

Involvement of Glucosamine 6 Phosphate Isomerase 2 (GNPDA2) Overproduction in β-Amyloid- and Tau P301L-Driven Pathomechanisms
 , , and
, , and 
Abstract
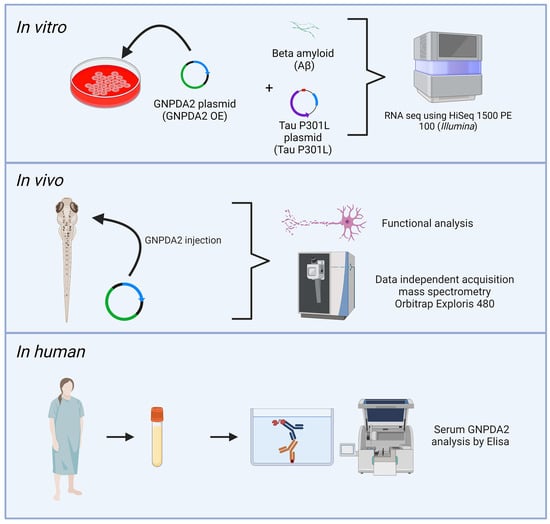
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Immunoprecipitation
2.4. RNA Sequencing (RNA-Seq) and Data Analysis
2.5. Zebrafish Studies
2.6. RNA Injection, Evaluation of Toxicity and Determination of GNPDA2 Protein Expression
2.7. RNA Injection and Evaluation of the Phenotype Associated with TAU P301L Overexpression
2.8. Axonal Motoneuron Extension
2.9. Detection of Neuronal Death
2.10. Detection of TAU Phosphorylation
2.11. Embryo Collection and Sample Preparation for Proteomic Analysis
2.12. Mass Spectrometry
2.13. Data Analysis
2.14. Bioinformatic Analysis
2.15. Enzyme-Linked Immunosorbent Assay
3. Results
3.1. GNPDA2 Interactome Is Partially Associated with Intraciliary Transport in NECs
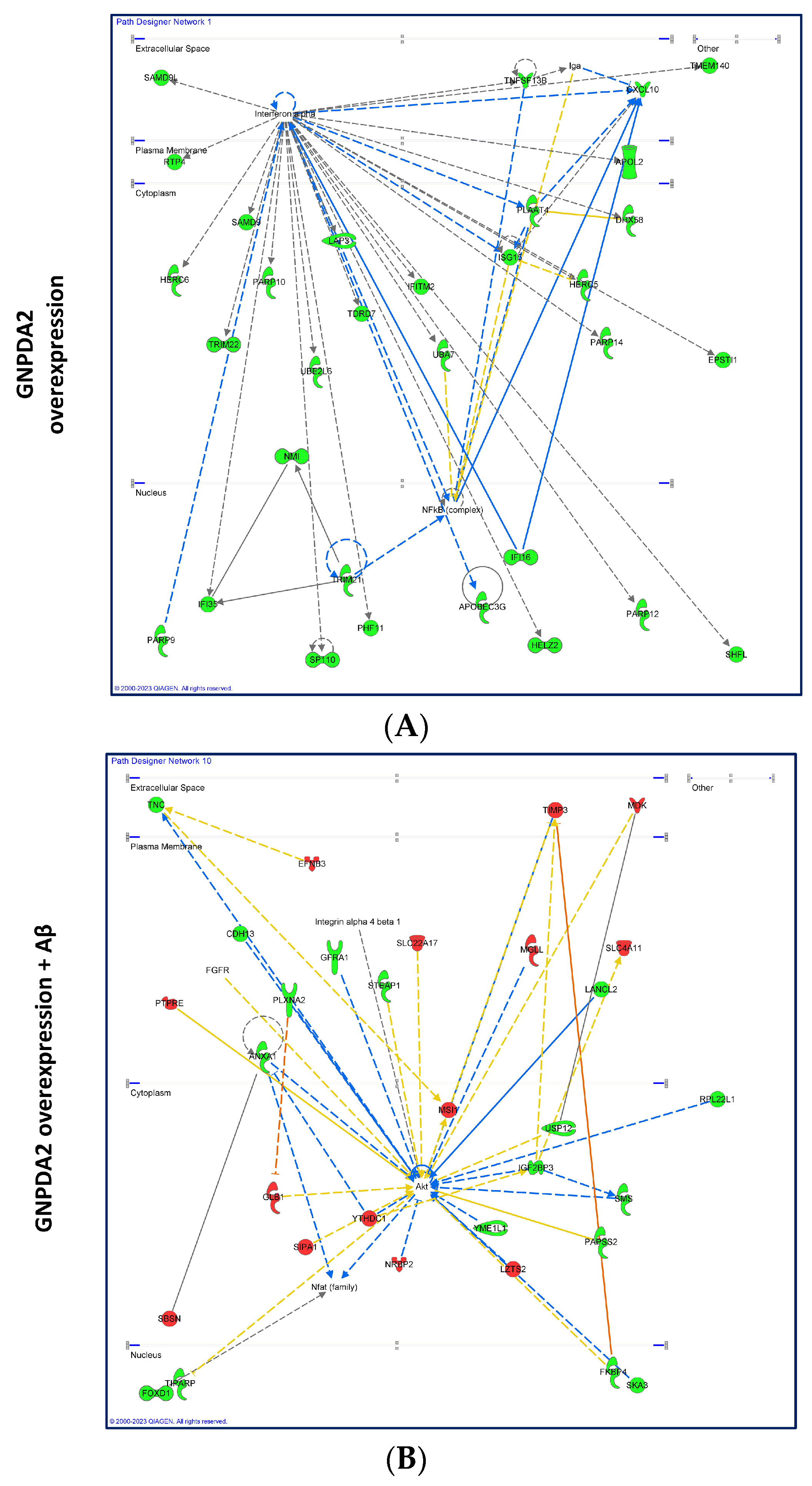
3.2. Dissimilar Transcriptomic Variability in GNPDA2-Overexpressing NECs upon Neuropathological Insult
3.3. Effects of Human GNPDA2 Overexpression in h.Tau P301L Transgenic Zebrafish Embryos
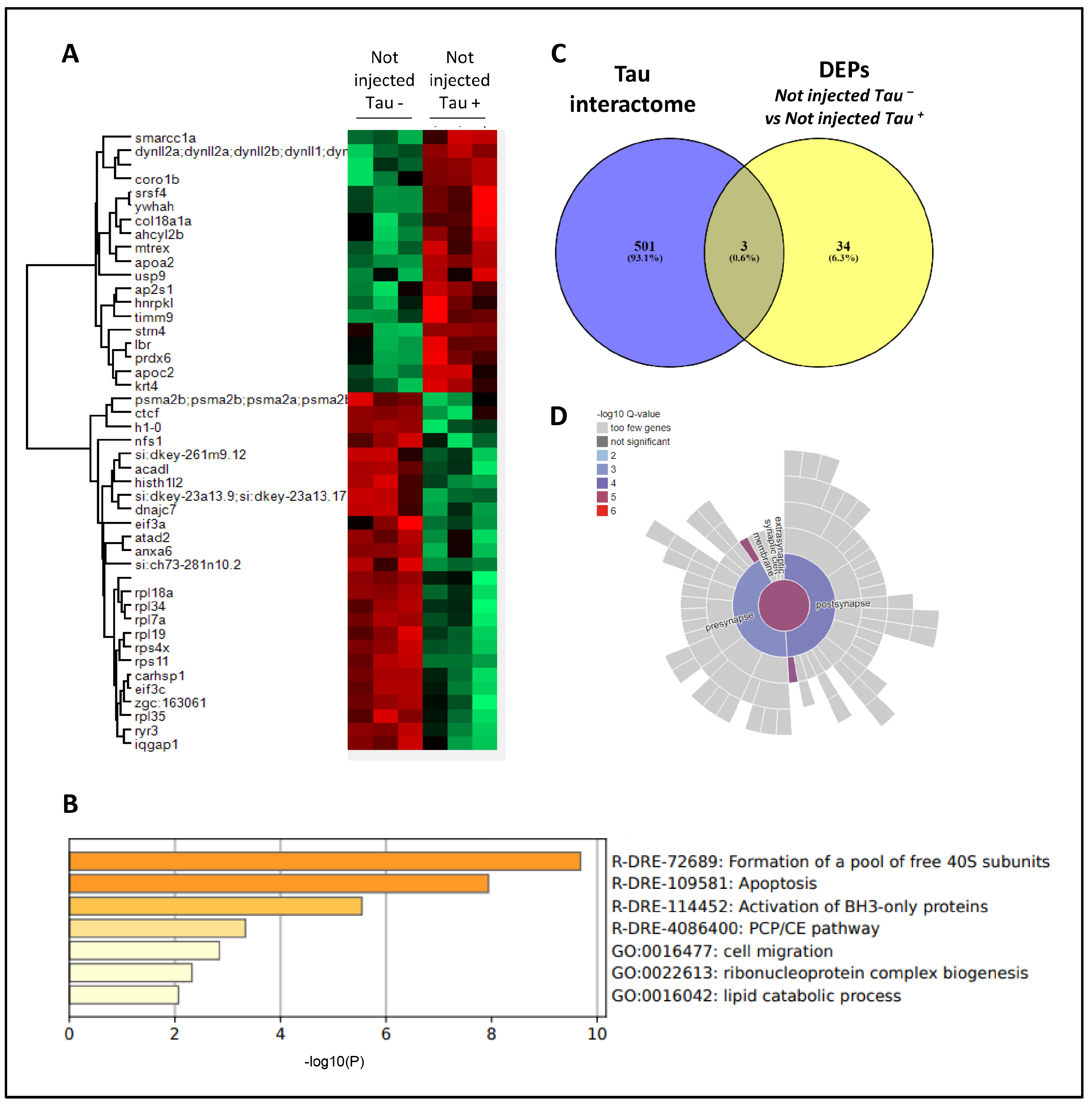
3.4. Proteomic Analysis Revealed Widespread Alterations in GNPDA2-Overexpressing h.Tau P301L Zebrafish Embryos
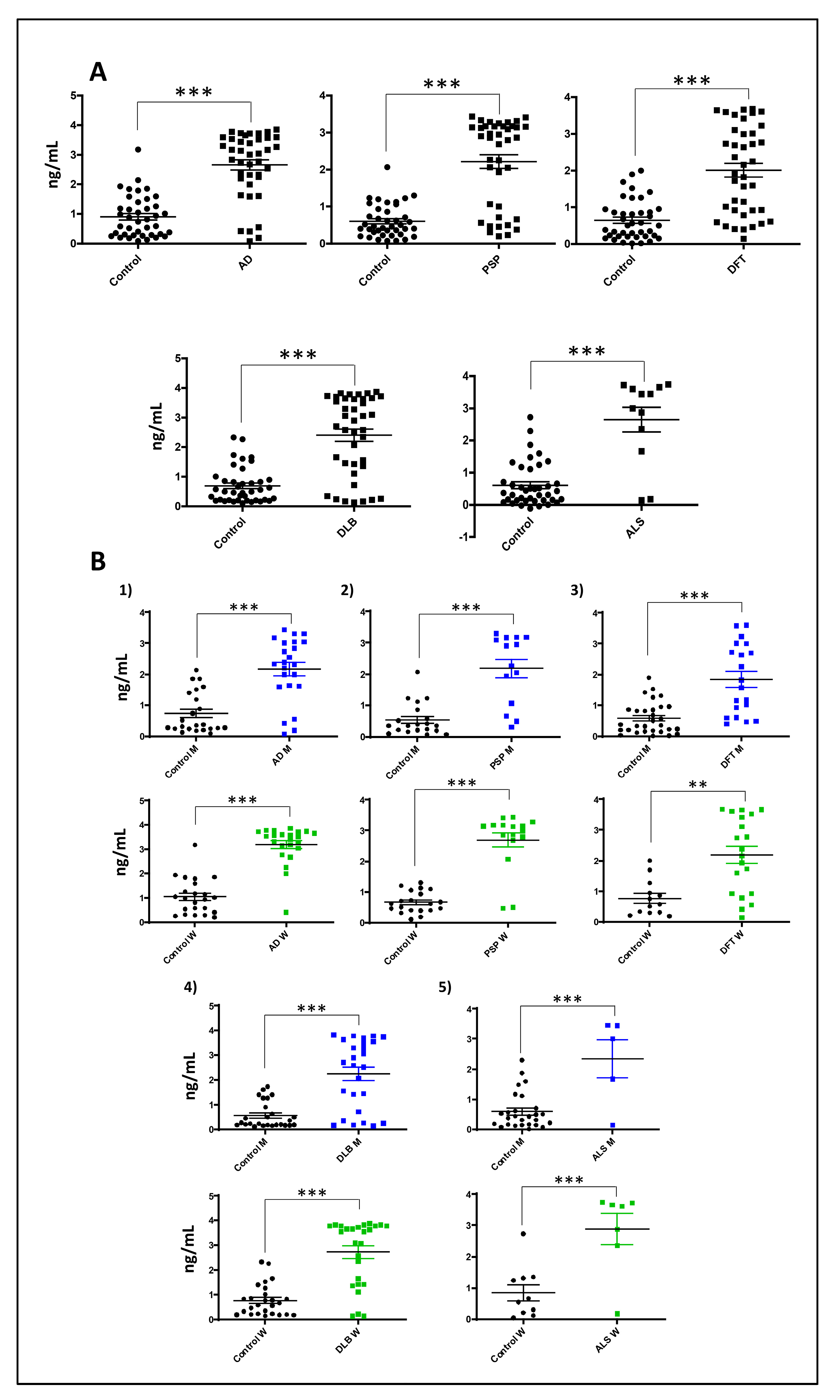
3.5. Increased Serum GNPDA2 Protein Levels across Neurodegenerative Proteinopathies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- 2023 Alzheimer’s disease facts and figures. Alzheimers Dement 2023, 19, 1598–1695. [CrossRef] [PubMed]
- Li, X.; Feng, X.; Sun, X.; Hou, N.; Han, F.; Liu, Y. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2019. Front. Aging Neurosci. 2022, 14, 937486. [Google Scholar] [CrossRef] [PubMed]
- Doty, R.L. Olfactory dysfunction in neurodegenerative diseases: Is there a common pathological substrate? Lancet Neurol. 2017, 16, 478–488. [Google Scholar] [CrossRef]
