
Multidimensional Landscape of SA-AKI Revealed by Integrated Proteomics and Metabolomics Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Analysis of Renal Function
2.3. Quantitative Real-Time PCR (qPCR)
2.4. Proteomics Analysis
2.5. LC-MS/MS Analysis
2.6. Peptide and Protein Identification
2.7. Metabolomics Analysis
2.8. Statistical Methods and Bioinformatics Analysis
3. Results
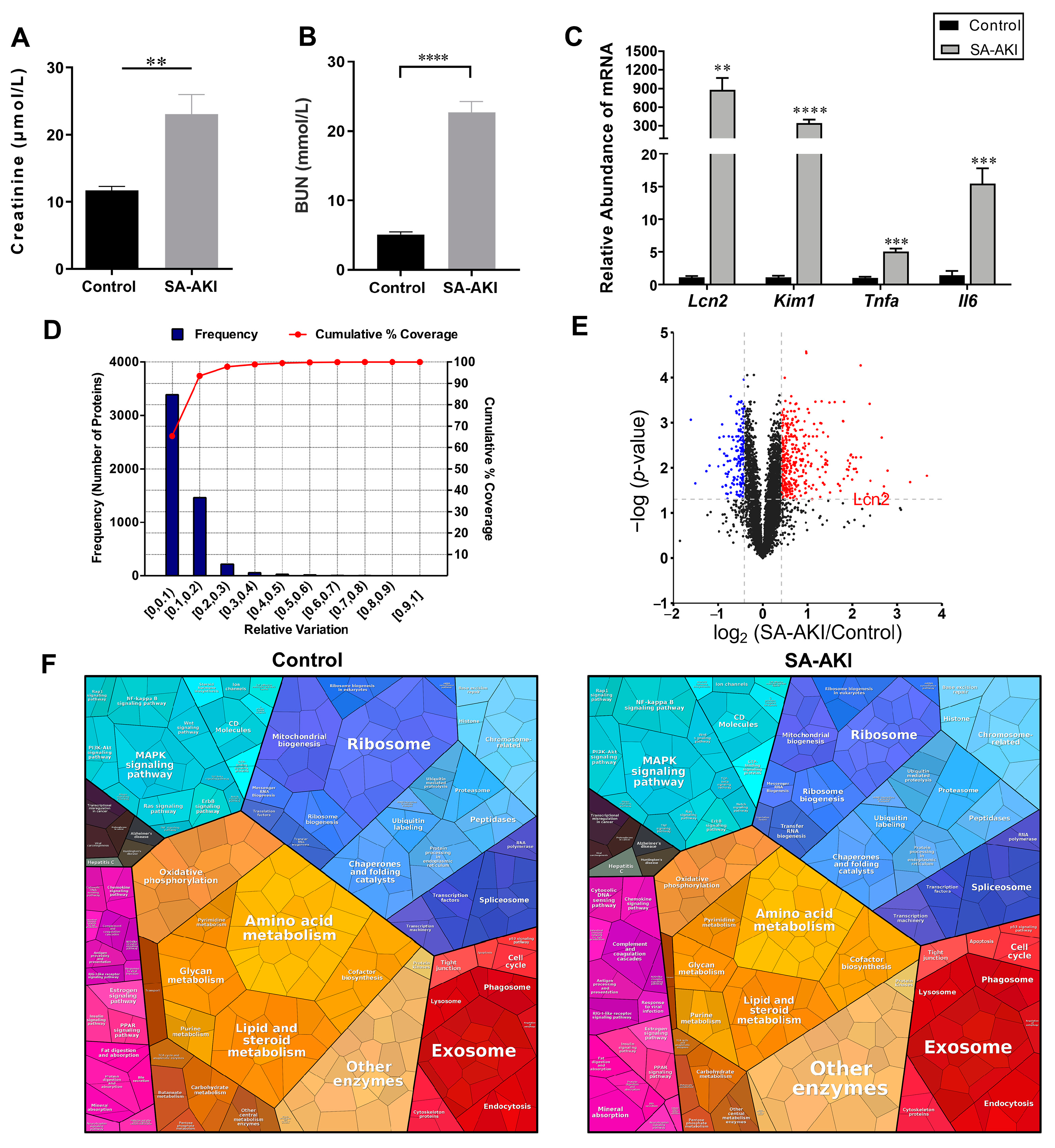
3.1. The Global Proteomic View of the Septic Kidneys
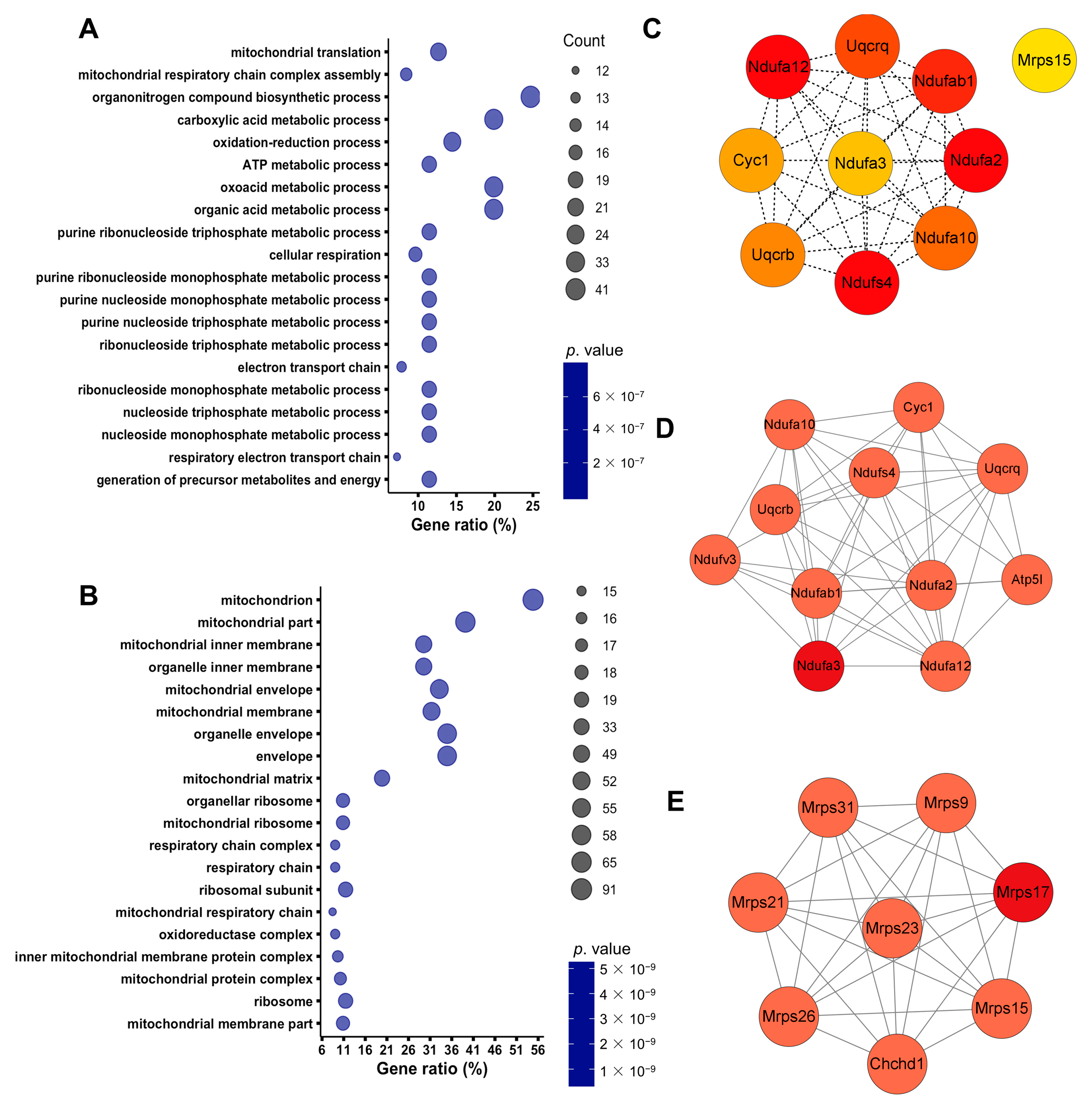
3.2. Septic Injury Impaired Mitochondrial Function in Kidneys
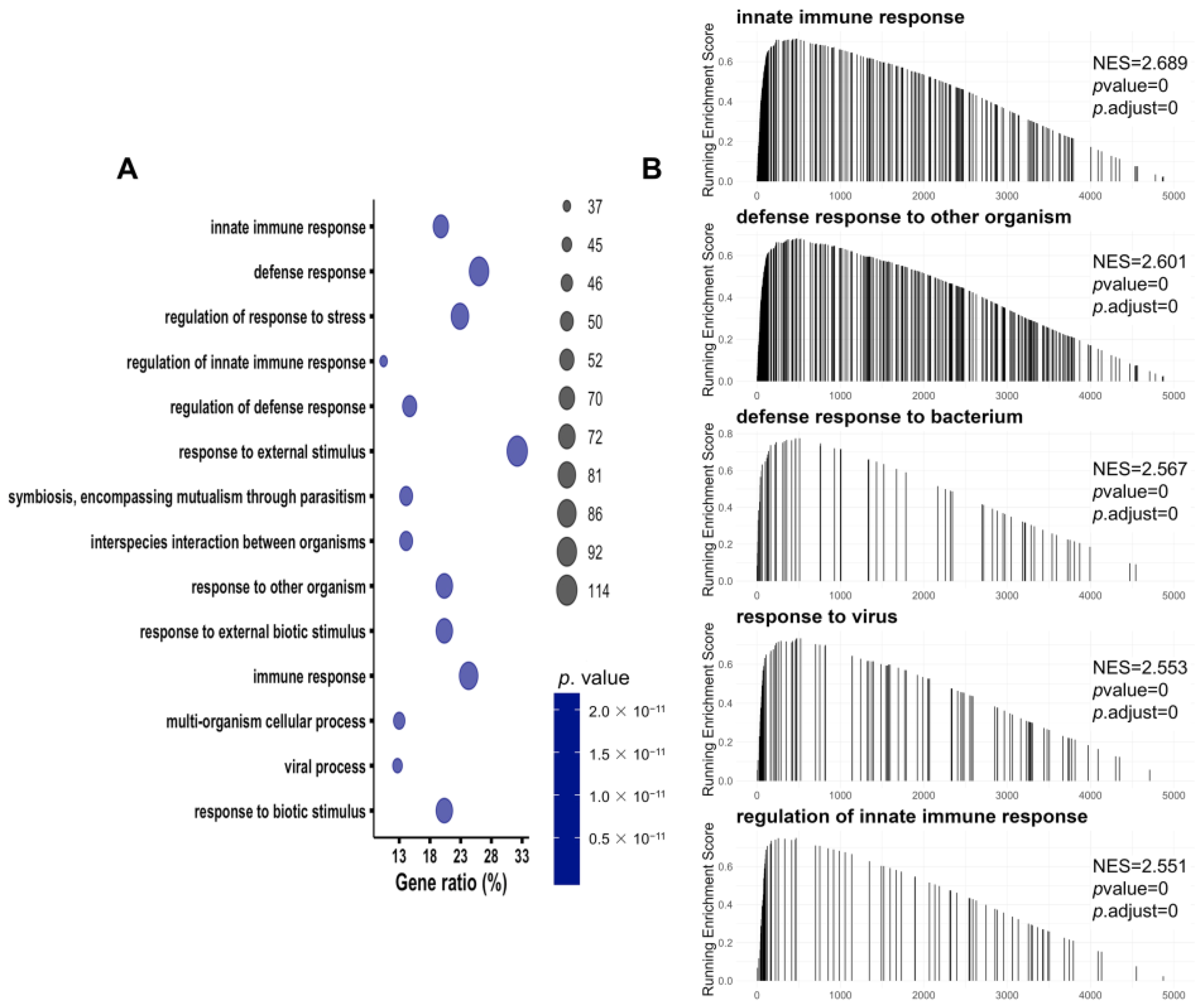
3.3. Immune-Related Pathways Were Significantly Activated in SA-AKI Kidneys
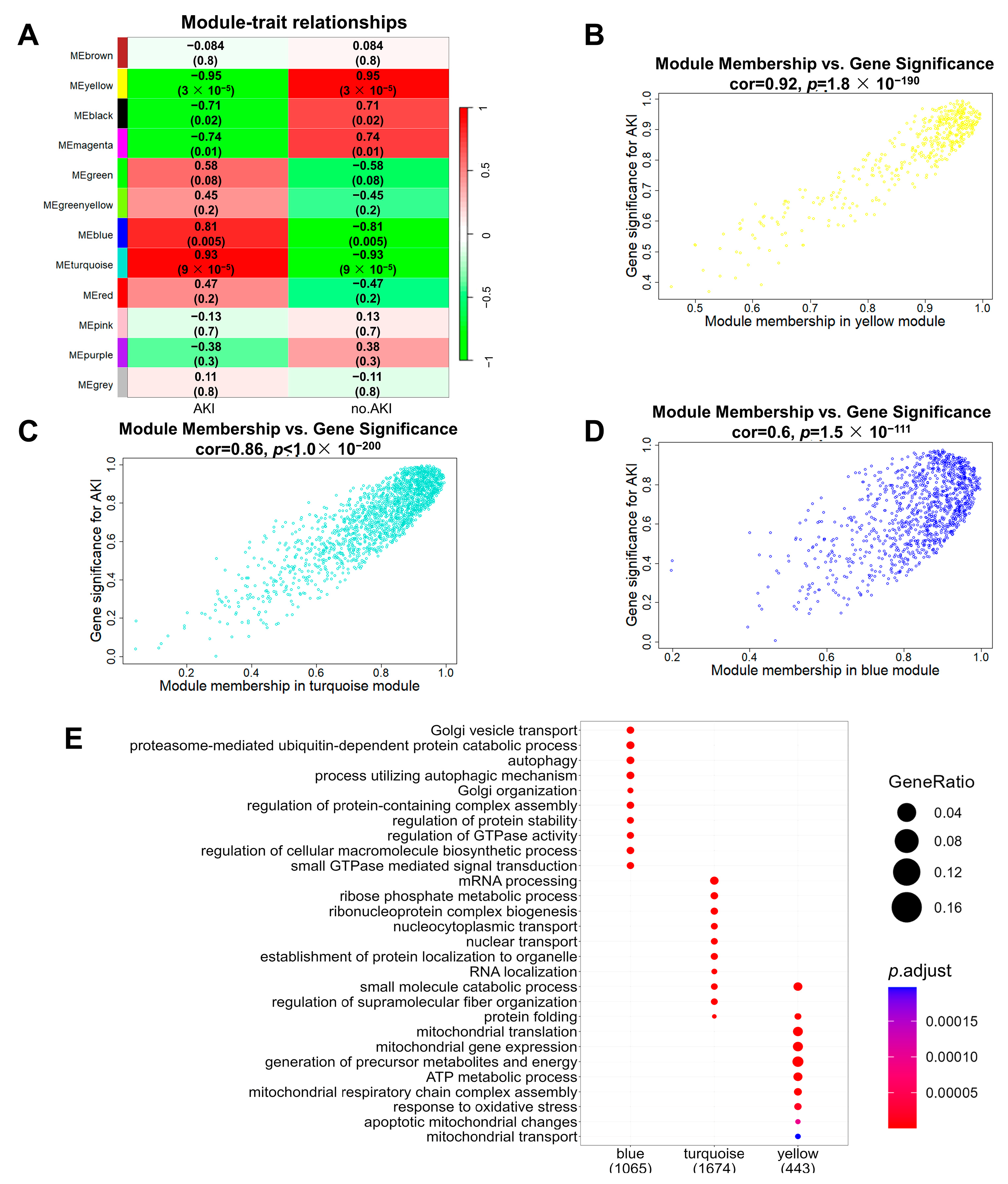
3.4. Construction of SA-AKI Protein Co-Expression Networks
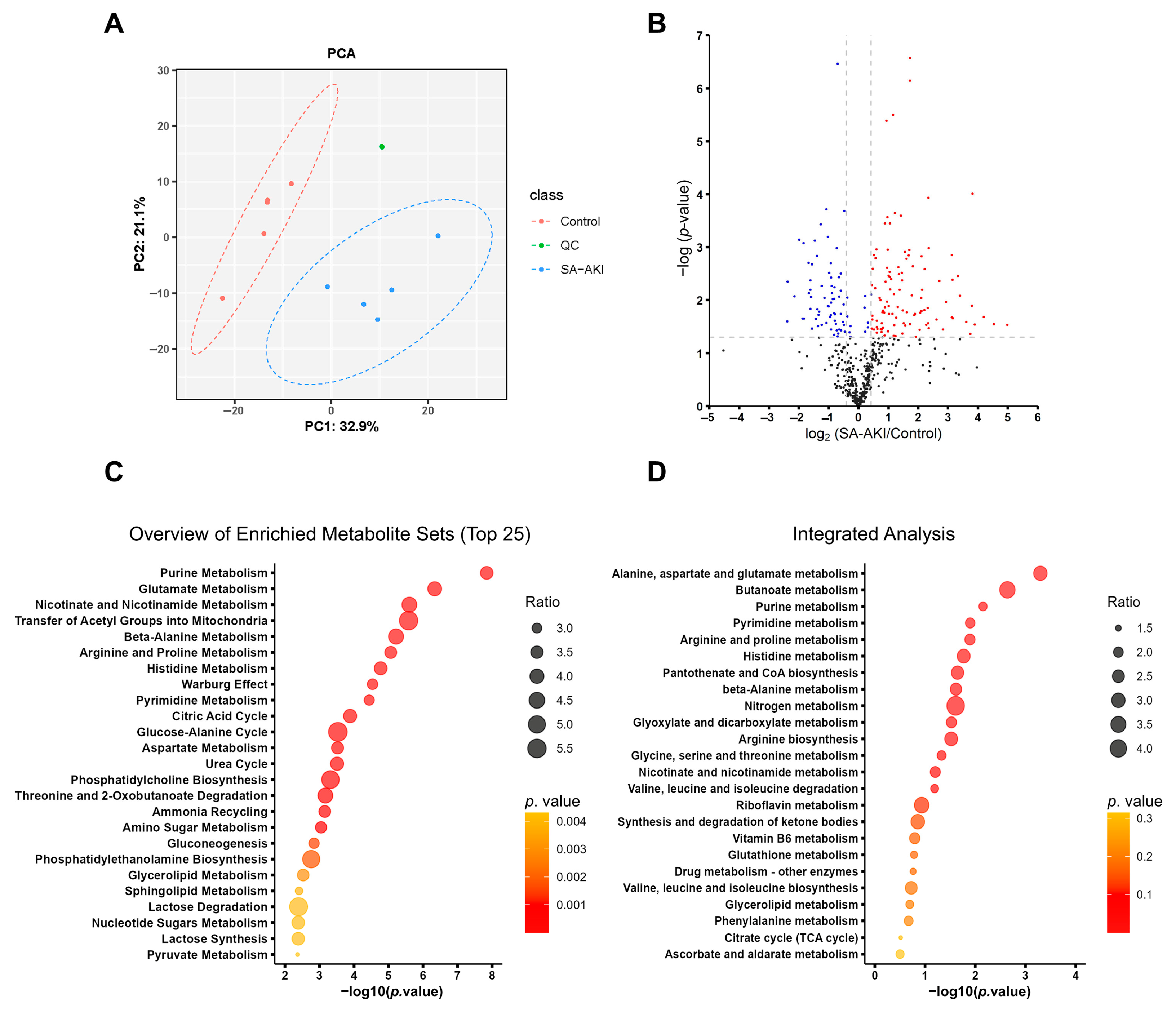
3.5. SA-AKI Induced Metabolic Disorders
