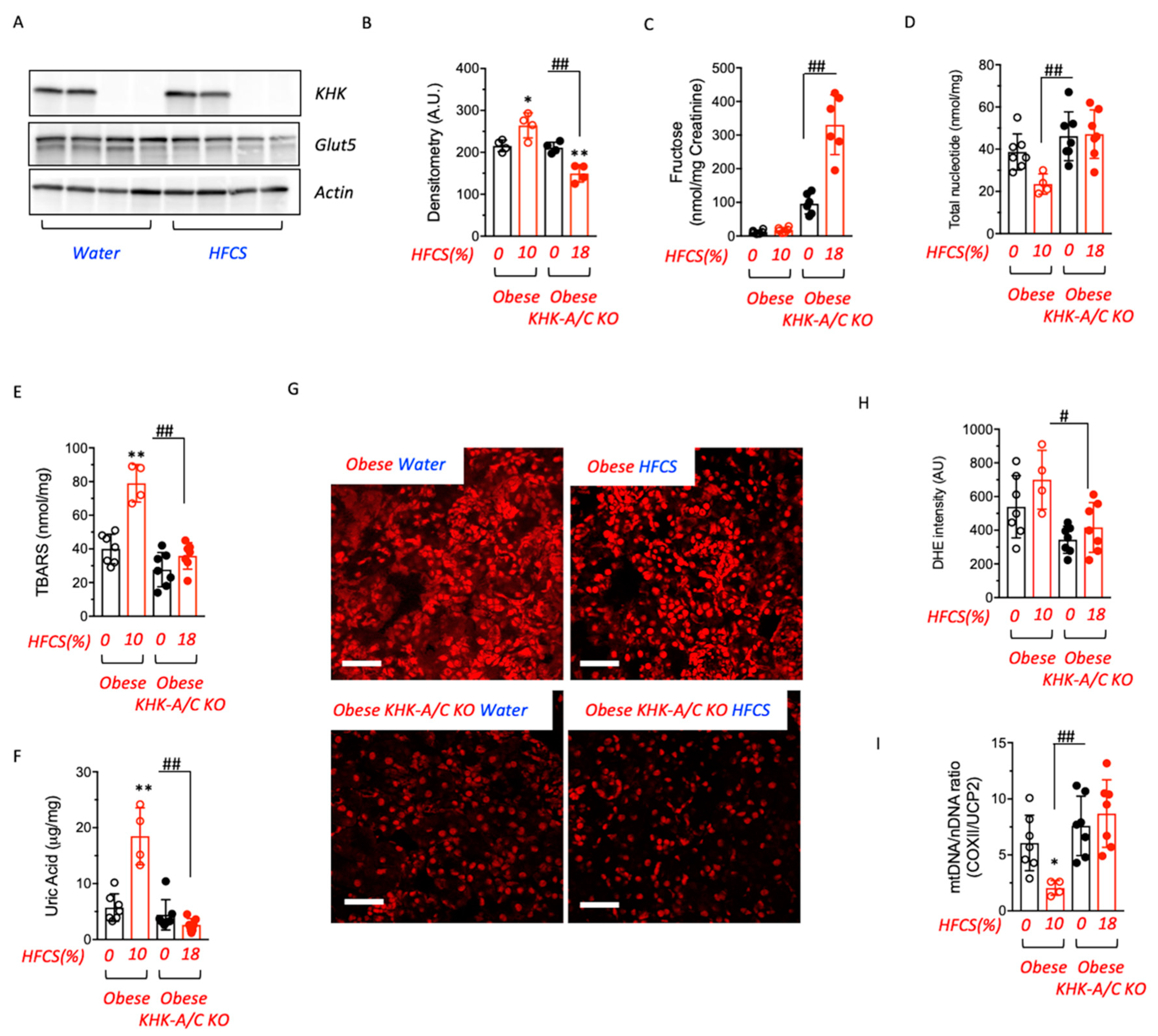
3.3. Blockade of Fructose Metabolism Protects against HFCS-Induced Kidney Dysfunction and Injury in Obese Mice
3.4. Fructose Metabolism Controls Fructose Transport in the Kidney
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hirode, G.; Wong, R.J. Trends in the Prevalence of Metabolic Syndrome in the United States, 2011–2016. JAMA 2020, 323, 2526–2528. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Muntner, P.; Hamm, L.L.; Jones, D.W.; Batuman, V.; Fonseca, V.; Whelton, P.K.; He, J. The metabolic syndrome and chronic kidney disease in U.S. adults. Ann. Intern. Med. 2004, 140, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Andres-Hernando, A.; Lanaspa, M.A.; Kuwabara, M.; Orlicky, D.J.; Cicerchi, C.; Bales, E.; Garcia, G.E.; Roncal-Jimenez, C.A.; Sato, Y.; Johnson, R.J. Obesity causes renal mitochondrial dysfunction and energy imbalance and accelerates chronic kidney disease in mice. Am. J. Physiol. Renal Physiol. 2019, 317, F941–F948. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Shafiu, M.; Sundaram, S.; Le, M.; Ishimoto, T.; Sautin, Y.Y.; Lanaspa, M.A. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 2013, 62, 3307–3315. [Google Scholar] [CrossRef]
- Perez-Pozo, S.E.; Schold, J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Johnson, R.J.; Lillo, J.L. Excessive fructose intake induces the features of metabolic syndrome in healthy adult men: Role of uric acid in the hypertensive response. Int. J. Obes. 2010, 34, 454–461. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Cicerchi, C.; Garcia, G.; Li, N.; Roncal-Jimenez, C.A.; Rivard, C.J.; Hunter, B.; Andres-Hernando, A.; Ishimoto, T.; Sanchez-Lozada, L.G.; et al. Counteracting roles of AMP deaminase and AMP kinase in the development of fatty liver. PLoS ONE 2012, 7, e48801. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef]
- Johnson, R.J.; Stenvinkel, P.; Andrews, P.; Sanchez-Lozada, L.G.; Nakagawa, T.; Gaucher, E.; Andres-Hernando, A.; Rodriguez-Iturbe, B.; Jimenez, C.R.; Garcia, G.; et al. Fructose metabolism as a common evolutionary pathway of survival associated with climate change, food shortage and droughts. J. Intern. Med. 2020, 287, 252–262. [Google Scholar] [CrossRef]
- Sanchez-Lozada, L.G.; Tapia, E.; Jimenez, A.; Bautista, P.; Cristobal, M.; Nepomuceno, T.; Soto, V.; Avila-Casado, C.; Nakagawa, T.; Johnson, R.J.; et al. Fructose-induced metabolic syndrome is associated with glomerular hypertension and renal microvascular damage in rats. Am. J. Physiol. Renal Physiol. 2007, 292, F423–F429. [Google Scholar] [CrossRef]
- Andres-Hernando, A.; Jensen, T.J.; Kuwabara, M.; Orlicky, D.J.; Cicerchi, C.; Li, N.; Roncal-Jimenez, C.A.; Garcia, G.E.; Ishimoto, T.; Maclean, P.S.; et al. Vasopressin mediates fructose-induced metabolic syndrome by activating the V1b receptor. JCI Insight 2021, 6, e140848. [Google Scholar] [CrossRef]