- Zou, Y.M.; Lu, D.; Liu, L.P.; Zhang, H.H.; Zhou, Y.Y. Olfactory dysfunction in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2016, 12, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Attems, J.; Walker, L.; Jellinger, K.A. Olfactory bulb involvement in neurodegenerative diseases. Acta Neuropathol. 2014, 127, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L. Alzheimer’s disease. Nature 2009, 461, 895–897. [Google Scholar] [CrossRef] [PubMed]
- Afridi, R.; Rahman, M.H.; Suk, K. Implications of glial metabolic dysregulation in the pathophysiology of neurodegenerative diseases. Neurobiol. Dis. 2022, 174, 105874. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Hu, Y.; Wang, B.; Wang, S.; Zhang, X. Metabolic Dysregulation Contributes to the Progression of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 530219. [Google Scholar] [CrossRef] [PubMed]
- Raut, S.; Bhalerao, A.; Powers, M.; Gonzalez, M.; Mancuso, S.; Cucullo, L. Hypometabolism, Alzheimer’s Disease, and Possible Therapeutic Targets: An Overview. Cells 2023, 12, 2019. [Google Scholar] [CrossRef] [PubMed]
- Colavitta, M.F.; Grasso, L.; Barrantes, F.J. Environmental Enrichment in Murine Models and Its Translation to Human Factors Improving Conditions in Alzheimer Disease. J. Prev. Alzheimer’s Dis. 2023, 10, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Dodart, J.C.; Mathis, C.; Bales, K.R.; Paul, S.M.; Ungerer, A. Early regional cerebral glucose hypometabolism in transgenic mice overexpressing the V717F beta-amyloid precursor protein. Neurosci. Lett. 1999, 277, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Park, J.; Han, I.O. Hexosamine biosynthetic pathway and O-GlcNAc cycling of glucose metabolism in brain function and disease. Am. J. Physiol. Physiol. 2023, 325, C981–C998. [Google Scholar] [CrossRef] [PubMed]
- Lachén-Montes, M.; González-Morales, A.; Iloro, I.; Elortza, F.; Ferrer, I.; Gveric, D.; Fernández-Irigoyen, J.; Santamaría, E. Un-veiling the olfactory proteostatic disarrangement in Parkinson’s disease by proteome-wide profiling. Neurobiol. Aging 2019, 73, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Paneque, A.; Fortus, H.; Zheng, J.; Werlen, G.; Jacinto, E. The Hexosamine Biosynthesis Pathway: Regulation and Function. Genes 2023, 14, 933. [Google Scholar] [CrossRef] [PubMed]