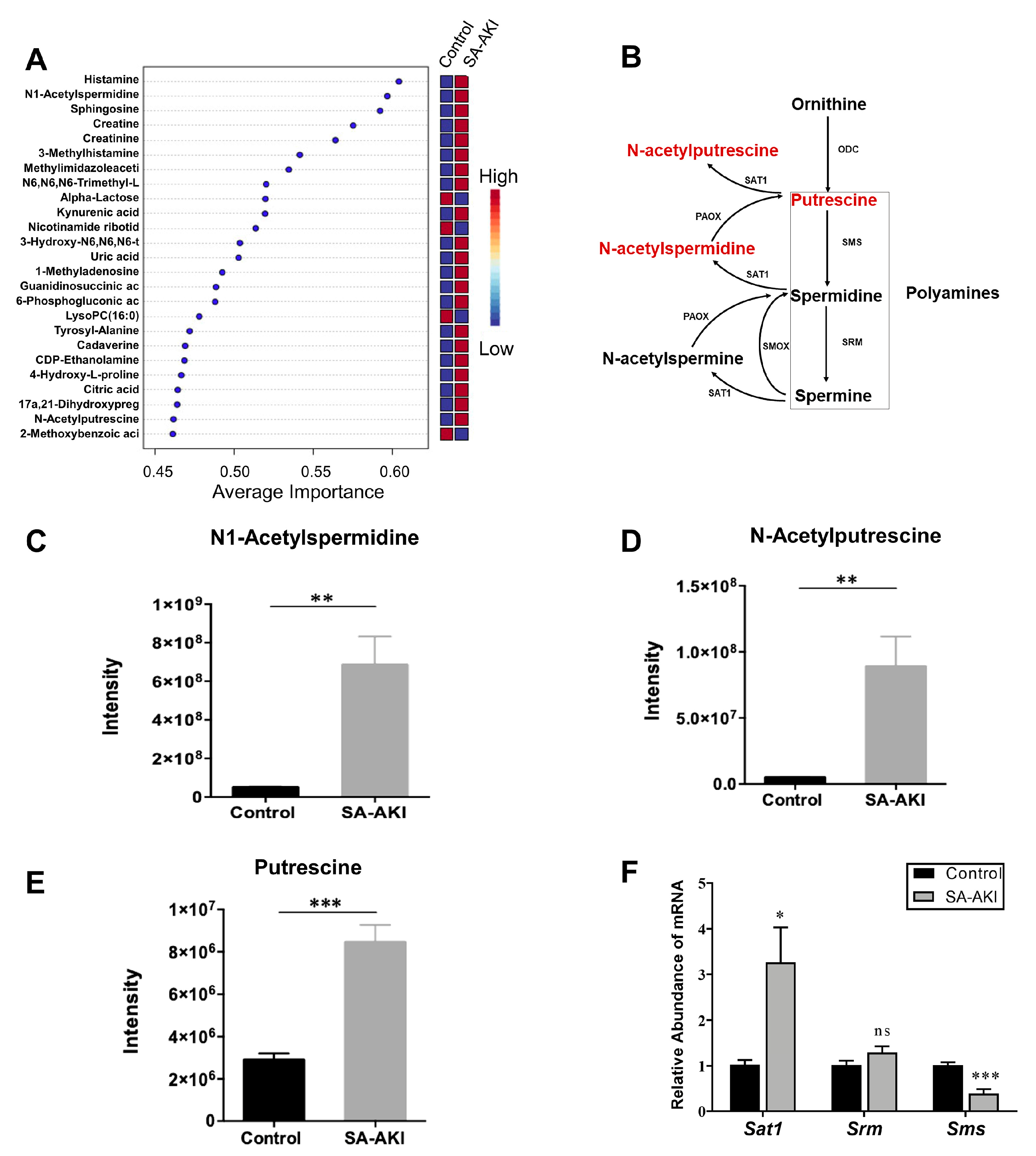
3.6. Catabolic Products of Polyamines Accumulated in SA-AKI Kidneys
4. Discussion
5. Study Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bellomo, R.; Kellum, J.A.; Ronco, C.; Wald, R.; Martensson, J.; Maiden, M.; Bagshaw, S.M.; Glassford, N.J.; Lankadeva, Y.; Vaara, S.T.; et al. Acute kidney injury in sepsis. Intensive Care Med. 2017, 43, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Godin, M.; Murray, P.; Mehta, R.L. Clinical Approach to the Patient With AKI and Sepsis. Semin. Nephrol. 2015, 35, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Uchino, S.; Kellum, J.A.; Bellomo, R.; Doig, G.S.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; et al. Acute renal failure in critically ill patients: A multinational, multicenter study. JAMA 2005, 294, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef]
