

Stress-Activated Protein Kinases in Intervertebral Disc Degeneration: Unraveling the Impact of JNK and p38 MAPK

Abstract
1. Introduction
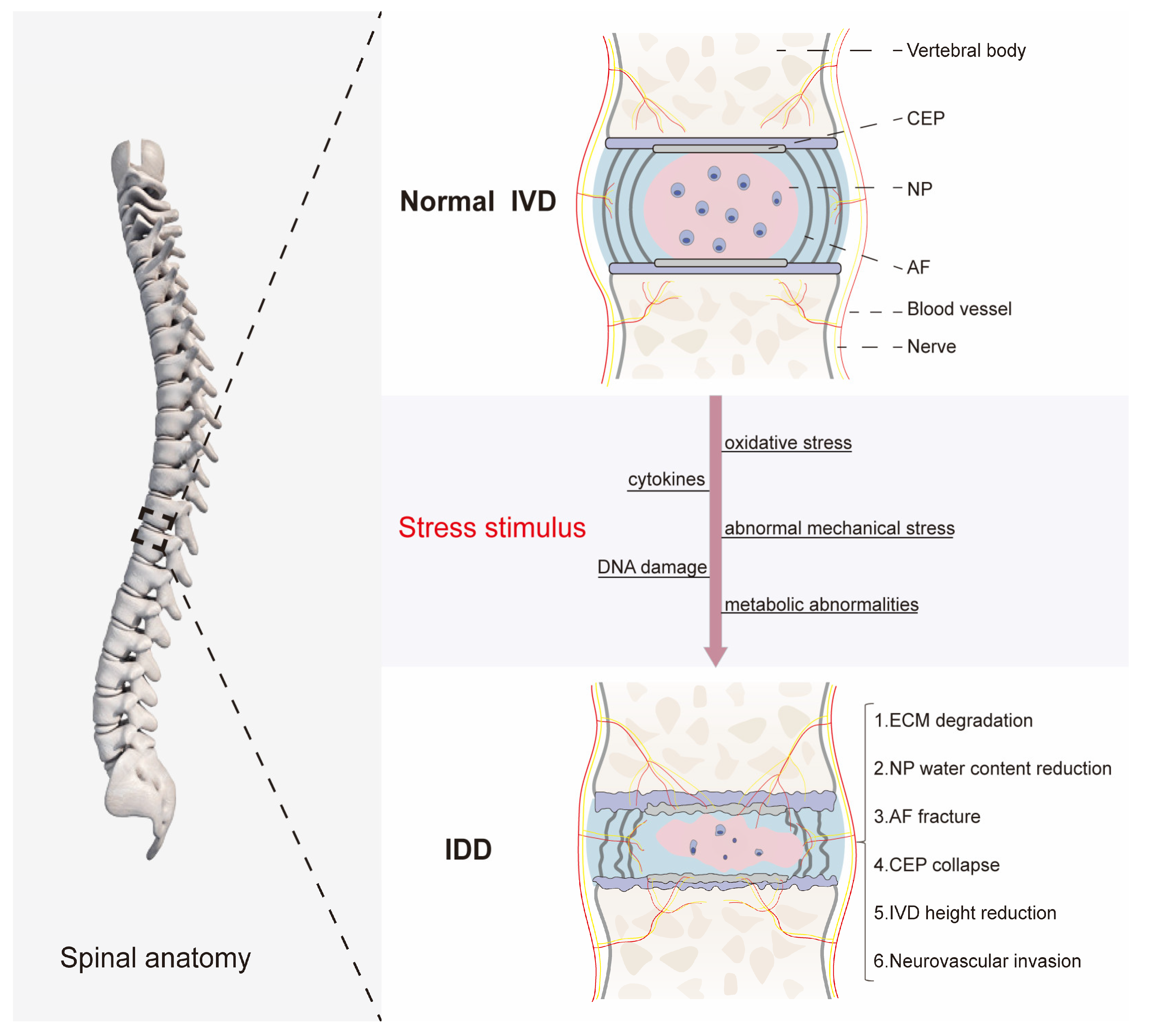
2. The Basic Structure and Function of the IVD
3. The Pathological Mechanism of IDD
4. Activation and Signaling of JNK and p38 MAPK
4.1. JNK
4.2. p38 MAPK
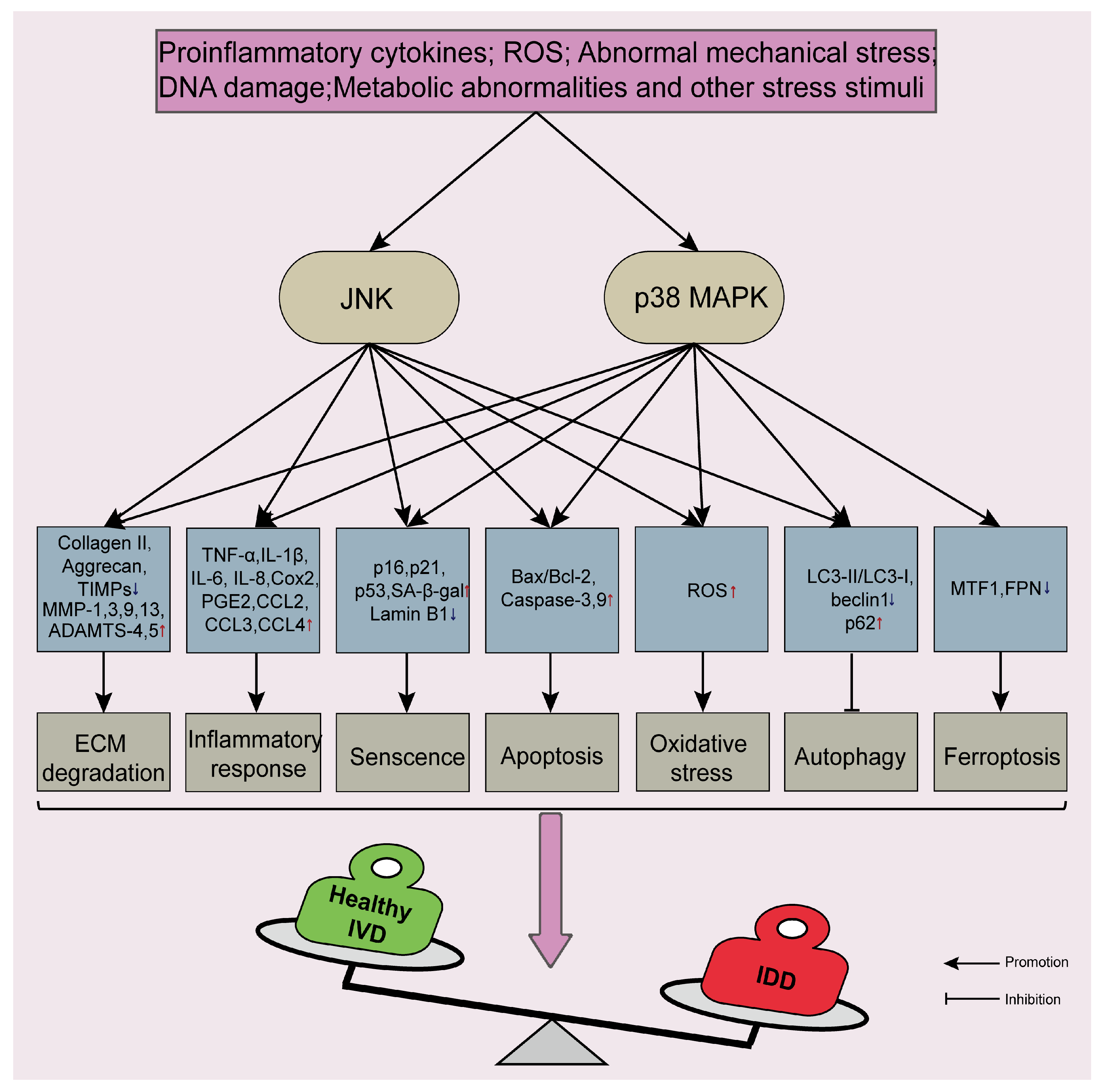
5. JNK and p38 MAPK Signaling Pathways Are Involved in the Process of IDD
5.1. JNK and p38 MAPK Signaling Pathways Disrupt ECM Metabolic Balance and Reduce ECM Content
5.2. JNK and p38 MAPK Signaling Pathways Regulate the Inflammatory Response
5.3. JNK and p38 MAPK Signaling Pathways Accelerate Cellular Senescence
5.4. JNK and p38 MAPK Signaling Pathways Promote Apoptosis
5.5. JNK and p38 MAPK Signaling Pathways Aggravate Oxidative Stress Injury and Lead to Ferroptosis
5.6. JNK and p38 MAPK Signaling Pathways Regulate Autophagy
6. Regulation of p38 MAPK by Non-Coding RNAs in IDD
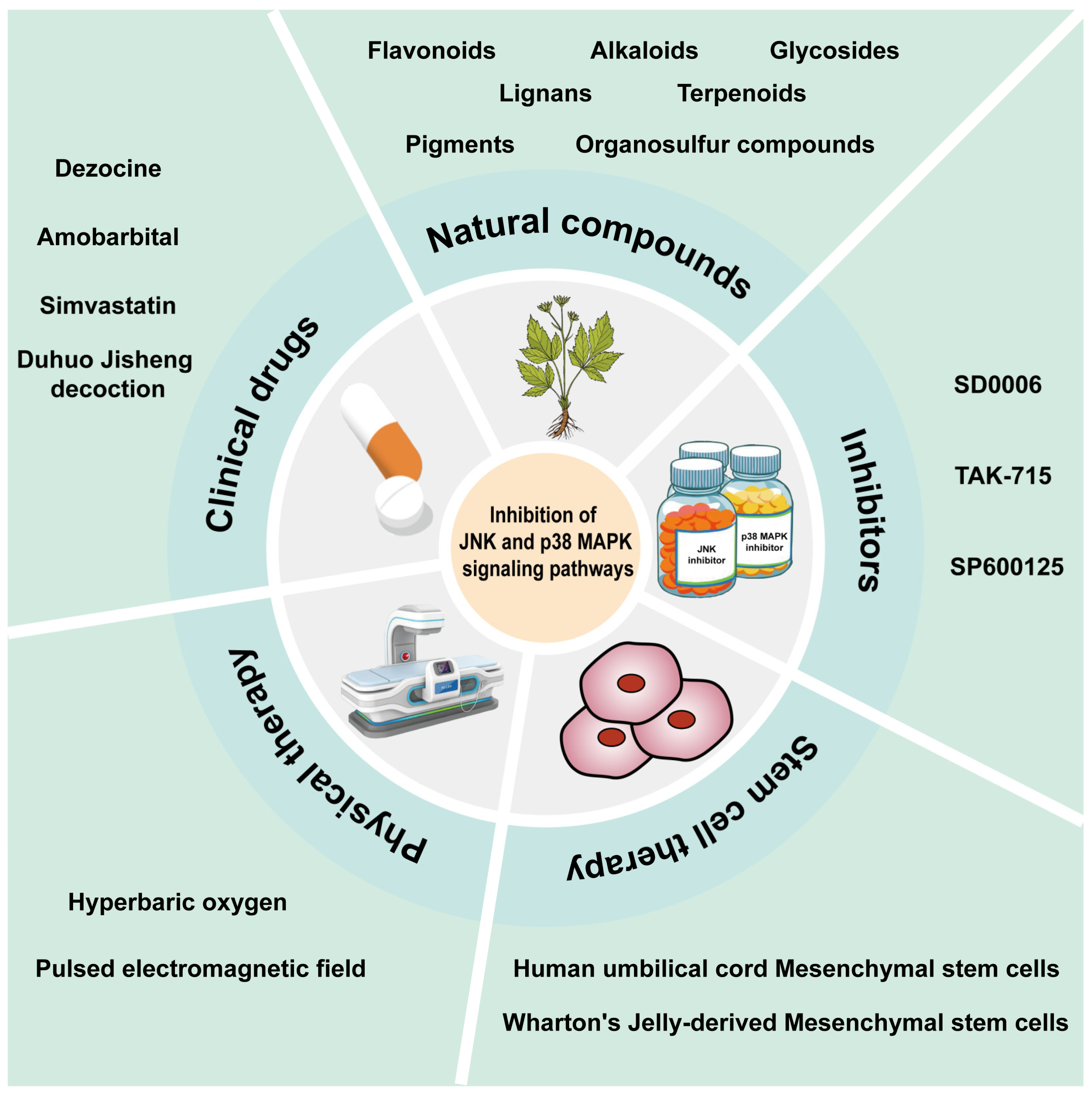
7. Potential Therapeutic Strategies Targeting JNK and p38 MAPK Signaling Pathways
7.1. Clinical Drugs
7.2. Natural Compounds
7.2.1. Flavonoids
7.2.2. Alkaloids
7.2.3. Lignans
7.2.4. Pigments
7.2.5. Terpenoids
7.2.6. Glycosides and Organosulfur Compounds
7.3. Inhibitors
7.4. Stem Cell Therapy
7.5. Physical Therapy
8. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cieza, A.; Causey, K.; Kamenov, K.; Hanson, S.W.; Chatterji, S.; Vos, T. Global estimates of the need for rehabilitation based on the Global Burden of Disease study 2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2021, 396, 2006–2017. [Google Scholar] [CrossRef]
- Hartvigsen, J.; Hancock, M.J.; Kongsted, A.; Louw, Q.; Ferreira, M.L.; Genevay, S.; Hoy, D.; Karppinen, J.; Pransky, G.; Sieper, J.; et al. What low back pain is and why we need to pay attention. Lancet 2018, 391, 2356–2367. [Google Scholar] [CrossRef]
- Binch, A.L.A.; Fitzgerald, J.C.; Growney, E.A.; Barry, F. Cell-based strategies for IVD repair: Clinical progress and translational obstacles. Nat. Rev. Rheumatol. 2021, 17, 158–175. [Google Scholar] [CrossRef]
- Zhao, L.; Manchikanti, L.; Kaye, A.D.; Abd-Elsayed, A. Treatment of Discogenic Low Back Pain: Current Treatment Strategies and Future Options-a Literature Review. Curr. Pain. Headache Rep. 2019, 23, 86. [Google Scholar] [CrossRef]
- Risbud, M.V.; Shapiro, I.M. Role of cytokines in intervertebral disc degeneration: Pain and disc content. Nat. Rev. Rheumatol. 2014, 10, 44–56. [Google Scholar] [CrossRef]
- Desmoulin, G.T.; Pradhan, V.; Milner, T.E. Mechanical Aspects of Intervertebral Disc Injury and Implications on Biomechanics. Spine 2020, 45, E457–E464. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, L.; Gao, Y.; Zou, X.; Wei, F. Oxidative Stress and Intervertebral Disc Degeneration: Pathophysiology, Signaling Pathway, and Therapy. Oxid. Med. Cell Longev. 2022, 2022, 1984742. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Fernandez, C.; Francisco, V.; Pino, J.; Mera, A.; Gonzalez-Gay, M.A.; Gomez, R.; Lago, F.; Gualillo, O. Molecular Relationships among Obesity, Inflammation and Intervertebral Disc Degeneration: Are Adipokines the Common Link? Int. J. Mol. Sci. 2019, 20, 2030. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhang, Y.; Wang, K.; Song, Y.; Tu, J.; Kang, L.; Zhao, K.; Wu, X.; Luo, R.; Yang, C. The involvement of regulated in development and DNA damage response 1 (REDD1) in the pathogenesis of intervertebral disc degeneration. Exp. Cell Res. 2018, 372, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Wilson, S.; Belham, C.M.; Robinson, C.J.; Scott, P.H.; Gould, G.W.; Plevin, R. Stress-activated protein kinases: Activation, regulation and function. Cell Signal 1997, 9, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef] [PubMed]
- Kasuya, Y.; Kim, J.D.; Hatano, M.; Tatsumi, K.; Matsuda, S. Pathophysiological Roles of Stress-Activated Protein Kinases in Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 22, 6041. [Google Scholar] [CrossRef] [PubMed]
- Cicuendez, B.; Ruiz-Garrido, I.; Mora, A.; Sabio, G. Stress kinases in the development of liver steatosis and hepatocellular carcinoma. Mol. Metab. 2021, 50, 101190. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, I.; Leiva, M.; Sabio, G. The role of stress kinases in metabolic disease. Nat. Rev. Endocrinol. 2020, 16, 697–716. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Bioactive lipids in intervertebral disc degeneration and its therapeutic implications. Biosci. Rep. 2019, 39, BSR20192117. [Google Scholar] [CrossRef]
- Tian, Y.; Yuan, W.; Fujita, N.; Wang, J.; Wang, H.; Shapiro, I.M.; Risbud, M.V. Inflammatory cytokines associated with degenerative disc disease control aggrecanase-1 (ADAMTS-4) expression in nucleus pulposus cells through MAPK and NF-kappaB. Am. J. Pathol. 2013, 182, 2310–2321. [Google Scholar] [CrossRef]
- Daniels, J.; Binch, A.A.; Le Maitre, C.L. Inhibiting IL-1 signaling pathways to inhibit catabolic processes in disc degeneration. J. Orthop. Res. 2017, 35, 74–85. [Google Scholar] [CrossRef]