- Seiler, N. Ammonia and Alzheimer’s disease. Neurochem. Int. 2002, 41, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Adlimoghaddam, A.; Sabbir, M.G.; Albensi, B.C. Ammonia as a Potential Neurotoxic Factor in Alzheimer’s Disease. Front. Mol. Neurosci. 2016, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.W.; Yang, L.; Dai, X.L.; Jiang, Z.F.; Huang, H.C. Roles of O-GlcNAcylation on amyloid-β precursor protein processing, tau phosphorylation, and hippocampal synapses dysfunction in Alzheimer’s disease. Neurol. Res. 2016, 38, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shan, X.; Yuzwa, S.A.; Vocadlo, D.J. The emerging link between O-GlcNAc and Alzheimer disease. J. Biol. Chem. 2014, 289, 34472–34481. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lai, M.K.P.; Arumugam, T.V.; Jo, D.G. O-GlcNAcylation as a Therapeutic Target for Alzheimer’s Disease. NeuroMolecular Med. 2020, 22, 171–193. [Google Scholar] [CrossRef] [PubMed]
- Pinho, T.S.; Verde, D.M.; Correia, S.C.; Cardoso, S.M.; Moreira, P.I. O-GlcNAcylation and neuronal energy status: Implications for Alzheimer’s disease. Ageing Res. Rev. 2018, 46, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Almén, M.S.; Fredriksson, R.; Schiöth, H.B. What model organisms and interactomics can reveal about the ge-netics of human obesity. Cell. Mol. Life Sci. 2012, 69, 3819–3834. [Google Scholar] [CrossRef] [PubMed]
- Yılmaz, B.; Gezmen Karadağ, M. The current review of adolescent obesity: The role of genetic factors. J. Pediatr. Endocrinol. Metab. 2021, 34, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.W.; Zhang, M.X.; Wu, L.J.; Fu, L.W.; Zhao, X.Y.; Mi, J. Association between rs10938397 polymorphism in GNPDA2 and obesity in children at different stages of development. Zhonghua Liu Xing Bing Xue Za Zhi 2018, 39, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Nakamura, M.; Nakamura, T.; Matsuo, T.; Nakata, Y.; Kamohara, S.; Miyatake, N.; Kotani, K.; Komatsu, R.; Itoh, N.; et al. Association between obesity and polymorphisms in SEC16B, TMEM18, GNPDA2, BDNF, FAIM2 and MC4R in a Japanese population. J. Hum. Genet. 2009, 54, 727–731. [Google Scholar] [CrossRef]
