
A Structural Investigation of the Interaction between a GC-376-Based Peptidomimetic PROTAC and Its Precursor with the Viral Main Protease of Coxsackievirus B3

 , ,
, ,  , ,
, ,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Expression and Purification of CVB3 3CPro
2.2. Analytical Gel Filtration
2.3. Crystallization, Data Collection and Structure Solution
2.4. NMR Spectroscopy
2.5. Enzyme Inhibition Kinetics Assay
3. Results
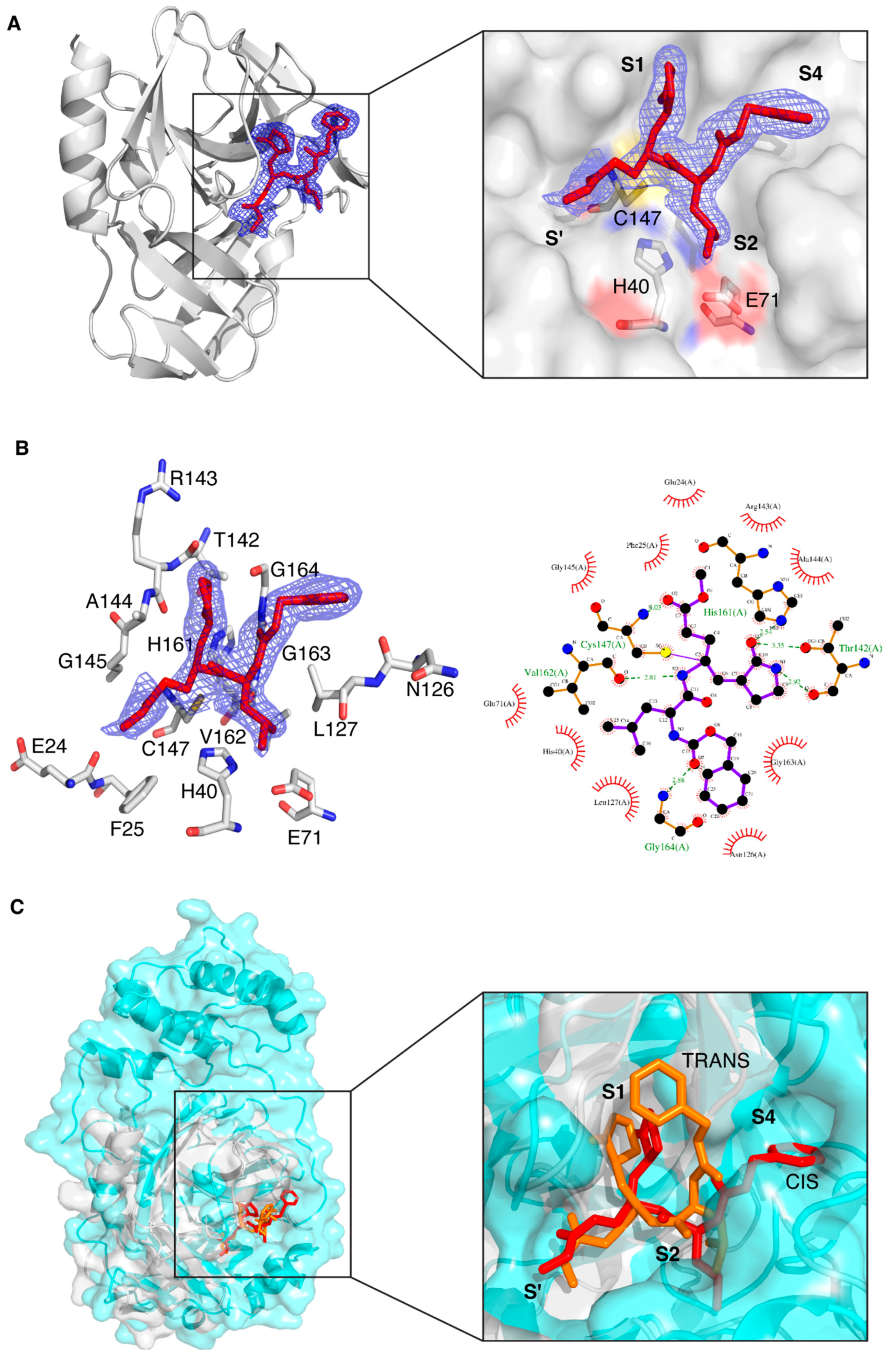
3.1. Crystal Structure of CVB3 3CPro in Complex with GC-376 PROTAC Precursor
3.2. Structural Characterization of CVB3 3CPro by Solution NMR
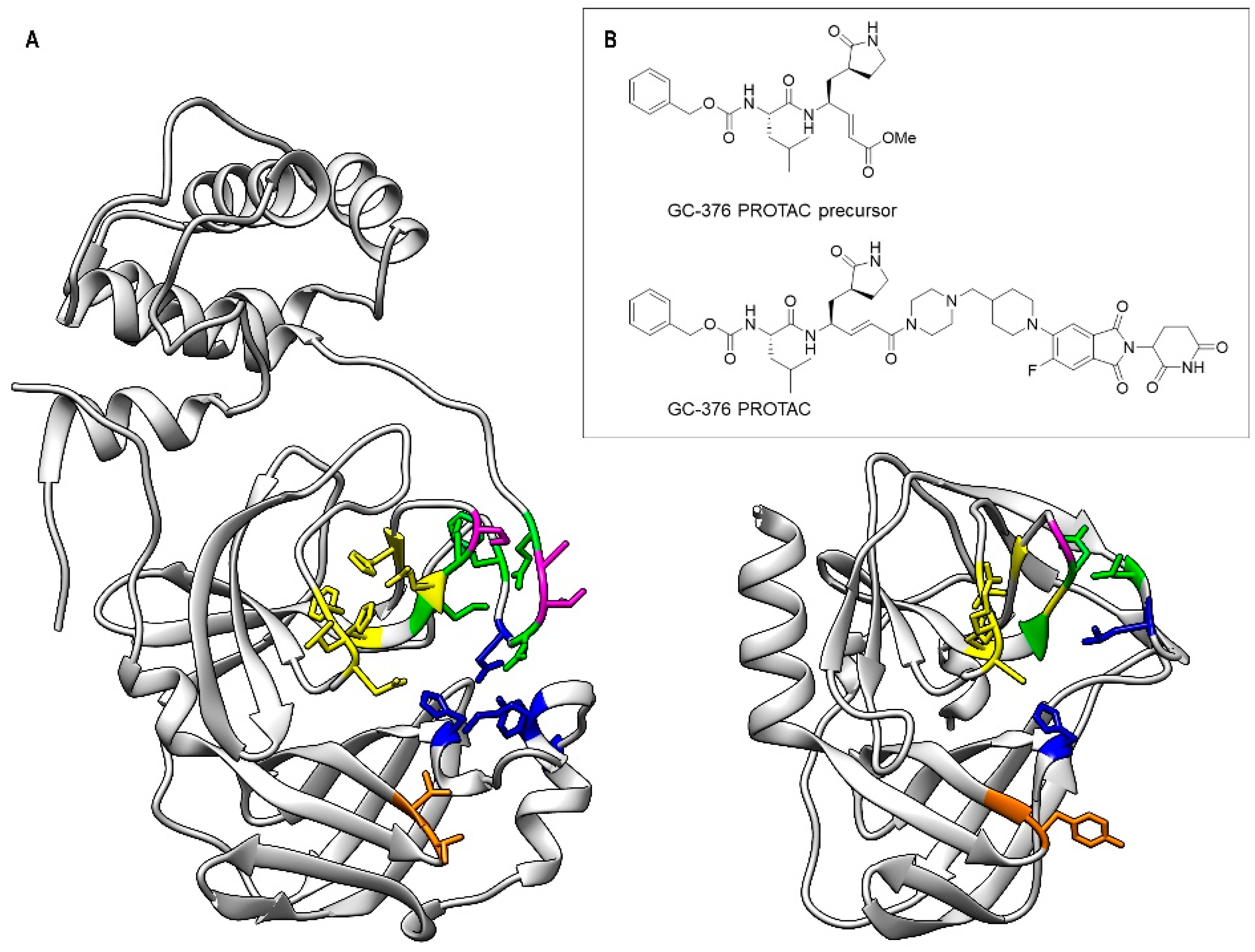
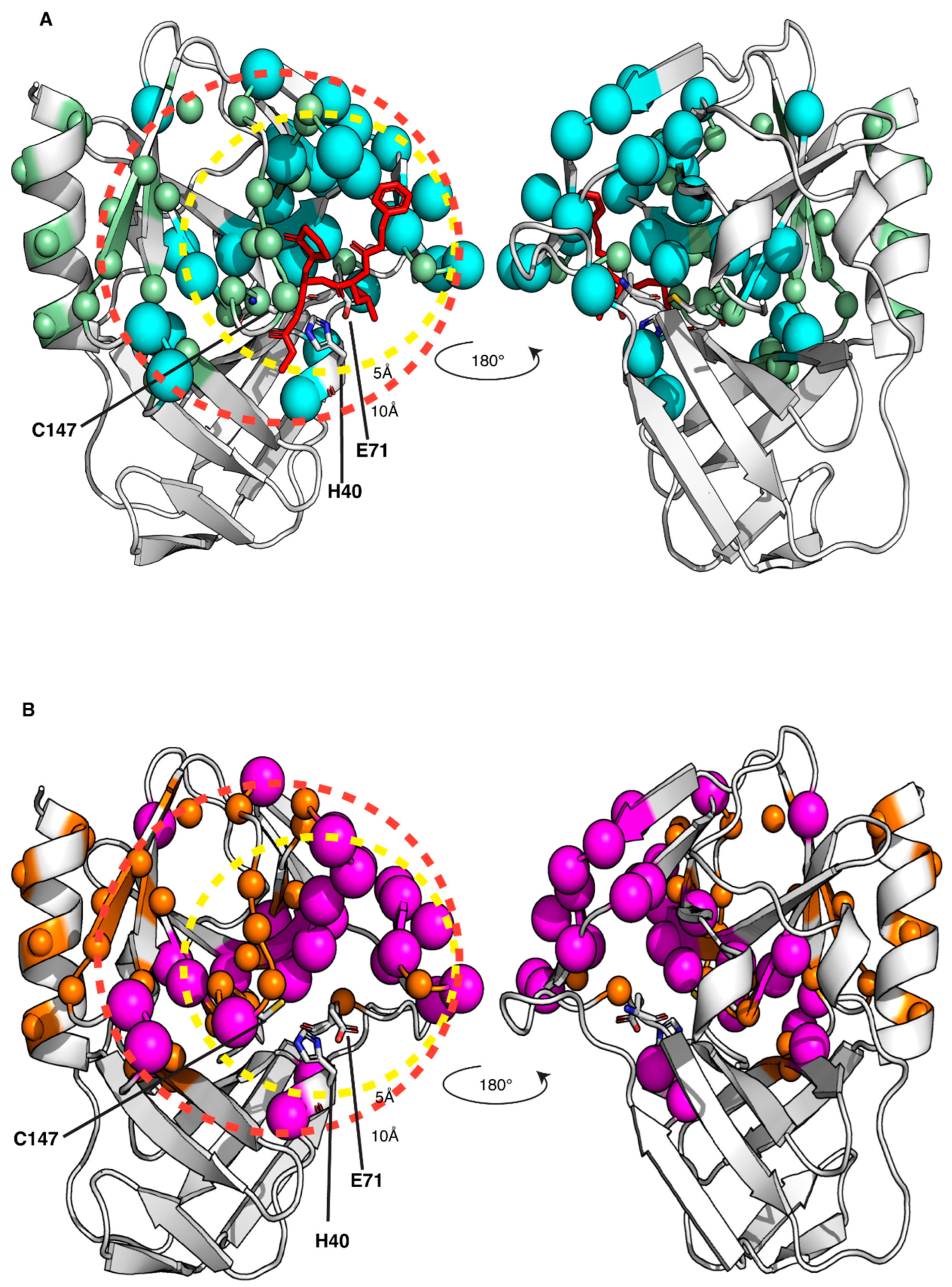
3.3. Mapping the Interaction of the GC-376 PROTAC and Its Precursor with CVB3 3CPro by Solution NMR
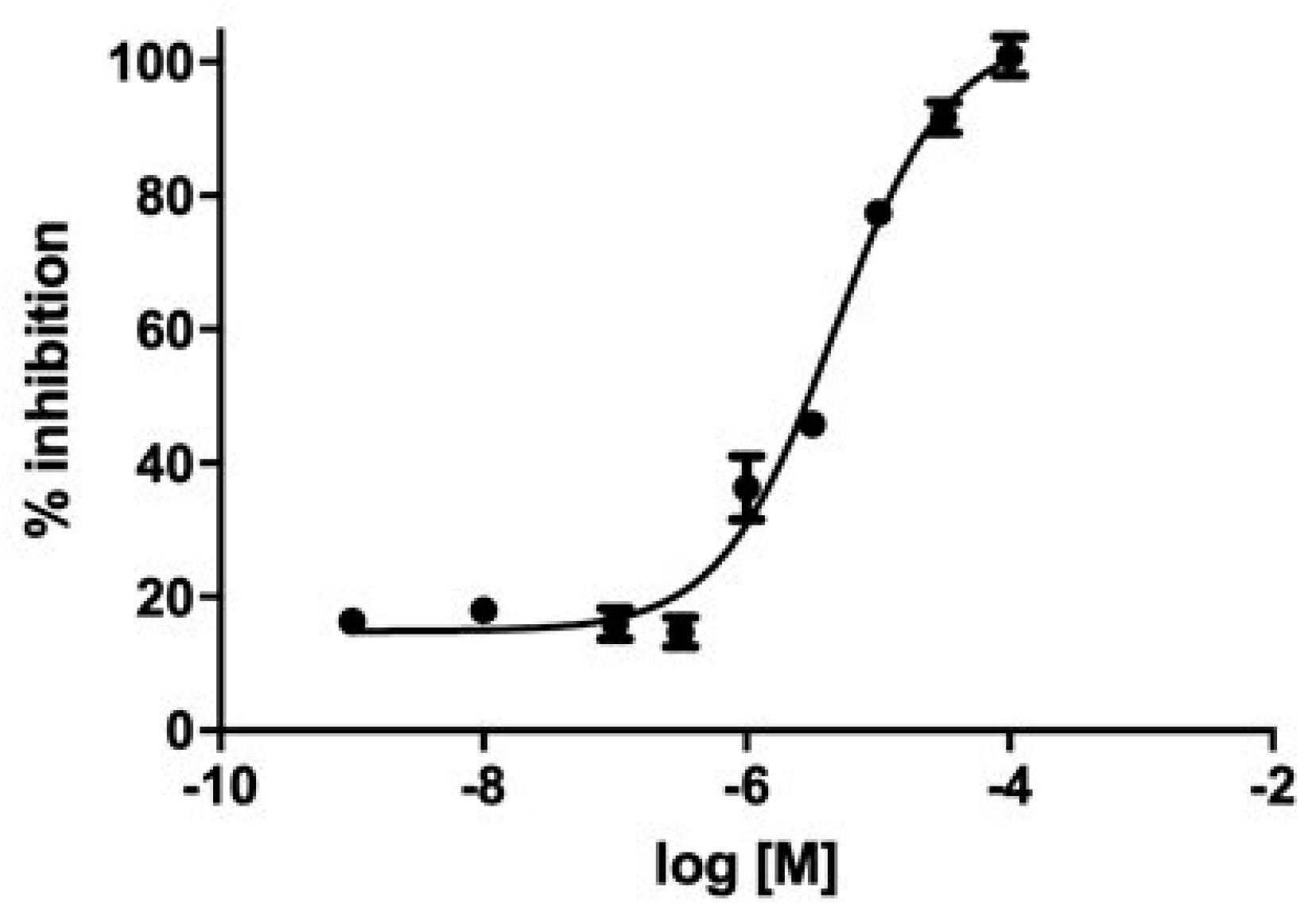
3.4. Enzyme Inhibition Kinetics of GC-376-PROTAC and Its Precursor
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Morreale, F.E.; Walden, H. Types of Ubiquitin Ligases. Cell 2016, 165, 248–248.e241. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Schulman, B.A.; Harper, J.W. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef]
- Li, K.; Crews, C.M. PROTACs: Past, present and future. Chem. Soc. Rev. 2022, 51, 5214–5236. [Google Scholar] [CrossRef] [PubMed]
- Ciulli, A.H.O. PROTAC Degraders Mechanism, Recent Advances, and Future Challenges. In Protein Homeostasis in Drug Discovery: A Chemical Biology Perspective; Jones, M.K.L.H., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2022; pp. 317–356. [Google Scholar]
- Toure, M.; Crews, C.M. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew. Chem. Int. Ed. Engl. 2016, 55, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Alugubelli, Y.R.; Xiao, J.; Khatua, K.; Kumar, S.; Sun, L.; Ma, Y.; Ma, X.R.; Vulupala, V.R.; Atla, S.; Blankenship, L.R.; et al. Discovery of First-in-Class PROTAC Degraders of SARS-CoV-2 Main Protease. J. Med. Chem. 2024, 67, 6495–6507. [Google Scholar] [CrossRef] [PubMed]
