
Combined Pharmacological Modulation of Translational and Transcriptional Activity Signaling Pathways as a Promising Therapeutic Approach in Children with Myocardial Changes



Abstract
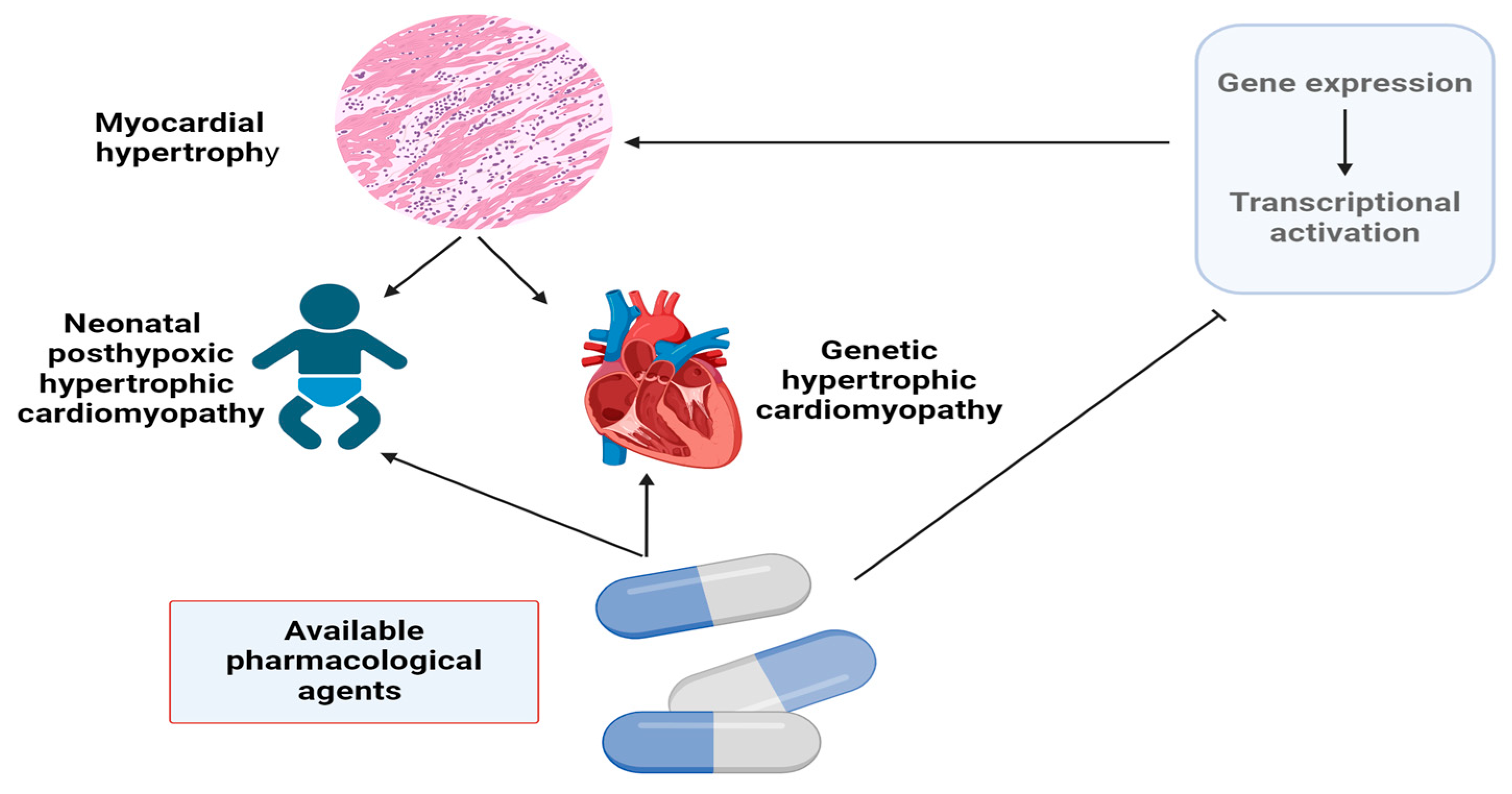
:1. Introduction
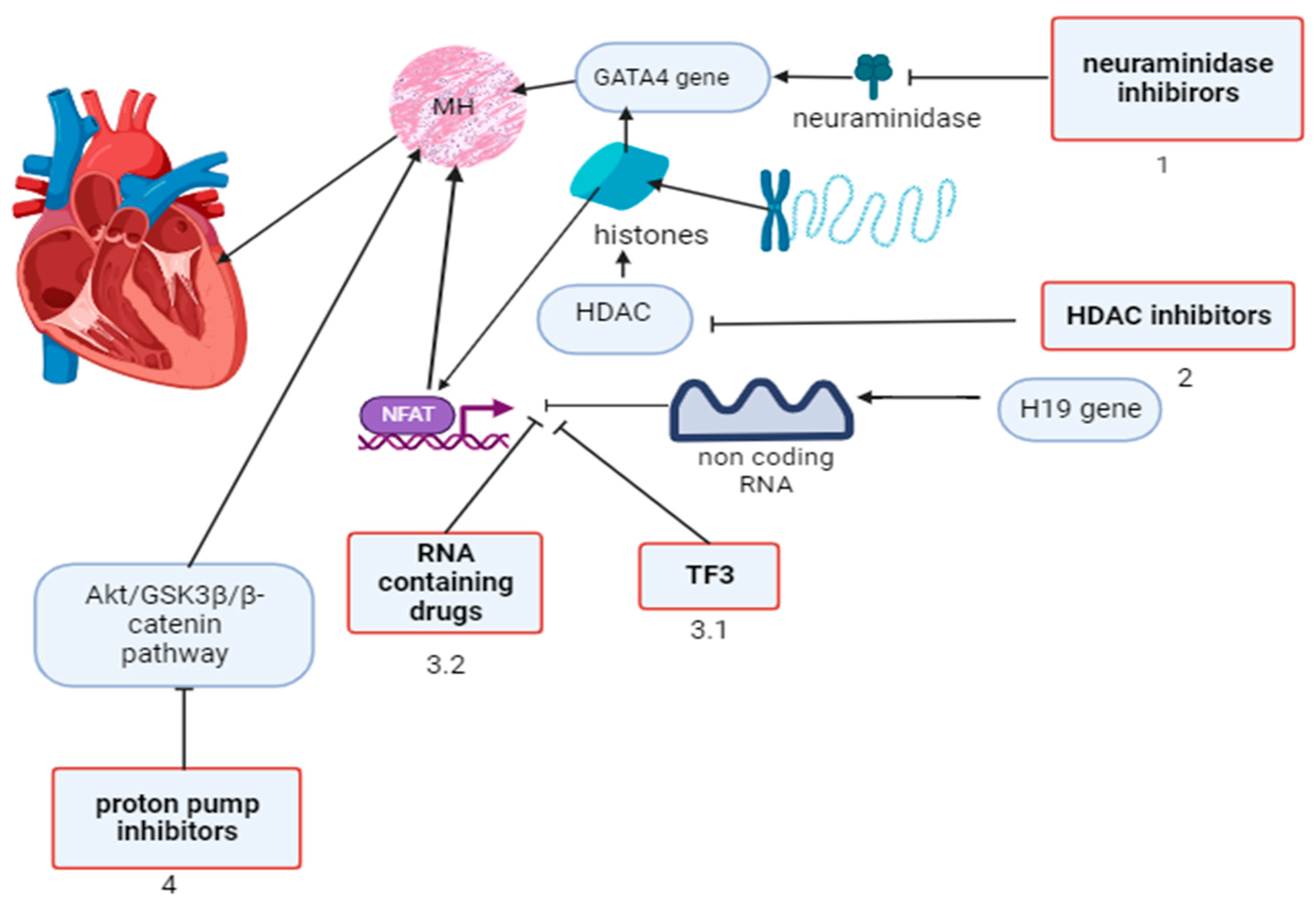
2. Regulatory Factors of Gene Expression in Myocardial Hypertrophy and Approaches to Pharmacological Modulation
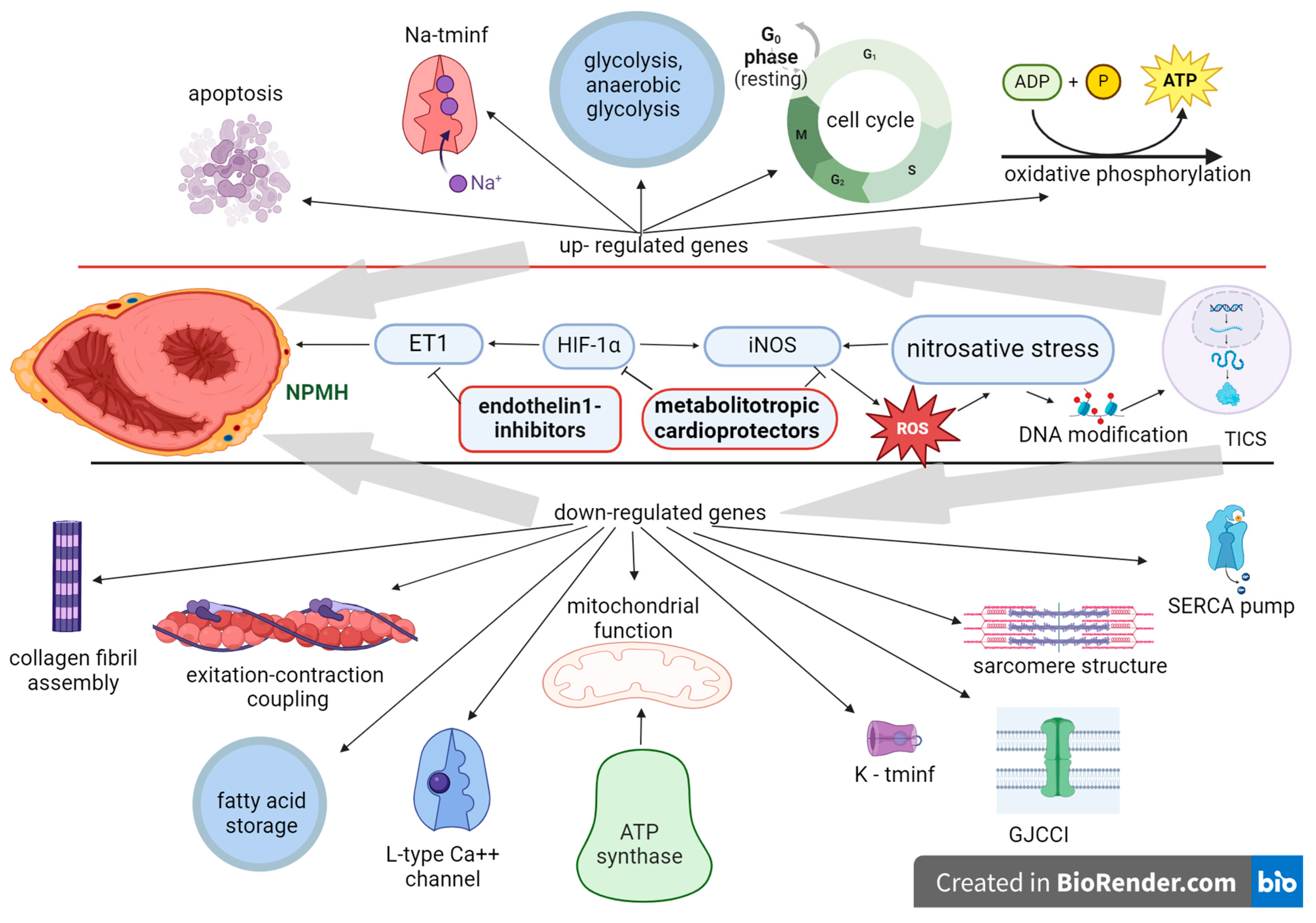
3. Neonatal Posthypoxic Myocardial Hypertrophy, Factors of Gene Expression Regulation and Pharmacological Modulation
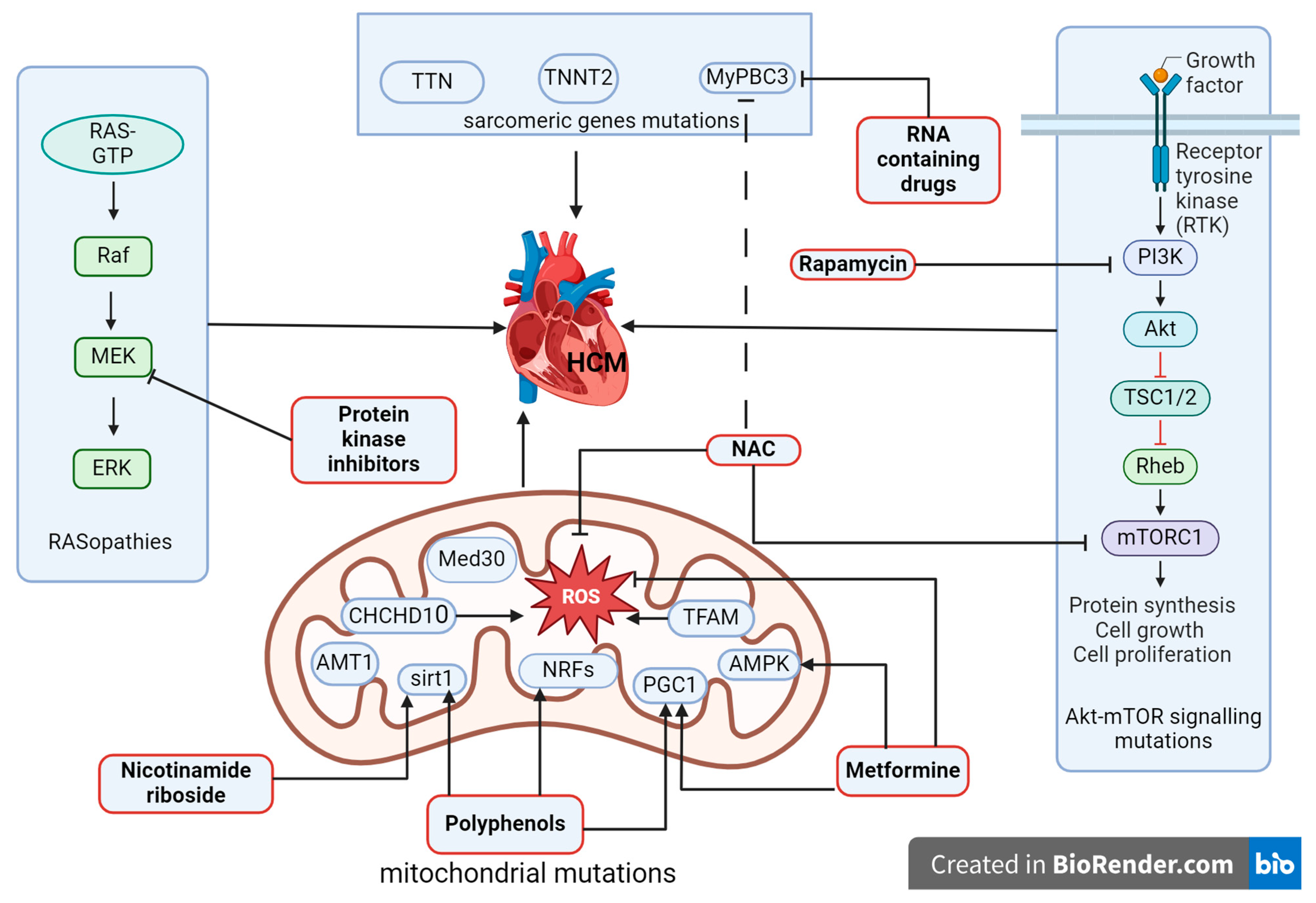
4. Hypertrophic Cardiomyopathy, Transcriptional Signaling Pathways, and Options for the Treatment
5. Conclusions, Discussion, and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Liu, L.; Hu, J.; Lei, H.; Qin, H.; Wang, C.; Gui, Y.; Xu, D. Regulatory T Cells in Pathological Cardiac Hypertrophy: Mechanisms and Therapeutic Potential. Cardiovasc. Drugs Ther. 2023, 37, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.M.; Nikolova, A.P. A Surprising Noncanonical Role for Calcineurin in Pressure-Induced Cardiac Hypertrophy. J. Am. Coll. Cardiol. 2018, 71, 668–669. [Google Scholar] [CrossRef] [PubMed]
- Mognol, G.P.; Carneiro, F.R.; Robbs, B.K.; Faget, D.V.; Viola, J.P. Cell cycle and apoptosis regulation by NFAT transcription factors: New roles for an old player. Cell Death Dis. 2016, 7, e2199. [Google Scholar] [CrossRef] [PubMed]
- Gunawan, F.; Gentile, A.; Gauvrit, S.; Stainier, D.Y.R.; Bensimon-Brito, A. Nfatc1 Promotes Interstitial Cell Formation During Cardiac Valve Development in Zebrafish. Circ. Res. 2020, 126, 968–984. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, M.G.; Linneweh, M.; Liebscher, S.; Van Handel, B.; Layland, S.L.; Schenke-Layland, K. Endocardial-to-mesenchymal transformation and mesenchymal cell colonization at the onset of human cardiac valve development. Development 2016, 143, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Kamenshchyk, A.; Gonchar, M.; Oksenych, V.; Kamyshnyi, A. Association of Myocardial Changes and Gene Expression of the NFATC1 and NFATC4-Calcineurin Signaling Pathway in Children with Bicuspid Aortic Valve. Children 2023, 10, 1434. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Vigil-Garcia, M.; Demkes, C.J.; Eding, J.E.C.; Versteeg, D.; de Ruiter, H.; Perini, I.; Kooijman, L.; Gladka, M.M.; Asselbergs, F.W.; Vink, A.; et al. Gene expression profiling of hypertrophic cardiomyocytes identifies new players in pathological remodelling. Cardiovasc. Res. 2021, 117, 1532–1545. [Google Scholar] [CrossRef] [PubMed]