- Paquet, D.; Bhat, R.; Sydow, A.; Mandelkow, E.M.; Berg, S.; Hellberg, S.; Fälting, J.; Distel, M.; Köster, R.W.; Schmid, B.; et al. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J. Clin. Investig. 2009, 119, 1382–1395. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput se-quencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Align-ment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. Feature Counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Lachén-Montes, M.; González-Morales, A.; Zelaya, M.V.; Pérez-Valderrama, E.; Ausín, K.; Ferrer, I.; Fernández-Irigoyen, J.; Santamaría, E. Olfactory bulb neuroproteomics reveals a chronological perturbation of survival routes and a disruption of pro-hibitin complex during Alzheimer’s disease progression. Sci. Rep. 2017, 7, 9115. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.F.; Chaves, R.S.; Silva, C.M.; Chaves, J.C.S.; Melo, K.P.; Ferrari, M.F.R. BDNF trafficking and signaling impairment during early neurodegeneration is prevented by moderate physical activity. IBRO Rep. 2016, 1, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; Sturchio, A.; Espay, A. The shift to a proteinopenia paradigm in neurodegeneration. Hand. Clin. Neurol. 2023, 193, 23–32. [Google Scholar] [CrossRef]
- Dahl, R.; Mygind, N. Anatomy, physiology and function of the nasal cavities in health and disease. Adv. Drug Deliv. Rev. 1998, 29, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Yeo, S.; Baek, S.; Jung, H.J.; Lee, M.S.; Choi, S.H.; Choe, Y. Abnormal accumulation of extracellular vesicles in hippo-campal dystrophic axons and regulation by the primary cilia in Alzheimer’s disease. Acta Neuropathol. Commun. 2023, 11, 142. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.G.; Zukin, R.S. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 2007, 8, 413–426. [Google Scholar] [CrossRef]