- Grifagni, D.; Lenci, E.; De Santis, A.; Orsetti, A.; Barracchia, C.G.; Tedesco, F.; Bellini Puglielli, R.; Lucarelli, F.; Lauriola, A.; Assfalg, M.; et al. Development of a GC-376 Based Peptidomimetic PROTAC as a Degrader of 3-Chymotrypsin-like Protease of SARS-CoV-2. ACS Med. Chem. Lett. 2024, 15, 250–257. [Google Scholar] [CrossRef]
- Desantis, J.; Bazzacco, A.; Eleuteri, M.; Tuci, S.; Bianconi, E.; Macchiarulo, A.; Mercorelli, B.; Loregian, A.; Goracci, L. Design, synthesis, and biological evaluation of first-in-class indomethacin-based PROTACs degrading SARS-CoV-2 main protease and with broad-spectrum antiviral activity. Eur. J. Med. Chem. 2024, 268, 116202. [Google Scholar] [CrossRef]
- Sang, X.; Wang, J.; Zhou, J.; Xu, Y.; An, J.; Warshel, A.; Huang, Z. A Chemical Strategy for the Degradation of the Main Protease of SARS-CoV-2 in Cells. J. Am. Chem. Soc. 2023, 145, 27248–27253. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Kim, Y.; Lovell, S.; Tiew, K.C.; Mandadapu, S.R.; Alliston, K.R.; Battaile, K.P.; Groutas, W.C.; Chang, K.O. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 2012, 86, 11754–11762. [Google Scholar] [CrossRef]
- Ito, T. Protein degraders—From thalidomide to new PROTACs. J. Biochem. 2024, 175, 507–519. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Braggio, E.; Shi, C.X.; Bruins, L.A.; Schmidt, J.E.; Van Wier, S.; Chang, X.B.; Bjorklund, C.C.; Fonseca, R.; Bergsagel, P.L.; et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011, 118, 4771–4779. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, A.K.; Kang, J.; Havens, C.G.; Conklin, T.; Ning, Y.; Wu, L.; Ito, T.; Ando, H.; Waldman, M.F.; Thakurta, A.; et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br. J. Haematol. 2014, 164, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Girardini, M.; Maniaci, C.; Hughes, S.J.; Testa, A.; Ciulli, A. Cereblon versus VHL: Hijacking E3 ligases against each other using PROTACs. Bioorg. Med. Chem. 2019, 27, 2466–2479. [Google Scholar] [CrossRef]