- Park, J.J.; Moon, H.J.; Park, J.H.; Kwon, T.H.; Park, Y.K.; Kim, J.H. Induction of proinflammatory cytokine production in intervertebral disc cells by macrophage-like THP-1 cells requires mitogen-activated protein kinase activity. J. Neurosurg. Spine 2016, 24, 167–175. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, W.; Wang, P.; Hu, B.; Lv, X.; Chen, S.; Wang, B.; Shao, Z. Activation of HSP70 impedes tert-butyl hydroperoxide (t-BHP)-induced apoptosis and senescence of human nucleus pulposus stem cells via inhibiting the JNK/c-Jun pathway. Mol. Cell Biochem. 2021, 476, 1979–1994. [Google Scholar] [CrossRef]
- Pang, L.; Li, P.; Zhang, R.; Xu, Y.; Song, L.; Zhou, Q. Role of p38-MAPK pathway in the effects of high-magnitude compression on nucleus pulposus cell senescence in a disc perfusion culture. Biosci. Rep. 2017, 37, BSR20170718. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Ying, J.; Zhang, Y.; Su, L.; Cheng, S.; Ruan, D. RhTSG-6 inhibits IL-1beta-induced extracellular matrix degradation and apoptosis by suppressing the p38, and JNK pathways in nucleus pulposus cells. Folia Histochem. Cytobiol. 2020, 58, 227–234. [Google Scholar] [CrossRef]
- Shan, L.; Yang, D.; Zhu, D.; Feng, F.; Li, X. High glucose promotes annulus fibrosus cell apoptosis through activating the JNK and p38 MAPK pathways. Biosci. Rep. 2019, 39, BSR20190853. [Google Scholar] [CrossRef]
- Dimozi, A.; Mavrogonatou, E.; Sklirou, A.; Kletsas, D. Oxidative stress inhibits the proliferation, induces premature senescence and promotes a catabolic phenotype in human nucleus pulposus intervertebral disc cells. Eur. Cell Mater. 2015, 30, 89–102; discussion 103. [Google Scholar] [CrossRef]
- Lu, S.; Song, Y.; Luo, R.; Li, S.; Li, G.; Wang, K.; Liao, Z.; Wang, B.; Ke, W.; Xiang, Q.; et al. Ferroportin-Dependent Iron Homeostasis Protects against Oxidative Stress-Induced Nucleus Pulposus Cell Ferroptosis and Ameliorates Intervertebral Disc Degeneration In Vivo. Oxid. Med. Cell Longev. 2021, 2021, 6670497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, C.; Li, Y.; Xu, H. Mechanism of the Mitogen-Activated Protein Kinases/Mammalian Target of Rapamycin Pathway in the Process of Cartilage Endplate Stem Cell Degeneration Induced by Tension Load. Glob. Spine J. 2022, 13, 21925682221085226. [Google Scholar] [CrossRef]
- Zhang, S.; Liang, W.; Abulizi, Y.; Xu, T.; Cao, R.; Xun, C.; Zhang, J.; Sheng, W. Quercetin Alleviates Intervertebral Disc Degeneration by Modulating p38 MAPK-Mediated Autophagy. Biomed. Res. Int. 2021, 2021, 6631562. [Google Scholar] [CrossRef] [PubMed]
- Newell, N.; Little, J.P.; Christou, A.; Adams, M.A.; Adam, C.J.; Masouros, S.D. Biomechanics of the human intervertebral disc: A review of testing techniques and results. J. Mech. Behav. Biomed. Mater. 2017, 69, 420–434. [Google Scholar] [CrossRef]
- Chan, W.C.; Sze, K.L.; Samartzis, D.; Leung, V.Y.; Chan, D. Structure and biology of the intervertebral disk in health and disease. Orthop. Clin. N. Am. 2011, 42, 447–464. [Google Scholar] [CrossRef]
- Iatridis, J.C.; Weidenbaum, M.; Setton, L.A.; Mow, V.C. Is the nucleus pulposus a solid or a fluid? Mechanical behaviors of the nucleus pulposus of the human intervertebral disc. Spine 1996, 21, 1174–1184. [Google Scholar] [CrossRef]
- Jackson, A.R.; Yuan, T.Y.; Huang, C.Y.; Brown, M.D.; Gu, W.Y. Nutrient transport in human annulus fibrosus is affected by compressive strain and anisotropy. Ann. Biomed. Eng. 2012, 40, 2551–2558. [Google Scholar] [CrossRef]
- Berg-Johansen, B.; Han, M.; Fields, A.J.; Liebenberg, E.C.; Lim, B.J.; Larson, P.E.; Gunduz-Demir, C.; Kazakia, G.J.; Krug, R.; Lotz, J.C. Cartilage Endplate Thickness Variation Measured by Ultrashort Echo-Time MRI Is Associated with Adjacent Disc Degeneration. Spine 2018, 43, E592–E600. [Google Scholar] [CrossRef]
- Giers, M.B.; Munter, B.T.; Eyster, K.J.; Ide, G.D.; Newcomb, A.; Lehrman, J.N.; Belykh, E.; Byvaltsev, V.A.; Kelly, B.P.; Preul, M.C.; et al. Biomechanical and Endplate Effects on Nutrient Transport in the Intervertebral Disc. World Neurosurg. 2017, 99, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Guilak, F.; Ting-Beall, H.P.; Baer, A.E.; Trickey, W.R.; Erickson, G.R.; Setton, L.A. Viscoelastic properties of intervertebral disc cells. Identification of two biomechanically distinct cell populations. Spine 1999, 24, 2475–2483. [Google Scholar] [CrossRef]
- Guerrero, J.; Hackel, S.; Croft, A.S.; Hoppe, S.; Albers, C.E.; Gantenbein, B. The nucleus pulposus microenvironment in the intervertebral disc: The fountain of youth? Eur. Cell Mater. 2021, 41, 707–738. [Google Scholar] [CrossRef]
- Kibble, M.J.; Domingos, M.; Hoyland, J.A.; Richardson, S.M. Importance of Matrix Cues on Intervertebral Disc Development, Degeneration, and Regeneration. Int. J. Mol. Sci. 2022, 23, 6915. [Google Scholar] [CrossRef]
- Le Maitre, C.L.; Pockert, A.; Buttle, D.J.; Freemont, A.J.; Hoyland, J.A. Matrix synthesis and degradation in human intervertebral disc degeneration. Biochem. Soc. Trans. 2007, 35, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Luo, R.; Li, G.; Zhang, W.; Song, Y.; Yang, C. The Proteolysis of ECM in Intervertebral Disc Degeneration. Int. J. Mol. Sci. 2022, 23, 1715. [Google Scholar] [CrossRef] [PubMed]