- Chaklader, M.; Rothermel, B.A. Calcineurin in the heart: New horizons for an old friend. Cell Signal 2021, 87, 110134. [Google Scholar] [CrossRef]
- Bazgir, F.; Nau, J.; Nakhaei-Rad, S.; Amin, E.; Wolf, M.J.; Saucerman, J.J.; Lorenz, K.; Ahmadian, M.R. The Microenvironment of the Pathogenesis of Cardiac Hypertrophy. Cells 2023, 12, 1780. [Google Scholar] [CrossRef]
- Viereck, J.; Buhrke, A.; Foinquinos, A.; Chatterjee, S.; Kleeberger, J.A.; Xiao, K.; Janssen-Peters, H.; Batkai, S.; Ramanujam, D.; Kraft, T.; et al. Targeting muscle-enriched long non-coding RNA H19 reverses pathological cardiac hypertrophy. Eur. Heart J. 2020, 41, 3462–3474. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Luo, Y.; Song, J.; Tan, T.; Zhu, H. Non-coding RNAs and Pathological Cardiac Hypertrophy. Adv. Exp. Med. Biol. 2020, 1229, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lei, F.; Wang, X.M.; Deng, K.Q.; Ji, Y.X.; Zhang, Y.; Li, H.; Zhang, X.D.; Lu, Z.; Zhang, P. NULP1 Alleviates Cardiac Hypertrophy by Suppressing NFAT3 Transcriptional Activity. J. Am. Heart Assoc. 2020, 9, e016419. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xia, C.; Yang, Y.; Warusawitharana, H.K.; Liu, X.; Tu, Y. The Prevention Role of Theaflavin-3,3’-digallate in Angiotensin II Induced Pathological Cardiac Hypertrophy via CaN-NFAT Signal Pathway. Nutrients 2022, 14, 1391. [Google Scholar] [CrossRef]
- Han, Y.; Nie, J.; Wang, D.W.; Ni, L. Mechanism of histone deacetylases in cardiac hypertrophy and its therapeutic inhibitors. Front. Cardiovasc. Med. 2022, 9, 931475. [Google Scholar] [CrossRef] [PubMed]
- Kolski-Andreaco, A.; Balut, C.M.; Bertuccio, C.A.; Wilson, A.S.; Rivers, W.M.; Liu, X.; Gandley, R.E.; Straub, A.C.; Butterworth, M.B.; Binion, D.; et al. Histone deacetylase inhibitors (HDACi) increase expression of KCa2.3 (SK3) in primary microvascular endothelial cells. Am. J. Physiol. Cell Physiol. 2022, 322, C338–C353. [Google Scholar] [CrossRef] [PubMed]
- Chun, P. Therapeutic effects of histone deacetylase inhibitors on heart disease. Arch. Pharm. Res. 2020, 43, 1276–1296. [Google Scholar] [CrossRef] [PubMed]