- Hoste, E.A.J.; Bagshaw, S.M.; Bellomo, R.; Cely, C.M.; Colman, R.; Cruz, D.N.; Edipidis, K.; Forni, L.G.; Gomersall, C.D.; Govil, D.; et al. Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med. 2015, 41, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Gomez, H.; Kellum, J.A. Sepsis-induced acute kidney injury. Curr. Opin. Crit. Care 2016, 22, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Poston, J.T.; Koyner, J.L. Sepsis associated acute kidney injury. BMJ—Br. Med. J. 2019, 364, k4891. [Google Scholar] [CrossRef]
- Wheeler, D.S.; Devarajan, P.; Ma, Q.; Harmon, K.; Monaco, M.; Cvijanovich, N.; Wong, H.R. Serum neutrophil gelatinase-associated lipocalin (NGAL) as a marker of acute kidney injury in critically ill children with septic shock. Crit. Care Med. 2008, 36, 1297–1303. [Google Scholar] [CrossRef]
- Bell, M.; Larsson, A.; Venge, P.; Bellomo, R.; Martensson, J. Assessment of cell-cycle arrest biomarkers to predict early and delayed acute kidney injury. Dis. Mrk. 2015, 2015, 158658. [Google Scholar] [CrossRef]
- Tu, Y.; Wang, H.; Sun, R.; Ni, Y.; Ma, L.; Xv, F.; Hu, X.; Jiang, L.; Wu, A.; Chen, X.; et al. Urinary netrin-1 and KIM-1 as early biomarkers for septic acute kidney injury. Ren. Fail. 2014, 36, 1559–1563. [Google Scholar] [CrossRef]
- Peerapornratana, S.; Manrique-Caballero, C.L.; Gomez, H.; Kellum, J.A. Acute kidney injury from sepsis: Current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019, 96, 1083–1099. [Google Scholar] [CrossRef]
- Liu, E.T. Systems biology, integrative biology, predictive biology. Cell 2005, 121, 505–506. [Google Scholar] [CrossRef] [PubMed]
- Silverman, E.A.-O.; Schmidt, H.A.-O.; Anastasiadou, E.A.-O.; Altucci, L.A.-O.; Angelini, M.A.-O.; Badimon, L.A.-O.; Balligand, J.A.-O.; Benincasa, G.A.-O.; Capasso, G.A.-O.; Conte, F.A.