- Gersch, M.S.; Mu, W.; Cirillo, P.; Reungjui, S.; Zhang, L.; Roncal, C.; Sautin, Y.Y.; Johnson, R.J.; Nakagawa, T. Fructose, but not dextrose, accelerates the progression of chronic kidney disease. Am. J. Physiol. Renal Physiol. 2007, 293, F1256–F1261. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Kosugi, T.; Gersch, M.; Connor, T.; Sanchez-Lozada, L.G.; Lanaspa, M.A.; Roncal, C.; Perez-Pozo, S.E.; Johnson, R.J.; Nakagawa, T. Dietary fructose causes tubulointerstitial injury in the normal rat kidney. Am. J. Physiol. Renal Physiol. 2010, 298, F712–F720. [Google Scholar] [CrossRef] [PubMed]
- Shoham, D.A.; Durazo-Arvizu, R.; Kramer, H.; Luke, A.; Vupputuri, S.; Kshirsagar, A.; Cooper, R.S. Sugary soda consumption and albuminuria: Results from the National Health and Nutrition Examination Survey, 1999–2004. PLoS ONE 2008, 3, e3431. [Google Scholar] [CrossRef]
- Bomback, A.S.; Derebail, V.K.; Shoham, D.A.; Anderson, C.A.; Steffen, L.M.; Rosamond, W.D.; Kshirsagar, A.V. Sugar-sweetened soda consumption, hyperuricemia, and kidney disease. Kidney Int. 2010, 77, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Rebholz, C.M.; Young, B.A.; Katz, R.; Tucker, K.L.; Carithers, T.C.; Norwood, A.F.; Correa, A. Patterns of Beverages Consumed and Risk of Incident Kidney Disease. Clin. J. Am. Soc. Nephrol. 2019, 14, 49–56. [Google Scholar] [CrossRef]
- Cai, X.Y.; Zhang, N.H.; Cheng, Y.C.; Ge, S.W.; Xu, G. Sugar-sweetened beverage consumption and mortality of chronic kidney disease: Results from the US National Health and Nutrition Examination Survey, 1999–2014. Clin. Kidney J. 2022, 15, 718–726. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, B.; Sheng, L.T.; Pan, X.F.; Zhou, Y.; Zhu, J.; Li, X.; Yang, K.; Guo, K.; Zhang, X.; et al. Association between weight status, metabolic syndrome, and chronic kidney disease among middle-aged and elderly Chinese. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 2017–2026. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Andres-Hernando, A.; Rivard, C.J.; Dai, Y.; Berl, T. Hypertonic stress increases claudin-4 expression and tight junction integrity in association with MUPP1 in IMCD3 cells. Proc. Natl. Acad. Sci. USA 2008, 105, 15797–15802. [Google Scholar] [CrossRef]
- Taylor, S.R.; Ramsamooj, S.; Liang, R.J.; Katti, A.; Pozovskiy, R.; Vasan, N.; Hwang, S.K.; Nahiyaan, N.; Francoeur, N.J.; Schatoff, E.M.; et al. Dietary fructose improves intestinal cell survival and nutrient absorption. Nature 2021, 597, 263–267. [Google Scholar] [CrossRef]
- Cho, Y.E.; Kim, D.K.; Seo, W.; Gao, B.; Yoo, S.H.; Song, B.J. Fructose Promotes Leaky Gut, Endotoxemia, and Liver Fibrosis Through Ethanol-Inducible Cytochrome P450-2E1-Mediated Oxidative and Nitrative Stress. Hepatology 2021, 73, 2180–2195. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Andres-Hernando, A.; Orlicky, D.J.; Cicerchi, C.; Jang, C.; Li, N.; Milagres, T.; Kuwabara, M.; Wempe, M.F.; Rabinowitz, J.D.; et al. Ketohexokinase C blockade ameliorates fructose-induced metabolic dysfunction in fructose-sensitive mice. J. Clin. Investig. 2018, 128, 2226–2238. [Google Scholar] [CrossRef] [PubMed]