- Diehl, C.J.; Ciulli, A. Discovery of small molecule ligands for the von Hippel-Lindau (VHL) E3 ligase and their use as inhibitors and PROTAC degraders. Chem. Soc. Rev. 2022, 51, 8216–8257. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [PubMed]
- Marjomäki, V.; Kalander, K.; Hellman, M.; Permi, P. Enteroviruses and coronaviruses: Similarities and therapeutic targets. Expert Opin. Ther. Targets 2021, 25, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Qi, Z.; Wang, J. The Function and Mechanism of Enterovirus 71 (EV71) 3C Protease. Curr. Microbiol. 2020, 77, 1968–1975. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, O.H.; Svedin, E.; Kapell, S.; Nurminen, A.; Hytönen, V.P.; Flodström-Tullberg, M. Enteroviral proteases: Structure, host interactions and pathogenicity. Rev. Med. Virol. 2016, 26, 251–267. [Google Scholar] [CrossRef]
- Tan, J.; George, S.; Kusov, Y.; Perbandt, M.; Anemüller, S.; Mesters, J.R.; Norder, H.; Coutard, B.; Lacroix, C.; Leyssen, P.; et al. 3C protease of enterovirus 68: Structure-based design of Michael acceptor inhibitors and their broad-spectrum antiviral effects against picornaviruses. J. Virol. 2013, 87, 4339–4351. [Google Scholar] [CrossRef]
- Nikonov, O.S.; Chernykh, E.S.; Garber, M.B.; Nikonova, E.Y. Enteroviruses: Classification, Diseases They Cause, and Approaches to Development of Antiviral Drugs. Biochem. Biokhimiia 2017, 82, 1615–1631. [Google Scholar] [CrossRef] [PubMed]
- Maier, R.; Krebs, P.; Ludewig, B. Immunopathological basis of virus-induced myocarditis. Clin. Dev. Immunol. 2004, 11, 1–5. [Google Scholar] [CrossRef]
- Gauntt, C.; Huber, S. Coxsackievirus experimental heart diseases. Front. Biosci. 2003, 8, e23–e35. [Google Scholar] [CrossRef]