- Sivan, S.S.; Wachtel, E.; Roughley, P. Structure, function, aging and turnover of aggrecan in the intervertebral disc. Biochim. Biophys. Acta 2014, 1840, 3181–3189. [Google Scholar] [CrossRef]
- Zhang, G.Z.; Liu, M.Q.; Chen, H.W.; Wu, Z.L.; Gao, Y.C.; Ma, Z.J.; He, X.G.; Kang, X.W. NF-kappaB signalling pathways in nucleus pulposus cell function and intervertebral disc degeneration. Cell Prolif. 2021, 54, e13057. [Google Scholar] [CrossRef]
- Yang, B.; O’Connell, G.D. Intervertebral disc swelling maintains strain homeostasis throughout the annulus fibrosus: A finite element analysis of healthy and degenerated discs. Acta Biomater. 2019, 100, 61–74. [Google Scholar] [CrossRef]
- Hassan, C.R.; Lee, W.; Komatsu, D.E.; Qin, Y.X. Evaluation of nucleus pulposus fluid velocity and pressure alteration induced by cartilage endplate sclerosis using a poro-elastic finite element analysis. Biomech. Model. Mechanobiol. 2021, 20, 281–291. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. pp54 microtubule-associated protein 2 kinase. A novel serine/threonine protein kinase regulated by phosphorylation and stimulated by poly-L-lysine. J. Biol. Chem. 1990, 265, 17355–17363. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Banerjee, P.; Nikolakaki, E.; Dai, T.; Rubie, E.A.; Ahmad, M.F.; Avruch, J.; Woodgett, J.R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 1994, 369, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Barr, R.K.; Bogoyevitch, M.A. The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs). Int. J. Biochem. Cell Biol. 2001, 33, 1047–1063. [Google Scholar] [CrossRef] [PubMed]
- Nishina, H.; Wada, T.; Katada, T. Physiological roles of SAPK/JNK signaling pathway. J. Biochem. 2004, 136, 123–126. [Google Scholar] [CrossRef]
- Pua, L.J.W.; Mai, C.W.; Chung, F.F.; Khoo, A.S.; Leong, C.O.; Lim, W.M.; Hii, L.W. Functional Roles of JNK and p38 MAPK Signaling in Nasopharyngeal Carcinoma. Int. J. Mol. Sci. 2022, 23, 1108. [Google Scholar] [CrossRef]
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Derijard, B.; Davis, R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Krishna, M.; Narang, H. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol. Life Sci. 2008, 65, 3525–3544. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef]
- Brewster, J.L.; de Valoir, T.; Dwyer, N.D.; Winter, E.; Gustin, M.C. An osmosensing signal transduction pathway in yeast. Science 1993, 259, 1760–1763. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, J.D.; Bibbs, L.; Ulevitch, R.J. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994, 265, 808–811. [Google Scholar] [CrossRef]
- Eckert, R.L.; Efimova, T.; Balasubramanian, S.; Crish, J.F.; Bone, F.; Dashti, S. p38 Mitogen-activated protein kinases on the body surface--a function for p38 delta. J. Investig. Dermatol. 2003, 120, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Remy, G.; Risco, A.M.; Inesta-Vaquera, F.A.; Gonzalez-Teran, B.; Sabio, G.; Davis, R.J.; Cuenda, A. Differential activation of p38MAPK isoforms by MKK6 and MKK3. Cell Signal 2010, 22, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Cuenda, A.; Sanz-Ezquerro, J.J. p38gamma and p38delta: From Spectators to Key Physiological Players. Trends Biochem. Sci. 2017, 42, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Cao, P.; Gao, Y.; Wu, M.; Lin, Y.; Tian, Y.; Yuan, W. Differential expression of p38 MAPK alpha, beta, gamma, delta isoforms in nucleus pulposus modulates macrophage polarization in intervertebral disc degeneration. Sci. Rep. 2016, 6, 22182. [Google Scholar] [CrossRef]
- Rodriguez Limardo, R.G.; Ferreiro, D.N.; Roitberg, A.E.; Marti, M.A.; Turjanski, A.G. p38gamma activation triggers dynamical changes in allosteric docking sites. Biochemistry 2011, 50, 1384–1395. [Google Scholar] [CrossRef]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.P.; Fitzgibbon, M.J.; Caron, P.R.; Griffith, J.P.; Chen, W.; McCaffrey, P.G.; Chambers, S.P.; Su, M.S. Crystal structure of p38 mitogen-activated protein kinase. J. Biol. Chem. 1996, 271, 27696–27700. [Google Scholar] [CrossRef]
- Wang, Z.; Harkins, P.C.; Ulevitch, R.J.; Han, J.; Cobb, M.H.; Goldsmith, E.J. The structure of mitogen-activated protein kinase p38 at 2.1-A resolution. Proc. Natl. Acad. Sci. USA 1997, 94, 2327–2332. [Google Scholar] [CrossRef]
- Bardwell, A.J.; Frankson, E.; Bardwell, L. Selectivity of docking sites in MAPK kinases. J. Biol. Chem. 2009, 284, 13165–13173. [Google Scholar] [CrossRef]
- Martinez-Limon, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The p38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef]
- Chen, H.W.; Zhou, J.W.; Zhang, G.Z.; Luo, Z.B.; Li, L.; Kang, X.W. Emerging role and therapeutic implication of mTOR signalling in intervertebral disc degeneration. Cell Prolif. 2023, 56, e13338. [Google Scholar] [CrossRef]