- Heimerl, M.; Sieve, I.; Ricke-Hoch, M.; Erschow, S.; Battmer, K.; Scherr, M.; Hilfiker-Kleiner, D. Neuraminidase-1 promotes heart failure after ischemia/reperfusion injury by affecting cardiomyocytes and invading monocytes/macrophages. Basic. Res. Cardiol. 2020, 115, 62. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Q.; Ma, G.; Liu, J.F.; Cai, Y.Y.; Zhang, J.Y.; Wei, T.T.; Pan, A.; Jiang, S.; Xiao, Y.; Xiao, P.; et al. Neuraminidase 1 is a driver of experimental cardiac hypertrophy. Eur. Heart J. 2021, 42, 3770–3782. [Google Scholar] [CrossRef]
- Lin, H.; Li, Y.; Zhu, H.; Wang, Q.; Chen, Z.; Chen, L.; Zhu, Y.; Zheng, C.; Wang, Y.; Liao, W.; et al. Lansoprazole alleviates pressure overload-induced cardiac hypertrophy and heart failure in mice by blocking the activation of beta-catenin. Cardiovasc. Res. 2020, 116, 101–113. [Google Scholar] [CrossRef]
- Aye, C.Y.L.; Lewandowski, A.J.; Lamata, P.; Upton, R.; Davis, E.; Ohuma, E.O.; Kenworthy, Y.; Boardman, H.; Wopperer, S.; Packham, A.; et al. Disproportionate cardiac hypertrophy during early postnatal development in infants born preterm. Pediatr. Res. 2017, 82, 36–46. [Google Scholar] [CrossRef]
- Hutter, D.; Kingdom, J.; Jaeggi, E. Causes and mechanisms of intrauterine hypoxia and its impact on the fetal cardiovascular system: A review. Int. J. Pediatr. 2010, 2010, 401323. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, L.; Wang, C. Prenatal hypoxia-induced epigenomic and transcriptomic reprogramming in rat fetal and adult offspring hearts. Sci. Data 2019, 6, 238. [Google Scholar] [CrossRef] [PubMed]
- Romanowicz, J.; Guerrelli, D.; Dhari, Z.; Mulvany, C.; Reilly, M.; Swift, L.; Vasandani, N.; Ramadan, M.; Leatherbury, L.; Ishibashi, N.; et al. Chronic perinatal hypoxia delays cardiac maturation in a mouse model for cyanotic congenital heart disease. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1873–H1886. [Google Scholar] [CrossRef]
- Huang, L.; Chen, X.; Dasgupta, C.; Chen, W.; Song, R.; Wang, C.; Zhang, L. Foetal hypoxia impacts methylome and transcriptome in developmental programming of heart disease. Cardiovasc. Res. 2019, 115, 1306–1319. [Google Scholar] [CrossRef]
- Paradis, A.N.; Gay, M.S.; Wilson, C.G.; Zhang, L. Newborn hypoxia/anoxia inhibits cardiomyocyte proliferation and decreases cardiomyocyte endowment in the developing heart: Role of endothelin-1. PLoS ONE 2015, 10, e0116600. [Google Scholar] [CrossRef]