- Lugo, D.; Krogstad, P. Enteroviruses in the early 21st century: New manifestations and challenges. Curr. Opin. Pediatr. 2016, 28, 107–113. [Google Scholar] [CrossRef]
- Fan, W.; McDougal, M.B.; Schoggins, J.W. Enterovirus 3C Protease Cleaves TRIM7 To Dampen Its Antiviral Activity. J. Virol. 2022, 96, e0133222. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Donchenko, A.P.; Blinov, V.M.; Koonin, E.V. Cysteine proteases of positive strand RNA viruses and chymotrypsin-like serine proteases. A distinct protein superfamily with a common structural fold. FEBS Lett. 1989, 243, 103–114. [Google Scholar] [CrossRef]
- Fan, K.; Wei, P.; Feng, Q.; Chen, S.; Huang, C.; Ma, L.; Lai, B.; Pei, J.; Liu, Y.; Chen, J.; et al. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004, 279, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. α-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. Embo J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef] [PubMed]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Singh, E.; Khan, R.J.; Jha, R.K.; Amera, G.M.; Jain, M.; Singh, R.P.; Muthukumaran, J.; Singh, A.K. A comprehensive review on promising anti-viral therapeutic candidates identified against main protease from SARS-CoV-2 through various computational methods. J. Genet. Eng. Biotechnol. 2020, 18, 69. [Google Scholar] [CrossRef]
- Azevedo, P.; Camargo, P.G.; Constant, L.E.C.; Costa, S.D.S.; Silva, C.S.; Rosa, A.S.; Souza, D.D.C.; Tucci, A.R.; Ferreira, V.N.S.; Oliveira, T.K.F.; et al. Statine-based peptidomimetic compounds as inhibitors for SARS-CoV-2 main protease (SARS-CoV-2 Mpro). Sci. Rep. 2024, 14, 8991. [Google Scholar] [CrossRef] [PubMed]
- Hayek-Orduz, Y.; Vásquez, A.F.; Villegas-Torres, M.F.; Caicedo, P.A.; Achenie, L.E.K.; González Barrios, A.F. Novel covalent and non-covalent complex-based pharmacophore models of SARS-CoV-2 main protease (M(pro)) elucidated by microsecond MD simulations. Sci. Rep. 2022, 12, 14030. [Google Scholar] [CrossRef] [PubMed]