- Ouyang, Z.H.; Wang, W.J.; Yan, Y.G.; Wang, B.; Lv, G.H. The PI3K/Akt pathway: A critical player in intervertebral disc degeneration. Oncotarget 2017, 8, 57870–57881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.Z.; Chen, H.W.; Deng, Y.J.; Liu, M.Q.; Wu, Z.L.; Ma, Z.J.; He, X.G.; Gao, Y.C.; Kang, X.W. BRD4 Inhibition Suppresses Senescence and Apoptosis of Nucleus Pulposus Cells by Inducing Autophagy during Intervertebral Disc Degeneration: An In Vitro and In Vivo Study. Oxid. Med. Cell Longev. 2022, 2022, 9181412. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.J.; Liao, H.Y.; Bai, D.Y.; Wang, Z.Q.; Xie, X.W. MAPK /ERK signaling pathway: A potential target for the treatment of intervertebral disc degeneration. Biomed. Pharmacother. 2021, 143, 112170. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.G.; Song, J.X.; Cheng, J.F.; Zhang, P.Z.; Wang, H.; Liu, P.; Lu, K.; Zhong, M. JNK phosphorylation promotes degeneration of cervical endplate chondrocytes through down-regulation of the expression of ANK in humans. Chin. Med. J. 2013, 126, 2067–2073. [Google Scholar] [CrossRef] [PubMed]
- Vo, N.V.; Hartman, R.A.; Yurube, T.; Jacobs, L.J.; Sowa, G.A.; Kang, J.D. Expression and regulation of metalloproteinases and their inhibitors in intervertebral disc aging and degeneration. Spine J. Off. J. N. Am. Spine Soc. 2013, 13, 331–341. [Google Scholar] [CrossRef]
- Liu, C.; Yang, H.; Gao, F.; Li, X.; An, Y.; Wang, J.; Jin, A. Resistin Promotes Intervertebral Disc Degeneration by Upregulation of ADAMTS-5 Through p38 MAPK Signaling Pathway. Spine 2016, 41, 1414–1420. [Google Scholar] [CrossRef]
- Li, Z.; Yu, X.; Liang, J.; Wu, W.K.; Yu, J.; Shen, J. Leptin downregulates aggrecan through the p38-ADAMST pathway in human nucleus pulposus cells. PLoS ONE 2014, 9, e109595. [Google Scholar] [CrossRef]
- Miao, D.; Zhang, L. Leptin modulates the expression of catabolic genes in rat nucleus pulposus cells through the mitogen-activated protein kinase and Janus kinase 2/signal transducer and activator of transcription 3 pathways. Mol. Med. Rep. 2015, 12, 1761–1768. [Google Scholar] [CrossRef]
- Ding, W.; Zhao, C.; Cao, L.; Zhang, K.; Sun, W.; Xie, Y.; Li, H.; Zhao, J. Leptin induces terminal differentiation of rat annulus fibrosus cells via activation of MAPK signaling. Anat. Rec. 2013, 296, 1806–1812. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, P.; Ma, X. Rab7 delays intervertebral disc degeneration through the inhibition of the p38MAPK pathway. Biochem. Biophys. Res. Commun. 2019, 514, 835–841. [Google Scholar] [CrossRef]
- Kim, J.S.; Ellman, M.B.; An, H.S.; Yan, D.; van Wijnen, A.J.; Murphy, G.; Hoskin, D.W.; Im, H.J. Lactoferricin mediates anabolic and anti-catabolic effects in the intervertebral disc. J. Cell Physiol. 2012, 227, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Cheng, X.; Yuan, C.; Qian, J.; Wu, C.; Cao, C.; Yang, H.; Zhou, F.; Zou, J. Syndecan-4 is a Novel Therapeutic Target for Intervertebral Disc Degeneration via Suppressing JNK/p53 Pathway. Int. J. Biol. Sci. 2020, 16, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, Y.; Wu, Y.; Ni, C.; Jiang, P.; Li, J.; Mao, L.; Zheng, Q.; Yue, J. HtrA1 upregulates the expression of ADAMTS-5 in HNPCs via the ERK/NF-kappaB/JNK signaling pathway. Am. J. Transl. Res. 2019, 11, 5114–5121. [Google Scholar] [PubMed]
- Livshits, G.; Kalinkovich, A. Hierarchical, imbalanced pro-inflammatory cytokine networks govern the pathogenesis of chronic arthropathies. Osteoarthr. Cartil. 2018, 26, 7–17. [Google Scholar] [CrossRef]
- Sadowska, A.; Nic Hausmann, O.; Wuertz-Kozak, K. Inflammaging in the Intervertebral Disc. Clin. Transl. Neurosci. 2018, 2, 8. [Google Scholar] [CrossRef]
- Koroth, J.; Buko, E.O.; Abbott, R.; Johnson, C.P.; Ogle, B.M.; Stone, L.S.; Ellingson, A.M.; Bradley, E.W. Macrophages and Intervertebral Disc Degeneration. Int. J. Mol. Sci. 2023, 24, 1367. [Google Scholar] [CrossRef]
- Ni, L.; Zheng, Y.; Gong, T.; Xiu, C.; Li, K.; Saijilafu; Li, B.; Yang, H.; Chen, J. Proinflammatory macrophages promote degenerative phenotypes in rat nucleus pulpous cells partly through ERK and JNK signaling. J. Cell Physiol. 2019, 234, 5362–5371. [Google Scholar] [CrossRef]
- Li, Z.; Wang, X.; Pan, H.; Yang, H.; Li, X.; Zhang, K.; Wang, H.; Zheng, Z.; Liu, H.; Wang, J. Resistin promotes CCL4 expression through toll-like receptor-4 and activation of the p38-MAPK and NF-kappaB signaling pathways: Implications for intervertebral disc degeneration. Osteoarthr. Cartil. 2017, 25, 341–350. [Google Scholar] [CrossRef]
- Hiyama, A.; Hiraishi, S.; Sakai, D.; Mochida, J. CCAAT/enhancer binding protein beta regulates the expression of tumor necrosis factor-alpha in the nucleus pulposus cells. J. Orthop. Res. 2016, 34, 865–875. [Google Scholar] [CrossRef]