- Ferreiro, C.R.; Chagas, A.C.; Carvalho, M.H.; Dantas, A.P.; Jatene, M.B.; Bento De Souza, L.C.; Lemos Da Luz, P. Influence of hypoxia on nitric oxide synthase activity and gene expression in children with congenital heart disease: A novel pathophysiological adaptive mechanism. Circulation 2001, 103, 2272–2276. [Google Scholar] [CrossRef]
- Niu, Y.; Kane, A.D.; Lusby, C.M.; Allison, B.J.; Chua, Y.Y.; Kaandorp, J.J.; Nevin-Dolan, R.; Ashmore, T.J.; Blackmore, H.L.; Derks, J.B.; et al. Maternal Allopurinol Prevents Cardiac Dysfunction in Adult Male Offspring Programmed by Chronic Hypoxia During Pregnancy. Hypertension 2018, 72, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Sud, N.; Sharma, S.; Wiseman, D.A.; Harmon, C.; Kumar, S.; Venema, R.C.; Fineman, J.R.; Black, S.M. Nitric oxide and superoxide generation from endothelial NOS: Modulation by HSP90. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1444–L1453. [Google Scholar] [CrossRef]
- Gratton, J.P.; Fontana, J.; O’Connor, D.S.; Garcia-Cardena, G.; McCabe, T.J.; Sessa, W.C. Reconstitution of an endothelial nitric-oxide synthase (eNOS), hsp90, and caveolin-1 complex in vitro. Evidence that hsp90 facilitates calmodulin stimulated displacement of eNOS from caveolin-1. J. Biol. Chem. 2000, 275, 22268–22272. [Google Scholar] [CrossRef]
- Hula, N.; Liu, R.; Spaans, F.; Pasha, M.; Quon, A.; Kirschenman, R.; Cooke, C.M.; Davidge, S.T. The Long-Term Effects of Prenatal Hypoxia on Coronary Artery Function of the Male and Female Offspring. Biomedicines 2022, 10, 3019. [Google Scholar] [CrossRef] [PubMed]
- De Pascali, F.; Hemann, C.; Samons, K.; Chen, C.A.; Zweier, J.L. Hypoxia and reoxygenation induce endothelial nitric oxide synthase uncoupling in endothelial cells through tetrahydrobiopterin depletion and S-glutathionylation. Biochemistry 2014, 53, 3679–3688. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF-1 signaling pathway in hypoxia-ischemia (Review). Mol. Med. Rep. 2018, 18, 3547–3554. [Google Scholar] [CrossRef] [PubMed]