- Diggle, C.P.; Shires, M.; Leitch, D.; Brooke, D.; Carr, I.M.; Markham, A.F.; Hayward, B.E.; Asipu, A.; Bonthron, D.T. Ketohexokinase: Expression and localization of the principal fructose-metabolizing enzyme. J. Histochem. Cytochem. 2009, 57, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Andres-Hernando, A.; Orlicky, D.J.; Kuwabara, M.; Ishimoto, T.; Nakagawa, T.; Johnson, R.J.; Lanaspa, M.A. Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab. 2020, 32, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Lanaspa, M.A.; Le, M.T.; Garcia, G.E.; Diggle, C.P.; Maclean, P.S.; Jackman, M.R.; Asipu, A.; Roncal-Jimenez, C.A.; Kosugi, T.; et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 4320–4325. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, P.; Gersch, M.S.; Mu, W.; Scherer, P.M.; Kim, K.M.; Gesualdo, L.; Henderson, G.N.; Johnson, R.J.; Sautin, Y.Y. Ketohexokinase-dependent metabolism of fructose induces proinflammatory mediators in proximal tubular cells. J. Am. Soc. Nephrol. 2009, 20, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, W.; McRae, S.; Marek, G.; Wymer, D.; Pannu, V.; Baylis, C.; Johnson, R.J.; Sautin, Y.Y. Hyperuricemia as a mediator of the proinflammatory endocrine imbalance in the adipose tissue in a murine model of the metabolic syndrome. Diabetes 2011, 60, 1258–1269. [Google Scholar] [CrossRef] [PubMed]
- Roncal-Jimenez, C.A.; Lanaspa, M.A.; Rivard, C.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Jalal, D.; Andres-Hernando, A.; Tanabe, K.; Madero, M.; Li, N.; et al. Sucrose induces fatty liver and pancreatic inflammation in male breeder rats independent of excess energy intake. Metabolism 2011, 60, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lozada, L.G.; Andres-Hernando, A.; Garcia-Arroyo, F.E.; Cicerchi, C.; Li, N.; Kuwabara, M.; Roncal-Jimenez, C.A.; Johnson, R.J.; Lanaspa, M.A. Uric acid activates aldose reductase and the polyol pathway for endogenous fructose and fat production causing development of fatty liver in rats. J. Biol. Chem. 2019, 294, 4272–4281. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Cicerchi, C.; Li, N.; Roncal-Jimenez, C.A.; Ishimoto, T.; Le, M.; Garcia, G.E.; Thomas, J.B.; Rivard, C.J.; et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS ONE 2012, 7, e47948. [Google Scholar] [CrossRef]
- Johnson, R.J.; Rivard, C.; Lanaspa, M.A.; Otabachian-Smith, S.; Ishimoto, T.; Cicerchi, C.; Cheeke, P.R.; Macintosh, B.; Hess, T. Fructokinase, Fructans, Intestinal Permeability, and Metabolic Syndrome: An Equine Connection? J. Equine Vet. Sci. 2013, 33, 120–126. [Google Scholar] [CrossRef]