- Fàbrega-Ferrer, M.; Herrera-Morandé, A.; Muriel-Goñi, S.; Pérez-Saavedra, J.; Bueno, P.; Castro, V.; Garaigorta, U.; Gastaminza, P.; Coll, M. Structure and inhibition of SARS-CoV-1 and SARS-CoV-2 main proteases by oral antiviral compound AG7404. Antivir. Res. 2022, 208, 105458. [Google Scholar] [CrossRef] [PubMed]
- Göhl, M.; Zhang, L.; El Kilani, H.; Sun, X.; Zhang, K.; Brönstrup, M.; Hilgenfeld, R. From Repurposing to Redesign: Optimization of Boceprevir to Highly Potent Inhibitors of the SARS-CoV-2 Main Protease. Molecules 2022, 27, 4292. [Google Scholar] [CrossRef]
- Lockbaum, G.J.; Henes, M.; Lee, J.M.; Timm, J.; Nalivaika, E.A.; Thompson, P.R.; Kurt Yilmaz, N.; Schiffer, C.A. Pan-3C Protease Inhibitor Rupintrivir Binds SARS-CoV-2 Main Protease in a Unique Binding Mode. Biochemistry 2021, 60, 2925–2931. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Jochmans, D.; Xie, H.; Yang, H.; Li, J.; Su, H.; Chang, D.; Wang, J.; Peng, J.; Zhu, L.; et al. Design, Synthesis, and Biological Evaluation of Peptidomimetic Aldehydes as Broad-Spectrum Inhibitors against Enterovirus and SARS-CoV-2. J. Med. Chem. 2022, 65, 2794–2808. [Google Scholar] [CrossRef]
- Ramajayam, R.; Tan, K.P.; Liu, H.G.; Liang, P.H. Synthesis and evaluation of pyrazolone compounds as SARS-coronavirus 3C-like protease inhibitors. Bioorg. Med. Chem. 2010, 18, 7849–7854. [Google Scholar] [CrossRef] [PubMed]
- Ramajayam, R.; Tan, K.P.; Liang, P.H. Recent development of 3C and 3CL protease inhibitors for anti-coronavirus and anti-picornavirus drug discovery. Biochem. Soc. Trans. 2011, 39, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Mandadapu, S.R.; Weerawarna, P.M.; Prior, A.M.; Uy, R.A.; Aravapalli, S.; Alliston, K.R.; Lushington, G.H.; Kim, Y.; Hua, D.H.; Chang, K.O.; et al. Macrocyclic inhibitors of 3C and 3C-like proteases of picornavirus, norovirus, and coronavirus. Bioorg. Med. Chem. Lett. 2013, 23, 3709–3712. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Shin, J.S.; Shie, J.J.; Ku, K.B.; Kim, C.; Go, Y.Y.; Huang, K.F.; Kim, M.; Liang, P.H. Identification and evaluation of potent Middle East respiratory syndrome coronavirus (MERS-CoV) 3CL(Pro) inhibitors. Antivir. Res. 2017, 141, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Kuo, C.J.; Ko, T.P.; Hsu, M.F.; Tsui, Y.C.; Chang, S.C.; Yang, S.; Chen, S.J.; Chen, H.C.; Hsu, M.C.; et al. Structural basis of inhibition specificities of 3C and 3C-like proteases by zinc-coordinating and peptidomimetic compounds. J. Biol. Chem. 2009, 284, 7646–7655. [Google Scholar] [CrossRef]
- Kuo, C.J.; Liu, H.G.; Lo, Y.K.; Seong, C.M.; Lee, K.I.; Jung, Y.S.; Liang, P.H. Individual and common inhibitors of coronavirus and picornavirus main proteases. FEBS Lett. 2009, 583, 549–555. [Google Scholar] [CrossRef]
- Fili, S.; Valmas, A.; Christopoulou, M.; Spiliopoulou, M.; Nikolopoulos, N.; Lichière, J.; Logotheti, S.; Karavassili, F.; Rosmaraki, E.; Fitch, A.; et al. Coxsackievirus B3 protease 3C: Expression, purification, crystallization and preliminary structural insights. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 877–884. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D. Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Vagin, A.A.; Teplyakov, A. An approach to multi-copy search in molecular replacement. Acta Cryst. D 2000, 56, 1622–1624. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkòczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Cryst. D 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Grzesiek, S.; Bax, A. Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N-enriched proteins. J. Biomol. NMR 1993, 3, 185–204. [Google Scholar] [CrossRef]