- Li, J.K.; Nie, L.; Zhao, Y.P.; Zhang, Y.Q.; Wang, X.; Wang, S.S.; Liu, Y.; Zhao, H.; Cheng, L. IL-17 mediates inflammatory reactions via p38/c-Fos and JNK/c-Jun activation in an AP-1-dependent manner in human nucleus pulposus cells. J. Transl. Med. 2016, 14, 77. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Yan, Q.; Wang, Y.; Cheng, X.; Song, D.; Wu, C.; Yu, H.; Yang, H.; Zou, J. IL-10 delays the degeneration of intervertebral discs by suppressing the p38 MAPK signaling pathway. Free Radic. Biol. Med. 2020, 147, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Cai, F.; Shi, R.; Wang, X.H.; Wu, X.T. Aging and age related stresses: A senescence mechanism of intervertebral disc degeneration. Osteoarthr. Cartil. 2016, 24, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Hou, G.; Zhang, R.; Gan, Y.; Xu, Y.; Song, L.; Zhou, Q. High-magnitude compression accelerates the premature senescence of nucleus pulposus cells via the p38 MAPK-ROS pathway. Arthritis Res. Ther. 2017, 19, 209. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Yang, L.; He, S.; Xia, T. Nucleus pulposus cell senescence is regulated by substrate stiffness and is alleviated by LOX possibly through the integrin beta1-p38 MAPK signaling pathway. Exp. Cell Res. 2022, 417, 113230. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sun, C.; Chen, M.; Wang, B. Lumican silencing alleviates tumor necrosis factor-alpha-induced nucleus pulposus cell inflammation and senescence by inhibiting apoptosis signal regulating kinase 1/p38 signaling pathway via inactivating Fas ligand expression. Bioengineered 2021, 12, 6891–6901. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Yu, W.; Jiang, D. Acidic pH promotes nucleus pulposus cell senescence through activating the p38 MAPK pathway. Biosci. Rep. 2018, 38, BSR20181451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.B.; Hu, Y.C.; Cheng, P.; Zhou, H.Y.; Chen, X.Y.; Wu, D.; Zhang, R.H.; Yu, D.C.; Gao, X.D.; Shi, J.T.; et al. Targeted therapy for intervertebral disc degeneration: Inhibiting apoptosis is a promising treatment strategy. Int. J. Med. Sci. 2021, 18, 2799–2813. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Liu, H.; Li, Z.; Chen, F.; Wang, H.; Zheng, Z.; Wang, J. TNF-alpha enhances apoptosis by promoting chop expression in nucleus pulposus cells: Role of the MAPK and NF-kappaB pathways. J. Orthop. Res. 2019, 37, 697–705. [Google Scholar] [CrossRef]
- Wang, J.; Chen, H.; Cao, P.; Wu, X.; Zang, F.; Shi, L.; Liang, L.; Yuan, W. Inflammatory cytokines induce caveolin-1/beta-catenin signalling in rat nucleus pulposus cell apoptosis through the p38 MAPK pathway. Cell Prolif. 2016, 49, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Fang, H.; Zhao, L.; Zhang, C.; Zhang, L.; Tian, B. Mechano growth factor attenuates mechanical overload-induced nucleus pulposus cell apoptosis through inhibiting the p38 MAPK pathway. Biosci. Rep. 2019, 39, BSR20182462. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zhao, Q.; Chen, L.; Luo, Y.; Shen, L.; Cao, Z.; Wang, Q. UBR3 promotes inflammation and apoptosis via DUSP1/p38 pathway in the nucleus pulposus cells of patients with intervertebral disc degeneration. Hum. Cell 2022, 35, 792–802. [Google Scholar] [CrossRef]
- Lin, Y.; Jiao, Y.; Yuan, Y.; Zhou, Z.; Zheng, Y.; Xiao, J.; Li, C.; Chen, Z.; Cao, P. Propionibacterium acnes induces intervertebral disc degeneration by promoting nucleus pulposus cell apoptosis via the TLR2/JNK/mitochondrial-mediated pathway. Emerg. Microbes Infect. 2018, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, X.; Liu, G.; Zhu, M.; Wu, Y.; Jie, Z.; Xie, Z.; Wang, S.; Ma, Q.; Fan, S.; et al. Oxidative stress abrogates the degradation of KMT2D to promote degeneration in nucleus pulposus. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165888. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.B.; Yu, Y.B.; Wa, Q.B.; Zhou, J.W.; He, M.; Cen, Y. The role of quinazoline in ameliorating intervertebral disc degeneration by inhibiting oxidative stress and anti-inflammation via NF-kappaB/MAPKs signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2077–2086. [Google Scholar] [CrossRef]
- Han, Y.; Li, X.; Yan, M.; Yang, M.; Wang, S.; Pan, J.; Li, L.; Tan, J. Oxidative damage induces apoptosis and promotes calcification in disc cartilage endplate cell through ROS/MAPK/NF-kappaB pathway: Implications for disc degeneration. Biochem. Biophys. Res. Commun. 2019, 516, 1026–1032. [Google Scholar] [CrossRef] [PubMed]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef]
- Kritschil, R.; Scott, M.; Sowa, G.; Vo, N. Role of autophagy in intervertebral disc degeneration. J. Cell Physiol. 2022, 237, 1266–1284. [Google Scholar] [CrossRef]
- Xu, K.; Chen, W.; Wang, X.; Peng, Y.; Liang, A.; Huang, D.; Li, C.; Ye, W. Autophagy attenuates the catabolic effect during inflammatory conditions in nucleus pulposus cells, as sustained by NF-kappaB and JNK inhibition. Int. J. Mol. Med. 2015, 36, 661–668. [Google Scholar] [CrossRef]
- Li, Z.; Wang, J.; Deng, X.; Huang, D.; Shao, Z.; Ma, K. Compression stress induces nucleus pulposus cell autophagy by inhibition of the PI3K/AKT/mTOR pathway and activation of the JNK pathway. Connect. Tissue Res. 2021, 62, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Chen, Z.; Wang, X.; Zhang, Y.; Guo, X.; Xu, Z.; Yang, H.; Hao, D. The potential mechanisms and application prospects of non-coding RNAs in intervertebral disc degeneration. Front. Endocrinol. 2022, 13, 1081185. [Google Scholar] [CrossRef]