- Du, Z.; Song, Y.; Chen, X.; Zhang, W.; Zhang, G.; Li, H.; Chang, L.; Wu, Y. Knockdown of astrocytic Grin2a aggravates β-amyloid-induced memory and cognitive deficits through regulating nerve growth factor. Aging Cell 2021, 20, e13437. [Google Scholar] [CrossRef] [PubMed]
- Guinez, C.; Mir, A.M.; Dehennaut, V.; Cacan, R.; Harduin-Lepers, A.; Michalski, J.C.; Lefebvre, T. Protein ubiquitination is modulated by O-GlcNAc glycosylation. FASEB J. 2008, 22, 2901–2911. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.B.; Nie, Y.; Yang, X. Regulation of protein degradation by O-GlcNAcylation: Crosstalk with ubiquitination. Mol. Cell. Proteom. 2013, 12, 3489–3497. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Figueiredo-Pereira, M.E. Ubiquitin/proteasome pathway impairment in neurodegeneration: Therapeutic implica-tions. Apoptosis 2010, 15, 1292–1311. [Google Scholar] [CrossRef]
- Limantoro, J.; de Liyis, B.G.; Sutedja, J.C. Akt signaling pathway: A potential therapy for Alzheimer’s disease through glycogen synthase kinase 3 beta inhibition. Egypt. J. Neurol. Psychiatry Neurosurg. 2023, 59, 147. [Google Scholar] [CrossRef]
- Lee, M.; Kim, D.W.; Khalmuratova, R.; Shin, S.H.; Kim, Y.M.; Han, D.H.; Kim, H.J.; Kim, D.Y.; Rhee, C.S.; Park, J.W.; et al. The IFN-γ-p38, ERK kinase axis exacerbates neutrophilic chronic rhinosinusitis by inducing the epithelial-to-mesenchymal transi-tion. Mucosal Immunol. 2019, 12, 601–611. [Google Scholar] [CrossRef]
- Li, T.; Li, Y.; Zhang, G. [The variations of p38 MAPK activity on lipopolysaccharide-induced inflammation of nasal epithelial cells and its significance in vitro]. Lin Chuang Er Bi Yan Hou Ke Za Zhi 2005, 19, 607–610. [Google Scholar]
- Dehvari, N.; Isacsson, O.; Winblad, B.; Cedazo-Minguez, A.; Cowburn, R.F. Presenilin regulates extracellular regulated kinase (Erk) activity by a protein kinase C alpha dependent mechanism. Neurosci. Lett. 2008, 436, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Zirrgiebel, U.; von Bohlen Und Halbach, O.; Strelau, J.; Laliberté, C.; Kaplan, D.R.; Unsicker, K. ERK activation promotes neuronal degeneration predominantly through plasma membrane damage and independently of caspase-3. J. Cell Biol. 2004, 165, 357–369. [Google Scholar] [CrossRef]