- Sanchez-Lozada, L.G.; Tapia, E.; Bautista-Garcia, P.; Soto, V.; Avila-Casado, C.; Vega-Campos, I.P.; Nakagawa, T.; Zhao, L.; Franco, M.; Johnson, R.J. Effects of febuxostat on metabolic and renal alterations in rats with fructose-induced metabolic syndrome. Am. J. Physiol. Renal Physiol. 2008, 294, F710–F718. [Google Scholar] [CrossRef] [PubMed]
- Glushakova, O.; Kosugi, T.; Roncal, C.; Mu, W.; Heinig, M.; Cirillo, P.; Sanchez-Lozada, L.G.; Johnson, R.J.; Nakagawa, T. Fructose induces the inflammatory molecule ICAM-1 in endothelial cells. J. Am. Soc. Nephrol. 2008, 19, 1712–1720. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhang, Z.; Gregg, E.W.; Flanders, W.D.; Merritt, R.; Hu, F.B. Added Sugar Intake and Cardiovascular Diseases Mortality among US Adults. JAMA Intern. Med. 2014, 174, 516–524. [Google Scholar] [CrossRef]
- Friedman, J.M. Leptin and the endocrine control of energy balance. Nat. Metab. 2019, 1, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Andres-Hernando, A.; Li, N.; Cicerchi, C.; Inaba, S.; Chen, W.; Roncal-Jimenez, C.; Le, M.T.; Wempe, M.F.; Milagres, T.; Ishimoto, T.; et al. Protective role of fructokinase blockade in the pathogenesis of acute kidney injury in mice. Nat. Commun. 2017, 8, 14181. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Ishimoto, T.; Cicerchi, C.; Tamura, Y.; Roncal-Jimenez, C.A.; Chen, W.; Tanabe, K.; Andres-Hernando, A.; Orlicky, D.J.; Finol, E.; et al. Endogenous fructose production and fructokinase activation mediate renal injury in diabetic nephropathy. J. Am. Soc. Nephrol. 2014, 25, 2526–2538. [Google Scholar] [CrossRef]
- Korczynska, J.; Czumaj, A.; Chmielewski, M.; Swierczynski, J.; Sledzinski, T. The Causes and Potential Injurious Effects of Elevated Serum Leptin Levels in Chronic Kidney Disease Patients. Int. J. Mol. Sci. 2021, 22, 4685. [Google Scholar] [CrossRef]
- Marchelek-Mysliwiec, M.; Wisniewska, M.; Nowosiad-Magda, M.; Safranow, K.; Kwiatkowska, E.; Banach, B.; Dolegowska, B.; Dolegowska, K.; Stepniewska, J.; Domanski, L.; et al. Association Between Plasma Concentration of Klotho Protein, Osteocalcin, Leptin, Adiponectin, and Bone Mineral Density in Patients with Chronic Kidney Disease. Horm. Metab. Res. 2018, 50, 816–821. [Google Scholar] [CrossRef]
- Alix, P.M.; Guebre-Egziabher, F.; Soulage, C.O. Leptin as an uremic toxin: Deleterious role of leptin in chronic kidney disease. Biochimie 2014, 105, 12–21. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Kuwabara, M.; Andres-Hernando, A.; Li, N.; Cicerchi, C.; Jensen, T.; Orlicky, D.J.; Roncal-Jimenez, C.A.; Ishimoto, T.; Nakagawa, T.; et al. High salt intake causes leptin resistance and obesity in mice by stimulating endogenous fructose production and metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, 3138–3143. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Ishimoto, T.; Li, N.; Cicerchi, C.; Orlicky, D.J.; Ruzycki, P.; Rivard, C.; Inaba, S.; Roncal-Jimenez, C.A.; Bales, E.S.; et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 2013, 4, 2434. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wild Type | KHK-A/C KO | Pound | Pound KHK-A/C KO | |||||||