- Li, G.; Tang, X.; Chen, H.; Sun, W.; Yuan, F. miR-148a inhibits pro-inflammatory cytokines released by intervertebral disc cells by regulating the p38/MAPK pathway. Exp. Ther. Med. 2018, 16, 2665–2669. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Chen, L. Inhibition of miR-27a suppresses the inflammatory response via the p38/MAPK pathway in intervertebral disc cells. Exp. Ther. Med. 2017, 14, 4572–4578. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Xue, J.; Peng, F. The regulatory activities of MALAT1 in the development of bone and cartilage diseases. Front Endocrinol 2022, 13, 1054827. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zeng, Q.; Li, D.; Ding, L.; Lu, W.; Bian, M.; Wu, J. Long non-coding RNA MALAT1 promotes high glucose-induced rat cartilage endplate cell apoptosis via the p38/MAPK signalling pathway. Mol. Med. Rep. 2020, 21, 2220–2226. [Google Scholar] [CrossRef]
- Zhu, F.; Duan, W.; Zhong, C.; Ji, B.; Liu, X. The protective effects of dezocine on interleukin-1beta-induced inflammation, oxidative stress and apoptosis of human nucleus pulposus cells and the possible mechanisms. Bioengineered 2022, 13, 1399–1410. [Google Scholar] [CrossRef]
- Seol, D.; Coleman, M.C.; Martin, J.A.; Song, I.; Jaidev, L.R.; Salem, A.K.; Lim, T.H. Targeting oxidative stress with amobarbital to prevent intervertebral disc degeneration: Part I. in vitro and ex vivo studies. Spine J. Off. J. N. Am. Spine Soc. 2021, 21, 1021–1030. [Google Scholar] [CrossRef]
- Tu, J.; Li, W.; Zhang, Y.; Wu, X.; Song, Y.; Kang, L.; Liu, W.; Wang, K.; Li, S.; Hua, W.; et al. Simvastatin Inhibits IL-1beta-Induced Apoptosis and Extracellular Matrix Degradation by Suppressing the NF-kB and MAPK Pathways in Nucleus Pulposus Cells. Inflammation 2017, 40, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Jin, S.; Huang, M.; Li, Y.; Wang, Z.; Wang, P.; Zhao, X.; Xia, P.; Feng, J. Duhuo jisheng decoction suppresses matrix degradation and apoptosis in human nucleus pulposus cells and ameliorates disc degeneration in a rat model. J. Ethnopharmacol. 2020, 250, 112494. [Google Scholar] [CrossRef]
- Li, B.; Yang, X.; Zhang, P.; Guo, J.; Rong, K.; Wang, X.; Cao, X.; Zhou, T.; Zhao, J. Engeletin Alleviates the Inflammation and Apoptosis in Intervertebral Disc Degeneration via Inhibiting the NF-kappaB and MAPK Pathways. J. Inflamm. Res. 2022, 15, 5767–5783. [Google Scholar] [CrossRef]
- Krupkova, O.; Sekiguchi, M.; Klasen, J.; Hausmann, O.; Konno, S.; Ferguson, S.J.; Wuertz-Kozak, K. Epigallocatechin 3-gallate suppresses interleukin-1beta-induced inflammatory responses in intervertebral disc cells in vitro and reduces radiculopathic pain in rats. Eur. Cell Mater. 2014, 28, 372–386. [Google Scholar] [CrossRef]
- Ge, J.; Zhou, Q.; Cheng, X.; Qian, J.; Yan, Q.; Wu, C.; Chen, Y.; Yang, H.; Zou, J. The protein tyrosine kinase inhibitor, Genistein, delays intervertebral disc degeneration in rats by inhibiting the p38 pathway-mediated inflammatory response. Aging 2020, 12, 2246–2260. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, Z.; Pang, Z.; Zhou, T.; Gu, Y. Acacetin Alleviates Inflammation and Matrix Degradation in Nucleus Pulposus Cells and Ameliorates Intervertebral Disc Degeneration in vivo. Drug Des. Devel Ther. 2020, 14, 4801–4813. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liao, Z.; Song, Y.; Yin, H.; Zhan, S.; Li, G.; Ma, L.; Lu, S.; Wang, K.; Li, S.; et al. Berberine ameliorates oxidative stress-induced apoptosis by modulating ER stress and autophagy in human nucleus pulposus cells. Life Sci. 2019, 228, 85–97. [Google Scholar] [CrossRef]
- Li, Y.; Li, K.; Hu, Y.; Xu, B.; Zhao, J. Piperine mediates LPS induced inflammatory and catabolic effects in rat intervertebral disc. Int. J. Clin. Exp. Pathol. 2015, 8, 6203–6213. [Google Scholar]
- Tang, P.; Gu, J.M.; Xie, Z.A.; Gu, Y.; Jie, Z.W.; Huang, K.M.; Wang, J.Y.; Fan, S.W.; Jiang, X.S.; Hu, Z.J. Honokiol alleviates the degeneration of intervertebral disc via suppressing the activation of TXNIP-NLRP3 inflammasome signal pathway. Free Radic. Biol. Med. 2018, 120, 368–379. [Google Scholar] [CrossRef]
- Li, K.; Li, Y.; Xu, B.; Mao, L.; Zhao, J. Sesamin inhibits lipopolysaccharide-induced inflammation and extracellular matrix catabolism in rat intervertebral disc. Connect. Tissue Res. 2016, 57, 347–359. [Google Scholar] [CrossRef]
- Li, K.; Li, Y.; Ma, Z.; Zhao, J. Crocin exerts anti-inflammatory and anti-catabolic effects on rat intervertebral discs by suppressing the activation of JNK. Int. J. Mol. Med. 2015, 36, 1291–1299. [Google Scholar] [CrossRef]
- Li, W.; Zhang, Y.; Xing, C.; Zhang, M. Tanshinone IIA represses inflammatory response and reduces radiculopathic pain by inhibiting IRAK-1 and NF-kappaB/p38/JNK signaling. Int. Immunopharmacol. 2015, 28, 382–389. [Google Scholar] [CrossRef]
- Dai, S.; Shi, X.; Qin, R.; Zhang, X.; Xu, F.; Yang, H. Sodium Tanshinone IIA Sulfonate Ameliorates Injury-Induced Oxidative Stress and Intervertebral Disc Degeneration in Rats by Inhibiting p38 MAPK Signaling Pathway. Oxid. Med. Cell Longev. 2021, 2021, 5556122. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhuang, J.; Wang, D.; Lv, L.; Zhu, F.; Yao, A.; Xu, T. Glycyrrhizin suppresses inflammation and cell apoptosis by inhibition of HMGB1 via p38/p-JUK signaling pathway in attenuating intervertebral disc degeneration. Am. J. Transl. Res. 2019, 11, 5105–5113. [Google Scholar] [PubMed]