- Lee, J.K.; Kim, N.J. Recent Advances in the Inhibition of p38 MAPK as a Potential Strategy for the Treatment of Alzheimer’s Disease. Molecules 2017, 22, 1287. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L.; Ammit, A.J. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 2010, 58, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Bachstetter, A.D.; Xing, B.; de Almeida, L.; Dimayuga, E.R.; Watterson, D.M.; Van Eldik, L.J. Microglial p38α MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Aβ). J. Neuroinflammation 2011, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N. Animal models of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006320. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Oguchi, K.; Okabe, S.; Kuno, J.; Terada, S.; Ohshima, T.; Sato-Yoshitake, R.; Takei, Y.; Noda, T.; Hirokawa, N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 1994, 369, 488–491. [Google Scholar] [CrossRef]
- Dawson, H.N.; Ferreira, A.; Eyster, M.V.; Ghoshal, N.; Binder, L.I.; Vitek, M.P. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 2001, 114, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; Morán, M.A.; Ferrer, I.; Avila, J.; Gómez-Ramos, P. Phosphorylated tau in neuritic plaques of APP(sw)/Tau (vlw) transgenic mice and Alzheimer disease. Acta Neuropathol. 2008, 116, 409–418. [Google Scholar] [CrossRef] [PubMed]
- López-González, I.; Aso, E.; Carmona, M.; Armand-Ugon, M.; Blanco, R.; Naudí, A.; Cabré, R.; Portero-Otin, M.; Pamplona, R.; Ferrer, I. Neuroinflammatory Gene Regulation, Mitochondrial Function, Oxidative Stress, and Brain Lipid Modifications with Disease Progression in Tau P301S Transgenic Mice as a Model of Frontotemporal Lobar Degeneration-Tau. J. Neuropathol. Exp. Neurol. 2015, 74, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.T.; Taylor, D.; Kneynsberg, A.; Bodea, L.G.; Götz, J. Altered ribosomal function and protein synthesis caused by tau. Acta Neuropathol. Commun. 