-O.; et al. Molecular networks in Network Medicine: Development and applications. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1489. [Google Scholar] [CrossRef] [PubMed]
- Panizza, E.; Regalado, B.D.; Wang, F.; Nakano, I.; Vacanti, N.M.; Cerione, R.A.; Antonyak, M.A. Proteomic analysis reveals microvesicles containing NAMPT as mediators of radioresistance in glioma. Life Sci. Alliance 2023, 6, e202201680. [Google Scholar] [CrossRef]
- Roka, B.; Tod, P.; Kaucsar, T.; Vizovisek, M.; Vidmar, R.; Turk, B.; Fonovic, M.; Szenasi, G.; Hamar, P. The Acute Phase Response Is a Prominent Renal Proteome Change in Sepsis in Mice. Int. J. Mol. Sci. 2019, 21, 200. [Google Scholar] [CrossRef]
- Star, B.S.; Boahen, C.K.; van der Slikke, E.C.; Quinten, V.M.; Ter Maaten, J.C.; Henning, R.H.; Kumar, V.; Bouma, H.R. Plasma proteomic characterization of the development of acute kidney injury in early sepsis patients. Sci. Rep. 2022, 12, 19705. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Platt, M.P.; Fu, H.; Gui, Y.; Wang, Y.; Gonzalez-Juarbe, N.; Zhou, D.; Yu, Y. Global Proteome and Phosphoproteome Characterization of Sepsis-induced Kidney Injury. Mol. Cell Proteom. 2020, 19, 2030–2047. [Google Scholar] [CrossRef]
- Hasson, D.; Goldstein, S.L.; Standage, S.W. The application of omic technologies to research in sepsis-associated acute kidney injury. Pediatr. Nephrol. 2021, 36, 1075–1086. [Google Scholar] [CrossRef]
- Chen, Q.; Liang, X.; Wu, T.; Jiang, J.; Jiang, Y.; Zhang, S.; Ruan, Y.; Zhang, H.; Zhang, C.; Chen, P.; et al. Integrative analysis of metabolomics and proteomics reveals amino acid metabolism disorder in sepsis. J. Transl. Med. 2022, 20, 123. [Google Scholar] [CrossRef]
- Primer Designing Tool. Available online: https://www.ncbi.nlm.nih.gov/tools/primer-blast/ (accessed on 18 July 2020).