- Jung, F.; Palmer, L.A.; Zhou, N.; Johns, R.A. Hypoxic regulation of inducible nitric oxide synthase via hypoxia inducible factor-1 in cardiac myocytes. Circ. Res. 2000, 86, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.; Gorbachova, S.; Pavlov, S.; Bukhtiyarova, N.; Puzyrenko, A.; Brek, O. Neurochemical Status of Nitric Oxide in the Settings of the Norm, Ishemic Event of Central Nervous System, and Pharmacological Bn Intervention. Georgian Med. News 2021, 315, 169–176. [Google Scholar]
- Dattilo, S.; Mancuso, C.; Koverech, G.; Di Mauro, P.; Ontario, M.L.; Petralia, C.C.; Petralia, A.; Maiolino, L.; Serra, A.; Calabrese, E.J.; et al. Heat shock proteins and hormesis in the diagnosis and treatment of neurodegenerative diseases. Immun. Ageing 2015, 12, 20. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Aliyeva, O.G.; Popazova, O.O.; Bukhtiyarova, N.V. Involvement of heat shock proteins HSP70 in the mechanisms of endogenous neuroprotection: The prospect of using HSP70 modulators. Front. Cell Neurosci. 2023, 17, 1131683. [Google Scholar] [CrossRef]
- Bogdan, C. Nitric oxide and the regulation of gene expression. Trends Cell Biol. 2001, 11, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Kroncke, K.D. Nitrosative stress and transcription. Biol. Chem. 2003, 384, 1365–1377. [Google Scholar] [CrossRef]
- Anavi, S.; Tirosh, O. iNOS as a metabolic enzyme under stress conditions. Free Radic. Biol. Med. 2020, 146, 16–35. [Google Scholar] [CrossRef]
- Bednov, A.; Espinoza, J.; Betancourt, A.; Vedernikov, Y.; Belfort, M.; Yallampalli, C. L-arginine prevents hypoxia-induced vasoconstriction in dual-perfused human placental cotyledons. Placenta 2015, 36, 1254–1259. [Google Scholar] [CrossRef]
- Popazova, O.; Belenichev, I.; Bukhtiyarova, N.; Ryzhenko, V.; Oksenych, V.; Kamyshnyi, A. Cardioprotective Activity of Pharmacological Agents Affecting NO Production and Bioavailability in the Early Postnatal Period after Intrauterine Hypoxia in Rats. Biomedicines 2023, 11, 2854. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Pimentel, D.R.; Remondino, A.; Sawyer, D.B.; Colucci, W.S. H(2)O(2) regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J. Mol. Cell Cardiol. 2003, 35, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Amin, J.K.; Xiao, L.; Pimental, D.R.; Pagano, P.J.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Reactive oxygen species mediate alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J. Mol. Cell Cardiol. 