- Xiang, Q.; Cheng, Z.; Wang, J.; Feng, X.; Hua, W.; Luo, R.; Wang, B.; Liao, Z.; Ma, L.; Li, G.; et al. Allicin Attenuated Advanced Oxidation Protein Product-Induced Oxidative Stress and Mitochondrial Apoptosis in Human Nucleus Pulposus Cells. Oxid. Med. Cell Longev. 2020, 2020, 6685043. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.D.; Guo, Z.G.; Deng, W.J.; Wang, J.G. SD0006 promotes nucleus pulposus cell proliferation via the p38MAPK/HDAC4 pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 10966–10974. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yao, D.; Li, Y.; Li, M.; Zeng, W.; Liao, Z.; Chen, E.; Lu, S.; Su, K.; Che, Z.; et al. TAK-715 alleviated IL-1beta-induced apoptosis and ECM degradation in nucleus pulposus cells and attenuated intervertebral disc degeneration ex vivo and in vivo. Arthritis Res. Ther. 2023, 25, 45. [Google Scholar] [CrossRef]
- Wang, X.; Wang, D.; Xia, P.; Cheng, K.; Wang, Q.; Wang, X.; Lin, Q.; Song, J.; Chen, A.; Li, X. Ultrasound-targeted simvastatin-loaded microbubble destruction promotes OA cartilage repair by modulating the cholesterol efflux pathway mediated by PPARgamma in rabbits. Bone Jt. Res. 2021, 10, 693–703. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, L.; Park, J.B.; Park, P.; Yang, V.C.; Hollister, S.J.; La Marca, F.; Lin, C.Y. Intradiscal injection of simvastatin retards progression of intervertebral disc degeneration induced by stab injury. Arthritis Res. Ther. 2009, 11, R172. [Google Scholar] [CrossRef]
- Chen, H.W.; Zhang, G.Z.; Liu, M.Q.; Zhang, L.J.; Kang, J.H.; Wang, Z.H.; Liu, W.Z.; Lin, A.X.; Kang, X.W. Natural Products of Pharmacology and Mechanisms in Nucleus Pulposus Cells and Intervertebral Disc Degeneration. Evid.-Based Complement. Altern. Med. Ecam 2021, 2021, 9963677. [Google Scholar] [CrossRef]
- Hayashi, Y.; Ohtori, S.; Yamashita, M.; Yamauchi, K.; Inoue, G.; Suzuki, M.; Orita, S.; Eguchi, Y.; Ochiai, N.; Kishida, S.; et al. Direct single injection of p38 mitogen-activated protein kinase inhibitor does not affect calcitonin gene-related peptide expression in dorsal root ganglion neurons innervating punctured discs in rats. Spine 2009, 34, 2843–2847. [Google Scholar] [CrossRef]
- Ito, T.; Ohtori, S.; Inoue, G.; Koshi, T.; Doya, H.; Ozawa, T.; Saito, T.; Moriya, H.; Takahashi, K. Glial phosphorylated p38 MAP kinase mediates pain in a rat model of lumbar disc herniation and induces motor dysfunction in a rat model of lumbar spinal canal stenosis. Spine 2007, 32, 159–167. [Google Scholar] [CrossRef]
- Xu, H.G.; Cheng, J.F.; Peng, H.X.; Lv, K.; Wang, H.; Liu, P.; Zhong, M.; Zhang, M.Y. JNK phosphorylation promotes natural degeneration of cervical endplate chondrocytes by down-regulating expression of ANK. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 2335–2344. [Google Scholar]
- Qi, L.; Wang, R.; Shi, Q.; Yuan, M.; Jin, M.; Li, D. Umbilical cord mesenchymal stem cell conditioned medium restored the expression of collagen II and aggrecan in nucleus pulposus mesenchymal stem cells exposed to high glucose. J. Bone Min. Metab. 2019, 37, 455–466. [Google Scholar] [CrossRef]
- Zhao, Y.; Qin, Y.; Wu, S.; Huang, D.; Hu, H.; Zhang, X.; Hao, D. Mesenchymal stem cells regulate inflammatory milieu within degenerative nucleus pulposus cells via p38 MAPK pathway. Exp. Ther. Med. 2020, 20, 22. [Google Scholar] [CrossRef]
- Niu, C.C.; Lin, S.S.; Yuan, L.J.; Lu, M.L.; Ueng, S.W.N.; Yang, C.Y.; Tsai, T.T.; Lai, P.L. Upregulation of miR-107 expression following hyperbaric oxygen treatment suppresses HMGB1/RAGE signaling in degenerated human nucleus pulposus cells. Arthritis Res. Ther. 2019, 21, 42. [Google Scholar] [CrossRef]
- Niu, C.C.; Yuan, L.J.; Chen, L.H.; Lin, S.S.; Tsai, T.T.; Liao, J.C.; Lai, P.L.; Chen, W.J. Beneficial effects of hyperbaric oxygen on human degenerated intervertebral disk cells via suppression of IL-1beta and p38 MAPK signal. J. Orthop. Res. 2011, 29, 14–19. [Google Scholar] [CrossRef]
- Niu, C.C.; Lin, S.S.; Yuan, L.J.; Chen, L.H.; Wang, I.C.; Tsai, T.T.; Lai, P.L.; Chen, W.J. Hyperbaric oxygen treatment suppresses MAPK signaling and mitochondrial apoptotic pathway in degenerated human intervertebral disc cells. J. Orthop. Res. 2013, 31, 204–209. [Google Scholar] [CrossRef]
- Markovic, L.; Wagner, B.; Crevenna, R. Effects of pulsed electromagnetic field therapy on outcomes associated with osteoarthritis: A systematic review of systematic reviews. Wien. Klin. Wochenschr. 2022, 134, 425–433. [Google Scholar] [CrossRef]
- Miller, S.L.; Coughlin, D.G.; Waldorff, E.I.; Ryaby, J.T.; Lotz, J.C. Pulsed electromagnetic field (PEMF) treatment reduces expression of genes associated with disc degeneration in human intervertebral disc cells. Spine J. Off. J. N. Am. Spine Soc. 2016, 16, 770–776. [Google Scholar] [CrossRef]
- Tang, X.; Coughlin, D.; Ballatori, A.; Berg-Johansen, B.; Waldorff, E.I.; Zhang, N.; Ryaby, J.T.; Aliston, T.; Lotz, J.C. Pulsed Electromagnetic Fields Reduce Interleukin-6 Expression in Intervertebral Disc Cells Via Nuclear Factor-kappabeta and Mitogen-Activated Protein Kinase p38 Pathways. Spine 2019, 44, E1290–E1297. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Name | Structural Formula | Dose/Concentration | Model Construction | Target/ Pathway | Function | References |