| Water | HFCS | Water | HFCS | Water | HFCS | Water | HFCS | |||
| Feature | Points | n = 7 | n = 7 | n = 7 | n = 7 | n = 6 | n = 4 | n = 6 | n = 6 | |
| (1) Renal Corpuscle | ||||||||||
| Mesangial expansion | None | 0 | x | x | x | x | x | x | x | x |
| (0–2) | Yes, central | 1–2 | ||||||||
| (0–2) | Yes, crescent | 1–2 | ||||||||
| Hypercellularity | None | 0 | x | x | x | x | x | x | ||
| (0–2) | Present | 1–2 | x | x | ||||||
| Inflammation | None | 0 | x | x | x | x | x | x | x | x |
| (0–2) | Moderate | 1 | ||||||||
| Severe | 2 | |||||||||
| Protein Casts in glomeruli (Vessels) | None | 0 | x | x | x | x | x | x | x | |
| (0–2) | Present | 2 | x | |||||||
| Protein Casts in glomerular urinary space | None | 0 | x | x | x | x | x | x | x | x |
| (0–2) | Present | 1–2 | ||||||||
| if yes, give approx % of RC with this | Check > 25RC | na | ||||||||
| thickened basement membrane | None | 0 | x | x | x | x | x | x | x | |
| (0–2) | Moderate | 1 | x | |||||||
| Severe | 2 | |||||||||
| renal corpuscle loss (0–2) | None (<10%) | 0 | x | x | x | x | x | x | x | x |
| Yes (>10%) | 2 | |||||||||
| afferent arteriole hyalinized/thickened | No | 0 | x | x | x | x | x | x | x | x |
| Yes | 1 | |||||||||
| metaplasia | Ct Only | Ct | ||||||||
| proxim c metaplas of renal corpus pariet c | <20% | 0 | x | x | x | x | x | x | x | x |
| (>180 degree or stratified) | >20% | 1 | ||||||||
| Bowman’s Capsule thickened | None | 0 | x | x | x | x | x | x | x | x |
| (0–2) | Present | 1–2 | ||||||||
| Renal corpuscle subtotal | 0 | 0 | 0 | 0 | 0.7 | 2.4 | 0 | 0 | ||
| (2) Cortex | ||||||||||
| Acute Tubular Necrosis (0–2) | None | 0 | x | x | x | x | x | x | x | x |
| Moderate | 1 | |||||||||
| Severe | 2 | |||||||||
| Any cortical casts, cell, protein | None | 0 | x | x | x | x | x | x | ||
| (0–2) | Moderate | 1 | x | x | ||||||
| Severe | 2 | |||||||||
| PCT Pathology (0–2) | None | 0 | x | x | x | x | x | x | ||
| Simplification | 1 | x | x | |||||||
| Necrosis | 2 | |||||||||
| PCT vacuolization present | Y/N | |||||||||
| Extent (a=little b=lots) | A,B,C,N | |||||||||
| Tubular Dilatation | None | 0 | x | x | x | |||||
| Present | 1 | x | x | x | x | x | ||||
| DCT Pathology (0–2) | None | 0 | x | x | x | x | x | x | x | x |
| Prot,disrupt | 1 | |||||||||
| Necrosis | 2 | |||||||||
| DCT Glycogen granules (0–1) | None | 0 | x | x | x | x | x | x | ||
| Present | 1 | x | x | |||||||
| Peritub/Interstit Inflamm | None | 0 | x | |||||||
| Near level of the AA (0–2) | Moderate | 1 | x | x | ||||||
| Severe | 2 | x | ||||||||
| Increase peritub interstit -tis fibrosis | None | 0 | x | x | x | x | x | x | x | |