2001, 33, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Hirotani, S.; Otsu, K.; Nishida, K.; Higuchi, Y.; Morita, T.; Nakayama, H.; Yamaguchi, O.; Mano, T.; Matsumura, Y.; Ueno, H.; et al. Involvement of nuclear factor-kappaB and apoptosis signal-regulating kinase 1 in G-protein-coupled receptor agonist-induced cardiomyocyte hypertrophy. Circulation 2002, 105, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Law, Y.M.; Asante-Korang, A.; Austin, E.D.; Dipchand, A.I.; Everitt, M.D.; Hsu, D.T.; Lin, K.Y.; Price, J.F.; Wilkinson, J.D.; et al. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation 2019, 140, e9–e68. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kumar, V.; Kim, J.J. Sarcomeric Gene Variants and Their Role with Left Ventricular Dysfunction in Background of Coronary Artery Disease. Biomolecules 2020, 10, 442. [Google Scholar] [CrossRef]
- Ramirez, C.D.; Padron, R. Familial hypertrophic cardiomyopathy: Genes, mutations and animal models. A review. Investig. Clin. 2004, 45, 69–99. [Google Scholar]
- Mearini, G.; Stimpel, D.; Geertz, B.; Weinberger, F.; Kramer, E.; Schlossarek, S.; Mourot-Filiatre, J.; Stoehr, A.; Dutsch, A.; Wijnker, P.J.; et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat. Commun. 2014, 5, 5515. [Google Scholar] [CrossRef]
- Jiang, J.; Wakimoto, H.; Seidman, J.G.; Seidman, C.E. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science 2013, 342, 111–114. [Google Scholar] [CrossRef]
- Monda, E.; Rubino, M.; Lioncino, M.; Di Fraia, F.; Pacileo, R.; Verrillo, F.; Cirillo, A.; Caiazza, M.; Fusco, A.; Esposito, A.; et al. Hypertrophic Cardiomyopathy in Children: Pathophysiology, Diagnosis, and Treatment of Non-sarcomeric Causes. Front. Pediatr. 2021, 9, 632293. [Google Scholar] [CrossRef]
- Pandit, B.; Sarkozy, A.; Pennacchio, L.A.; Carta, C.; Oishi, K.; Martinelli, S.; Pogna, E.A.; Schackwitz, W.; Ustaszewska, A.; Landstrom, A.; et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat. Genet. 2007, 39, 1007–1012. [Google Scholar] [CrossRef]
- Calcagni, G.; Adorisio, R.; Martinelli, S.; Grutter, G.; Baban, A.; Versacci, P.; Digilio, M.C.; Drago, F.; Gelb, B.D.; Tartaglia, M.; et al. Clinical Presentation and Natural History of Hypertrophic Cardiomyopathy in RASopathies. Heart Fail. Clin. 2018, 14, 225–235. [Google Scholar] [CrossRef]
- Wu, X.; Simpson, J.; Hong, J.H.; Kim, K.H.; Thavarajah, N.K.; Backx, P.H.; Neel, B.G.; Araki, T. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J. Clin. Investig. 2011, 121, 1009–1025. [Google Scholar] [CrossRef]