- Keller, R. The Computer Aided Resonance Assignment Tutorial; Cantina: Arth, Switzerland, 2004; pp. 1–81. [Google Scholar]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Farrow, N.A.; Muhandiram, R.; Singer, A.U.; Pascal, S.M.; Kay, C.M.; Gish, G.; Shoelson, S.E.; Pawson, T.; Forman-Kay, J.D.; Kay, L.E. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 1994, 33, 5984–6003. [Google Scholar] [CrossRef]
- Grzesiek, S.; Bax, A. The Importance of Not Saturating H2o in Protein Nmr—Application to Sensitivity Enhancement and Noe Measurements. J. Am. Chem. Soc. 1993, 115, 12593–12594. [Google Scholar] [CrossRef]
- Mandel, M.A.; Akke, M.; Palmer, A.G., III. Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J. Mol. Biol. 1995, 246, 144–163. [Google Scholar] [CrossRef]
- Garcia de la Torre, J.; Huertas, M.L.; Carrasco, B. HYDRONMR: Prediction of NMR relaxation of globular proteins from atomic-level structures and hydrodynamic calculations. J. Magn. Reson. 2000, 147, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.A.; Dragovich, P.S.; Webber, S.E.; Fuhrman, S.A.; Patick, A.K.; Zalman, L.S.; Hendrickson, T.F.; Love, R.A.; Prins, T.J.; Marakovits, J.T.; et al. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. USA 1999, 96, 11000–11007. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.O.; Hua, Y.; Luu, H.T.; Brown, E.L.; Chan, F.; Chu, S.S.; Dragovich, P.S.; Eastman, B.W.; Ferre, R.A.; Fuhrman, S.A.; et al. Structure-based design of a parallel synthetic array directed toward the discovery of irreversible inhibitors of human rhinovirus 3C protease. J. Med. Chem. 2002, 45, 2016–2023. [Google Scholar] [CrossRef] [PubMed]
- Golovanov, A.P.; Hautbergue, G.M.; Wilson, S.A.; Lian, L.Y. A simple method for improving protein solubility and long-term stability. J. Am. Chem. Soc. 2004, 126, 8933–8939. [Google Scholar] [CrossRef]