| Near level of the AA (0–2) | Moderate | 1 | x | |||||||
| Severe | 2 | |||||||||
| Brown Pigmented macs | None | 0 | x | x | x | x | x | x | x | |
| Present | 1 | x | ||||||||
| Hemosiderosis on collect duct | None | 0 | x | x | x | x | x | x | x | x |
| Present | 1 | |||||||||
| Peri-arcuate artery inflamm | None | 0 | x | x | x | x | x | x | x | |
| Around 10–50% of AA | Moderate | 1 | x | |||||||
| Around >50% of AA | Severe | 2 | ||||||||
| Cortex Subtotal | 0 | 1.1 | 0 | 0 | 1.8 | 3.1 | 0.8 | 1.6 | ||
| (3) Medulla | ||||||||||
| OM Casts (0–2) | None | 0 | x | x | x | x | ||||
| Present | 1–2 | x | x | x | x | |||||
| IM Casts (0–2) | None | 0 | x | x | x | x | ||||
| Present | 1–2 | x | x | x | ||||||
| Inflammation (0–2) | None | 0 | x | x | x | x | x | x | ||
| Present | 1–2 | x | x | |||||||
| Medulla subtotal | 0 | 0 | 0 | 0 | 1.1 | 1.6 | 0.7 | 1.2 | ||
| (4) Other | ||||||||||
| Peri-interlobar artery inflammation | None | 0 | x | x | x | x | x | x | x | |
| (0–2) | Moderate | 1 | x | |||||||
| Severe | 2 | |||||||||
| Pelvic Inflammation | None | 0 | x | x | x | x | x | x | x | x |
| (0–2) | Moderate | 1 | ||||||||
| Severe | 2 | |||||||||
| Other subtotal | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 | 0 | ||
| Summary | ||||||||||
| Scoring by Category | Wild type | KHK-A/C KO | Pound | Pound KHK-A/C KO | ||||||
| Water | HFCS | Water | HFCS | Water | HFCS | Water | HFCS | |||
| n = 7 | n = 7 | n = 7 | n = 7 | n = 7 | n = 6 | n = 7 | n = 7 | |||
| (1) Renal Corpuscle | 0 | 0 | 0 | 0 | 0.7 | 2.4 | 0 | 0 | ||
| (2) Renal Cortex | 0 | 0.5 | 0 | 0 | 1.8 | 3.1 | 0.8 | 1.6 | ||
| (3) Medulla | 0 | 0 | 0 | 0 | 1.1 | 1.6 | 0.7 | 1.2 | ||
| (4) Other | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 | 0 | ||
| Total | 0 | 0.5 | 0 | 0 | 3.6 | 7.6 | 1.5 | 2.8 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andres-Hernando, A.; Orlicky, D.J.; Cicerchi, C.; Kuwabara, M.; Garcia, G.E.; Nakagawa, T.; Sanchez-Lozada, L.G.; Johnson, R.J.; Lanaspa, M.A. High Fructose Corn Syrup Accelerates Kidney Disease and Mortality in Obese Mice with Metabolic Syndrome. Biomolecules 2023, 13, 780. https://doi.org/10.3390/biom13050780
Andres-Hernando A, Orlicky DJ, Cicerchi C, Kuwabara M, Garcia GE, Nakagawa T, Sanchez-Lozada LG, Johnson RJ, Lanaspa MA. High Fructose Corn Syrup Accelerates Kidney Disease and Mortality in Obese Mice with Metabolic Syndrome. Biomolecules. 2023; 13(5):780. https://doi.org/10.3390/biom13050780
Chicago/Turabian StyleAndres-Hernando, Ana, David J. Orlicky, Christina Cicerchi, Masanari Kuwabara, Gabriela E. Garcia, Takahiko Nakagawa, Laura Gabriela Sanchez-Lozada, Richard J. Johnson, and Miguel A. Lanaspa. 2023. "High Fructose Corn Syrup Accelerates Kidney Disease and Mortality in Obese Mice with Metabolic Syndrome" Biomolecules 13, no. 5: 780. https://doi.org/10.3390/biom13050780