- Marin, T.M.; Keith, K.; Davies, B.; Conner, D.A.; Guha, P.; Kalaitzidis, D.; Wu, X.; Lauriol, J.; Wang, B.; Bauer, M.; et al. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J. Clin. Investig. 2011, 121, 1026–1043. [Google Scholar] [CrossRef]
- Hahn, A.; Lauriol, J.; Thul, J.; Behnke-Hall, K.; Logeswaran, T.; Schanzer, A.; Bogurcu, N.; Garvalov, B.K.; Zenker, M.; Gelb, B.D.; et al. Rapidly progressive hypertrophic cardiomyopathy in an infant with Noonan syndrome with multiple lentigines: Palliative treatment with a rapamycin analog. Am. J. Med. Genet. A 2015, 167A, 744–751. [Google Scholar] [CrossRef]
- Zhang, J.; Cao, L.; Tan, Y.; Zheng, Y.; Gui, Y. N-acetylcysteine protects neonatal mice from ventricular hypertrophy induced by maternal obesity in a sex-specific manner. Biomed. Pharmacother. 2021, 133, 110989. [Google Scholar] [CrossRef]
- Zafarullah, M.; Li, W.Q.; Sylvester, J.; Ahmad, M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol. Life Sci. 2003, 60, 6–20. [Google Scholar] [CrossRef]
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent Advances in Mitochondrial Disease. Annu. Rev. Genomics Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef]
- Brunel-Guitton, C.; Levtova, A.; Sasarman, F. Mitochondrial Diseases and Cardiomyopathies. Can. J. Cardiol. 2015, 31, 1360–1376. [Google Scholar] [CrossRef]
- Menezes, M.J.; Riley, L.G.; Christodoulou, J. Mitochondrial respiratory chain disorders in childhood: Insights into diagnosis and management in the new era of genomic medicine. Biochim. Biophys. Acta 2014, 1840, 1368–1379. [Google Scholar] [CrossRef]
- Yang, J.; Chen, S.; Duan, F.; Wang, X.; Zhang, X.; Lian, B.; Kou, M.; Chiang, Z.; Li, Z.; Lian, Q. Mitochondrial Cardiomyopathy: Molecular Epidemiology, Diagnosis, Models, and Therapeutic Management. Cells 2022, 11, 3511. [Google Scholar] [CrossRef]
- Driver, C.; Bamitale, K.D.S.; Kazi, A.; Olla, M.; Nyane, N.A.; Owira, P.M.O. Cardioprotective Effects of Metformin. J. Cardiovasc. Pharmacol. 2018, 72, 121–127. [Google Scholar] [CrossRef]
- Dziubak, A.; Wojcicka, G.; Wojtak, A.; Beltowski, J. Metabolic Effects of Metformin in the Failing Heart. Int. J. Mol. Sci. 2018, 19, 2869. [Google Scholar] [CrossRef]