- Deutsch, E.W.; Bandeira, N.; Perez-Riverol, Y.A.-O.; Sharma, V.; Carver, J.J.; Mendoza, L.; Kundu, D.J.; Wang, S.; Bandla, C.; Kamatchinathan, S.; et al. The ProteomeXchange consortium at 10 years: 2023 update. (1362–4962 (Electronic)). Nucleic Acids Res. 2023, 51, D1539–D1548. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Tang, Y.; Li, M.; Wang, J.; Pan, Y.; Wu, F.-X. CytoNCA: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems 2015, 127, 67–72. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Walter, W.; Sanchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef]
- Lu, Y.; Pang, Z.; Xia, J. Comprehensive investigation of pathway enrichment methods for functional interpretation of LC–MS global metabolomics data. Brief. Bioinform. 2022, 24, bbac553. [Google Scholar] [CrossRef]
- Gan, C.S.; Chong, P.K.; Pham, T.K.; Wright, P.C. Technical, experimental, and biological variations in isobaric tags for relative and absolute quantitation (iTRAQ). J. Proteome Res. 2007, 6, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Liebermeister, W.; Noor, E.; Flamholz, A.; Davidi, D.; Bernhardt, J.; Milo, R. Visual account of protein investment in cellular functions. Proc. Natl. Acad. Sci. USA 2014, 111, 8488–8493. [Google Scholar] [CrossRef] [PubMed]
- DAVID: Functional Annotation Result Summary. Available online: https://david.ncifcrf.gov/summary.jsp (accessed on 10 February 2020).
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Di Cara, F.; Andreoletti, P.; Trompier, D.; Vejux, A.; Bulow, M.H.; Sellin, J.; Lizard, G.; Cherkaoui-Malki, M.; Savary, S. Peroxisomes in Immune Response and Inflammation. Int. J. Mol. Sci. 2019, 20, 3877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A General Framework for Weighted Gene Co-Expression Network Analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 1–45. [Google Scholar] [CrossRef]
- Alexander, M.P.; Mangalaparthi, K.K.; Madugundu, A.K.; Moyer, A.M.; Adam, B.A.; Mengel, M.; Singh, S.; Herrmann, S.M.; Rule, A.D.; Cheek, E.H.; et al. Acute Kidney Injury in Severe COVID-19 Has Similarities to Sepsis-Associated Kidney Injury: A Multi-Omics Study. Mayo Clin. Proc. 2021, 96, 2561–2575. [Google Scholar] [CrossRef]
- Doke, T.; Susztak, K. The multifaceted role of kidney tubule mitochondrial dysfunction in kidney disease development. Trends Cell Biol. 2022, 32, 841–853. [Google Scholar] [CrossRef]
- Hochachka, P.W.; Buck, L.T.; Doll, C.J.; Land, S.C. Unifying theory of hypoxia tolerance: Molecular metabolic defense and rescue mechanisms for surviving oxygen lack. Proc. Natl. Acad. Sci. USA 1996, 93, 9493–9498. [Google Scholar] [CrossRef]
- Kashani, K.; Al-Khafaji, A.; Ardiles, T.; Artigas, A.; Bagshaw, S.M.; Bell, M.; Bihorac, A.; Birkhahn, R.; Cely, C.M.; Chawla, L.S.; et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit. Care 2013, 17, 12. [Google Scholar] [CrossRef]
- Li, J.; Shi, X.; Chen, Z.; Xu, J.; Zhao, R.; Liu, Y.; Wen, Y.; Chen, L. Aldehyde dehydrogenase 2 alleviates mitochondrial dysfunction by promoting PGC-1alpha-mediated biogenesis in acute kidney injury. Cell Death Dis. 2023, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Lenis, Y.Y.; Elmetwally, M.A.; Maldonado-Estrada, J.G.; Bazer, F.W. Physiological importance of polyamines. Zygote 2017, 25, 244–255. [Google Scholar] [CrossRef]
- Zahedi, K.; Barone, S.L.; Xu, J.; Steinbergs, N.; Schuster, R.; Lentsch, A.B.; Amlal, H.; Wang, J.; Casero, R.A., Jr.; Soleimani, M. Hepatocyte-specific ablation of spermine/spermidine-N1-acetyltransferase gene reduces the severity of CCl4-induced acute liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G546–G560. [Google Scholar] [CrossRef]
- Ivanova, S.; Batliwalla, F.; Mocco, J.; Kiss, S.; Huang, J.; Mack, W.; Coon, A.; Eaton, J.W.; Al-Abed, Y.; Gregersen, P.K.; et al. Neuroprotection in cerebral ischemia by neutralization of 3-aminopropanal. Proc. Natl. Acad. Sci. USA 2002, 99, 5579–5584. [Google Scholar] [CrossRef]