- Kuo, C.J.; Shie, J.J.; Fang, J.M.; Yen, G.R.; Hsu, J.T.; Liu, H.G.; Tseng, S.N.; Chang, S.C.; Lee, C.Y.; Shih, S.R.; et al. Design, synthesis, and evaluation of 3C protease inhibitors as anti-enterovirus 71 agents. Bioorg. Med. Chem. 2008, 16, 7388–7398. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Zhu, Z.; Jiang, H.; Zou, X.; Zeng, X.; Wang, J.; Zeng, P.; Li, W.; Zhou, X.; Zhang, J.; et al. Structural Basis for Coronaviral Main Proteases Inhibition by the 3CLpro Inhibitor GC376. J. Mol. Biol. 2024, 436, 168474. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Feng, Y.; Li, W.; Liu, T.; Lv, X.; Tong, X.; Xi, G.; Ye, X.; Li, X. Development of novel antivrial agents that induce the degradation of the main protease of human-infecting coronaviruses. Eur. J. Med. Chem. 2024, 275, 116629. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Santis, A.; Grifagni, D.; Orsetti, A.; Lenci, E.; Rosato, A.; D’Onofrio, M.; Trabocchi, A.; Ciofi-Baffoni, S.; Cantini, F.; Calderone, V. A Structural Investigation of the Interaction between a GC-376-Based Peptidomimetic PROTAC and Its Precursor with the Viral Main Protease of Coxsackievirus B3. Biomolecules 2024, 14, 1260. https://doi.org/10.3390/biom14101260
De Santis A, Grifagni D, Orsetti A, Lenci E, Rosato A, D’Onofrio M, Trabocchi A, Ciofi-Baffoni S, Cantini F, Calderone V. A Structural Investigation of the Interaction between a GC-376-Based Peptidomimetic PROTAC and Its Precursor with the Viral Main Protease of Coxsackievirus B3. Biomolecules. 2024; 14(10):1260. https://doi.org/10.3390/biom14101260
Chicago/Turabian StyleDe Santis, Alessia, Deborah Grifagni, Andrea Orsetti, Elena Lenci, Antonio Rosato, Mariapina D’Onofrio, Andrea Trabocchi, Simone Ciofi-Baffoni, Francesca Cantini, and Vito Calderone. 2024. "A Structural Investigation of the Interaction between a GC-376-Based Peptidomimetic PROTAC and Its Precursor with the Viral Main Protease of Coxsackievirus B3" Biomolecules 14, no. 10: 1260. https://doi.org/10.3390/biom14101260