2021, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.T.; Benetatos, J.; van Roijen, M.; Bodea, L.G.; Götz, J. Decreased synthesis of ribosomal proteins in tauopathy revealed by non-canonical amino acid labelling. EMBO J. 2019, 38, e101174. [Google Scholar] [CrossRef] [PubMed]
- Lomonosova, E.; Chinnadurai, G. BH3-only proteins in apoptosis and beyond: An overview. Oncogene 2008, 27 (Suppl. S1), S2–S19. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; Henry, H.; Villard, A.M.; Zurich, M.G.; Loup, M.; Eilers, B.; Parlascino, G.; Matter, E.; Boulat, O.; Honegger, P.; et al. Ammonium-induced impairment of axonal growth is prevented through glial creatine. J. Neurosci. 2002, 22, 9810–9820. [Google Scholar] [CrossRef] [PubMed]
- Buzańska, L.; Zabłocka, B.; Dybel, A.; Domańska-Janik, K.; Albrecht, J. Delayed induction of apoptosis by ammonia in C6 glioma cells. Neurochem. Int. 2000, 37, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Sims, B.; Powers, R.E.; Sabina, R.L.; Theibert, A.B. Elevated adenosine monophosphate deaminase activity in Alzheimer’s disease brain. Neurobiol. Aging 1998, 19, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Clayton, J.A. Sex influences in neurological disorders: Case studies and perspectives. Dialog. Clin. Neurosci. 2016, 18, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Subramaniapillai, S.; Almey, A.; Natasha Rajah, M.; Einstein, G. Sex and gender differences in cognitive and brain reserve: Implications for Alzheimer’s disease in women. Front. Neuroendocr. 2021, 60, 100879. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Cao, J.; Hou, J.; Li, Y.; Huang, M.; Zhu, L.; Zhang, L.; Lee, Y.; Duarte, M.L.; Zhou, X.; et al. Sex specifc molecular networks and key drivers of Alzheimer’s disease. Mol. Neurodegener. 2023, 18, 39. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zhong, M.B.; Zhang, L.; Zhang, B.; Cai, D. Sex Differences in Alzheimer’s Disease: Insights from the Multiomics Landscape. Biol. Psychiatry 2022, 91, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Park, J.C.; Lim, H.; Byun, M.S.; Yi, D.; Byeon, G.; Jung, G.; Kim, Y.K.; Lee, D.Y.; Han, S.H.; Mook-Jung, I. Sex differences in the progression of glucose. Nature 2023, 55, 1023–1032. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | N° of Patients (Mean Age) | Male | Female |