- Poyhonen, M.J.; Takala, J.A.; Pitkanen, O.; Kari, A.; Alakuijala, L.A.; Eloranta, T.O. Differential-Effects of Sepsis and Trauma on Urinary-Excretion of Polyamines. Nutrition 1993, 9, 406–410. [Google Scholar] [PubMed]
- Bourgin, M.; Derosa, L.; Silva, C.A.C.; Goubet, A.G.; Dubuisson, A.; Danlos, F.X.; Grajeda-Iglesias, C.; Cerbone, L.; Geraud, A.; Laparra, A.; et al. Circulating acetylated polyamines correlate with Covid-19 severity in cancer patients. Aging 2021, 13, 20860–20885. [Google Scholar] [CrossRef] [PubMed]
- McCubbrey, A.L.; McManus, S.A.; McClendon, J.D.; Thomas, S.M.; Chatwin, H.B.; Reisz, J.A.; D’Alessandro, A.; Mould, K.J.; Bratton, D.L.; Henson, P.M.; et al. Polyamine import and accumulation causes immunomodulation in macrophages engulfing apoptotic cells. Cell Rep 2022, 38, 110222. [Google Scholar] [CrossRef]
- Puleston, D.J.; Buck, M.D.; Klein Geltink, R.I.; Kyle, R.L.; Caputa, G.; O’Sullivan, D.; Cameron, A.M.; Castoldi, A.; Musa, Y.; Kabat, A.M.; et al. Polyamines and eIF5A Hypusination Modulate Mitochondrial Respiration and Macrophage Activation. Cell Metab 2019, 30, 352–363. [Google Scholar] [CrossRef]
- Zhang, H.; Alsaleh, G.; Feltham, J.; Sun, Y.; Napolitano, G.; Riffelmacher, T.; Charles, P.; Frau, L.; Hublitz, P.; Yu, Z.; et al. Polyamines Control eIF5A Hypusination, TFEB Translation, and Autophagy to Reverse B Cell Senescence. Mol. Cell 2019, 76, 110–125. [Google Scholar] [CrossRef]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef]
- Bugarski, M.; Ghazi, S.; Polesel, M.; Martins, J.R.; Hall, A.M. Changes in NAD and Lipid Metabolism Drive Acidosis-Induced Acute Kidney Injury. J. Am. Soc. Nephrol. 2021, 32, 342–356. [Google Scholar] [CrossRef]
- Denzer, L.; Schroten, H.; Schwerk, C. From Gene to Protein-How Bacterial Virulence Factors Manipulate Host Gene Expression During Infection. Int. J. Mol. Sci. 2020, 21, 3730. [Google Scholar] [CrossRef]
- Selkrig, J.; Stanifer, M.; Mateus, A.; Mitosch, K.; Barrio-Hernandez, I.; Rettel, M.; Kim, H.; Voogdt, C.G.P.; Walch, P.; Kee, C.; et al. SARS-CoV-2 infection remodels the host protein thermal stability landscape. Mol. Syst. Biol. 2021, 17, e10188. [Google Scholar] [CrossRef]
- Nightingale, K.; Lin, K.M.; Ravenhill, B.J.; Davies, C.; Nobre, L.; Fielding, C.A.; Ruckova, E.; Fletcher-Etherington, A.; Soday, L.; Nichols, H.; et al. High-Definition Analysis of Host Protein Stability during Human Cytomegalovirus Infection Reveals Antiviral Factors and Viral Evasion Mechanisms. Cell Host Microbe 2018, 24, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Yin, Y.L.; Li, D.; Kim, S.W.; Wu, G. Amino acids and immune function. Br. J. Nutr. 2007, 98, 237–252. [Google Scholar] [CrossRef] [PubMed]
- McGaha, T.L.; Huang, L.; Lemos, H.; Metz, R.; Mautino, M.; Prendergast, G.C.; Mellor, A.L. Amino acid catabolism: A pivotal regulator of innate and adaptive immunity. Immunol. Rev. 2012, 249, 135–157. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.P.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Li, J.; Li, Y.; Shi, X.; Zhu, H.; Chen, L. Multidimensional Landscape of SA-AKI Revealed by Integrated Proteomics and Metabolomics Analysis. Biomolecules 2023, 13, 1329. https://doi.org/10.3390/biom13091329
Xu J, Li J, Li Y, Shi X, Zhu H, Chen L. Multidimensional Landscape of SA-AKI Revealed by Integrated Proteomics and Metabolomics Analysis. Biomolecules. 2023; 13(9):1329. https://doi.org/10.3390/biom13091329
Chicago/Turabian StyleXu, Jiatong, Jiaying Li, Yan Li, Xiaoxiao Shi, Huadong Zhu, and Limeng Chen. 2023. "Multidimensional Landscape of SA-AKI Revealed by Integrated Proteomics and Metabolomics Analysis" Biomolecules 13, no. 9: 1329. https://doi.org/10.3390/biom13091329