- Li, C.; Mu, N.; Gu, C.; Liu, M.; Yang, Z.; Yin, Y.; Chen, M.; Wang, Y.; Han, Y.; Yu, L.; et al. Metformin mediates cardioprotection against aging-induced ischemic necroptosis. Aging Cell 2020, 19, e13096. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhu, L.; Wang, X.; Jin, H. RNA-based therapeutics: An overview and prospectus. Cell Death Dis. 2022, 13, 644. [Google Scholar] [CrossRef]
- Palandri, C.; Santini, L.; Argiro, A.; Margara, F.; Doste, R.; Bueno-Orovio, A.; Olivotto, I.; Coppini, R. Pharmacological Management of Hypertrophic Cardiomyopathy: From Bench to Bedside. Drugs 2022, 82, 889–912. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Myocardial Changes in Children | Signaling Pathways | Modulating Pharmacological Agents |
|---|---|---|
| Myocardial hypertrophy in congenital heart diseases | GATA4 | Neuraminidase inhibitors |
| HDAC | HDAC inhibitors | |
| NFAT | RNA-containing drugs, theaflavin-3,3′-digallate | |
| Akt/GSK3β/β–catenin | Proton pump inhibitors | |
| Neonatal posthypoxic hypertrophic myocardial cardiomyopathy | ET1 | Endothelin 1 inhibitors |
| HIF1α | Metabolitotropic cardioprotectors | |
| Genetic hypertrophic cardiomyopathy | MyBP-C3 | RNA-containing drugs, N-acetylcysteine |
| Akt-mTOR | Rapamycin, N-acetylcysteine | |
| AMPK, PGC1 | Metformin, polyphenols | |
| NRFs, SIRT1 | Polyphenols, Nicotinamide riboside | |
| RAS/MAPK | Protein kinase inhibitors |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamenshchyk, A.; Belenichev, I.; Oksenych, V.; Kamyshnyi, O. Combined Pharmacological Modulation of Translational and Transcriptional Activity Signaling Pathways as a Promising Therapeutic Approach in Children with Myocardial Changes. Biomolecules 2024, 14, 477. https://doi.org/10.3390/biom14040477
Kamenshchyk A, Belenichev I, Oksenych V, Kamyshnyi O. Combined Pharmacological Modulation of Translational and Transcriptional Activity Signaling Pathways as a Promising Therapeutic Approach in Children with Myocardial Changes. Biomolecules. 2024; 14(4):477. https://doi.org/10.3390/biom14040477
Chicago/Turabian StyleKamenshchyk, Andrii, Igor Belenichev, Valentyn Oksenych, and Oleksandr Kamyshnyi. 2024. "Combined Pharmacological Modulation of Translational and Transcriptional Activity Signaling Pathways as a Promising Therapeutic Approach in Children with Myocardial Changes" Biomolecules 14, no. 4: 477. https://doi.org/10.3390/biom14040477