|---|---|---|---|---|---|---|---|
| Clinical drugs | Dezocine | 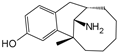 | 0.5 μg/mL, 1 μg/mL, 2 μg/mL | IL-1β (10 ng/mL) stimulates HNPCs for 24 h | p38 MAPK and ERK1/2 | Protect against IL-1β-induced inflammation, oxidative stress, and apoptosis of HNPCs | [109] |
| Amobarbital | 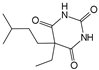 | 2.5 mmol/L | TBHP (50 μmol/L) stimulates rabbit NPCs for 1 h | ERK, JNK and p38 MAPK | Improved mitochondrial function and decreased TBHP-induced apoptosis, necrosis, and ROS production | [110] | |
| Simvastatin |  | 10 μmol/L, 20 μmol/L | IL-1β (10 ng/mL) stimulates HNPCs for 48 h | ERK, JNK and p38 MAPK | Inhibition of IL-1β-induced apoptosis and ECM degradation | [111] | |
| Duhuo Jisheng decoction | 200 μg/mL | HNPCs were subjected to 1 MPa static pressure | p38 MAPK | Attenuate pressure-induced apoptosis and increase ECM synthesis | [112] | ||
| Natural compounds | Engeletin | 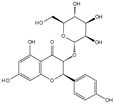 | 80 μmol/L | TNF-α (20 ng/mL) stimulates rat NPCs for 24 h | ERK, JNK, p38 MAPK and NF-κB | Inhibition of TNF-α-induced apoptosis, inflammation, and ECM degradation | [113] |
| Epigallocatechin 3-gallate |  | 10 μmol/L | IL-1β (10 ng/mL) stimulates HNPCs for 2 h | IRAK-1, p38 MAPK, JNK and NF-κB | Repress inflammatory responses and reduce MMPs | [114] | |
| Genistein |  | 10 ng/mL, 20 ng/mL, 30 ng/mL | IL-1β (10 ng/mL) stimulates rat NPCs for 24 h | P38 MAPK | Relieve IL-1β-induced generation of inflammatory mediators | [115] | |
| Quercetin | 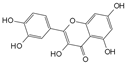 | 25 μmol/L | TBHP (50 μmol/L) stimulates rat NPCs for 24 h | P38 MAPK, mTOR | Protect against TBHP-induced rat NPC apoptosis and ECM degradation | [27] | |
| Acacetin |  | 0.3 μmol/L, 1 μmol/L | TBHP (50 μmol/L) stimulates rat NPCs for 24 h | Nrf2, p38 MAPK, JNK and ERK1/2 | Relieve TBHP-induced generation of inflammatory mediators and ECM degradation | [116] | |
| Berberine | 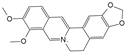 | 1 μmol/L, 2 μmol/L, 4 μmol/L, 8 μmol/L | H2O2 (300 μmol/L) stimulates HNPCs for 24 h | IRE1 and JNK | Prevent H2O2-induced apoptosis by modulating ER stress and autophagy | [117] | |
| Piperine |  | 10 μmol/L, 50 μmol/L, 100 μmol/L | LPS (10 μg/mL) stimulates rat NPCs for 24 h | JNK and NF-κB | Protect against LPS-induced inflammation and ECM catabolism | [118] | |
| Honokiol |  | 2.5 μmol/L, 5 μmol/L | H2O2 (500 μmol/L) stimulates rat NPCs for 24 h | TXNIP, NLRP3, JNK and NF-κB | Reduce H2O2-induced oxidative stress, apoptosis, and inflammatory responses | [119] | |
| Sesamin |  | 0.1 μmol/L, 0.5 μmol/L, 1 μmol/L | LPS (10 μg/mL) stimulates rat NPCs for 24 h | JNK | Inhibition of LPS-induced inflammatory responses and ECM catabolism | [120] | |
| Crocin | 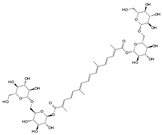 | 10 μmol/L, 50 μmol/L, 100 μmol/L | LPS (10 μg/mL) stimulates rat NPCs for 0.5 and 24 h | JNK | Repress LPS-induced inflammatory responses and reduce ECM catabolism | [121] | |
| Tanshinone IIA | 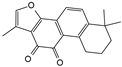 |
|
|
|
| [122,123] | |
| Glycyrrhizin |  | 100 μmol/L | IL-1β (10 ng/mL) stimulates HNPCs for 24 h | p38 MAPK, JNK and HMGB1 | Inhibition of IL-1β-induced cell apoptosis and inflammatory responses | [124] | |
| Allicin |  | 5 μmol/L, 10 μmol/L, 20 μmol/L | AOPP (400 μg/mL) stimulates HNPCs for 24 h | P38 MAPK | Attenuate AOPP-induced oxidative stress and mitochondrial apoptosis | [125] | |
| Inhibitors | SD0006 |  | 70 nmol/L | IL-1β (5 ng/mL) stimulates HNPCs for 24 h | p38 MAPK | Reduce IL-1β- induced cell proliferation inhibition and inflammatory responses | [126] |
| TAK-715 | 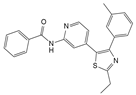 | 0.5 μmol/L, 1 μmol/L | IL-1β (10 ng/mL) stimulates rat NPCs for 48 h and IL-1β-induced ex vivo IVD culture model | P38 MAPK | Protect against IL-1β-induced rat NPC apoptosis and ECM degradation | [127] | |
| SP600125 | 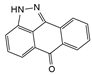 | 10 μmol/L | HAFCs and HNPCs were co-cultured with phorbol myristate acetate-stimulated macrophage-like THP-1 cells | JNK | Attenuate co-culture cells-induced inflammatory responses | [19] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Zhang, G.; Yang, Z.; Kang, X. Stress-Activated Protein Kinases in Intervertebral Disc Degeneration: Unraveling the Impact of JNK and p38 MAPK. Biomolecules 2024, 14, 393. https://doi.org/10.3390/biom14040393
Li L, Zhang G, Yang Z, Kang X. Stress-Activated Protein Kinases in Intervertebral Disc Degeneration: Unraveling the Impact of JNK and p38 MAPK. Biomolecules. 2024; 14(4):393. https://doi.org/10.3390/biom14040393
Chicago/Turabian StyleLi, Lei, Guangzhi Zhang, Zhili Yang, and Xuewen Kang. 2024. "Stress-Activated Protein Kinases in Intervertebral Disc Degeneration: Unraveling the Impact of JNK and p38 MAPK" Biomolecules 14, no. 4: 393. https://doi.org/10.3390/biom14040393
APA StyleLi, L., Zhang, G., Yang, Z., & Kang, X. (2024). Stress-Activated Protein Kinases in Intervertebral Disc Degeneration: Unraveling the Impact of JNK and p38 MAPK. Biomolecules, 14(4), 393. https://doi.org/10.3390/biom14040393