|---|---|---|---|
| Control | 71 (69.2) | 40 | 31 |
| Alzheimer’s disease | 40 (75.1) | 20 | 20 |
| Progressive supranuclear palsy | 40 (67.9) | 15 | 25 |
| Frontotemporal dementia | 40 (69.5) | 20 | 20 |
| Dementia with Lewy bodies | 12 (73.5) | 6 | 6 |
| Amyotrophic lateral sclerosis | 12 (57.8) | 5 | 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lachén-Montes, M.; Cartas-Cejudo, P.; Cortés, A.; Anaya-Cubero, E.; Peral, E.; Ausín, K.; Díaz-Peña, R.; Fernández-Irigoyen, J.; Santamaría, E. Involvement of Glucosamine 6 Phosphate Isomerase 2 (GNPDA2) Overproduction in β-Amyloid- and Tau P301L-Driven Pathomechanisms. Biomolecules 2024, 14, 394. https://doi.org/10.3390/biom14040394
Lachén-Montes M, Cartas-Cejudo P, Cortés A, Anaya-Cubero E, Peral E, Ausín K, Díaz-Peña R, Fernández-Irigoyen J, Santamaría E. Involvement of Glucosamine 6 Phosphate Isomerase 2 (GNPDA2) Overproduction in β-Amyloid- and Tau P301L-Driven Pathomechanisms. Biomolecules. 2024; 14(4):394. https://doi.org/10.3390/biom14040394
Chicago/Turabian StyleLachén-Montes, Mercedes, Paz Cartas-Cejudo, Adriana Cortés, Elena Anaya-Cubero, Erika Peral, Karina Ausín, Ramón Díaz-Peña, Joaquín Fernández-Irigoyen, and Enrique Santamaría. 2024. "Involvement of Glucosamine 6 Phosphate Isomerase 2 (GNPDA2) Overproduction in β-Amyloid- and Tau P301L-Driven Pathomechanisms" Biomolecules 14, no. 4: 394. https://doi.org/10.3390/biom14040394
APA StyleLachén-Montes, M., Cartas-Cejudo, P., Cortés, A., Anaya-Cubero, E., Peral, E., Ausín, K., Díaz-Peña, R., Fernández-Irigoyen, J., & Santamaría, E. (2024). Involvement of Glucosamine 6 Phosphate Isomerase 2 (GNPDA2) Overproduction in β-Amyloid- and Tau P301L-Driven Pathomechanisms. Biomolecules, 14(4), 394. https://doi.org/10.3390/biom14040394