
Advancements in the Application of Ribosomally Synthesized and Post-Translationally Modified Peptides (RiPPs)


Abstract
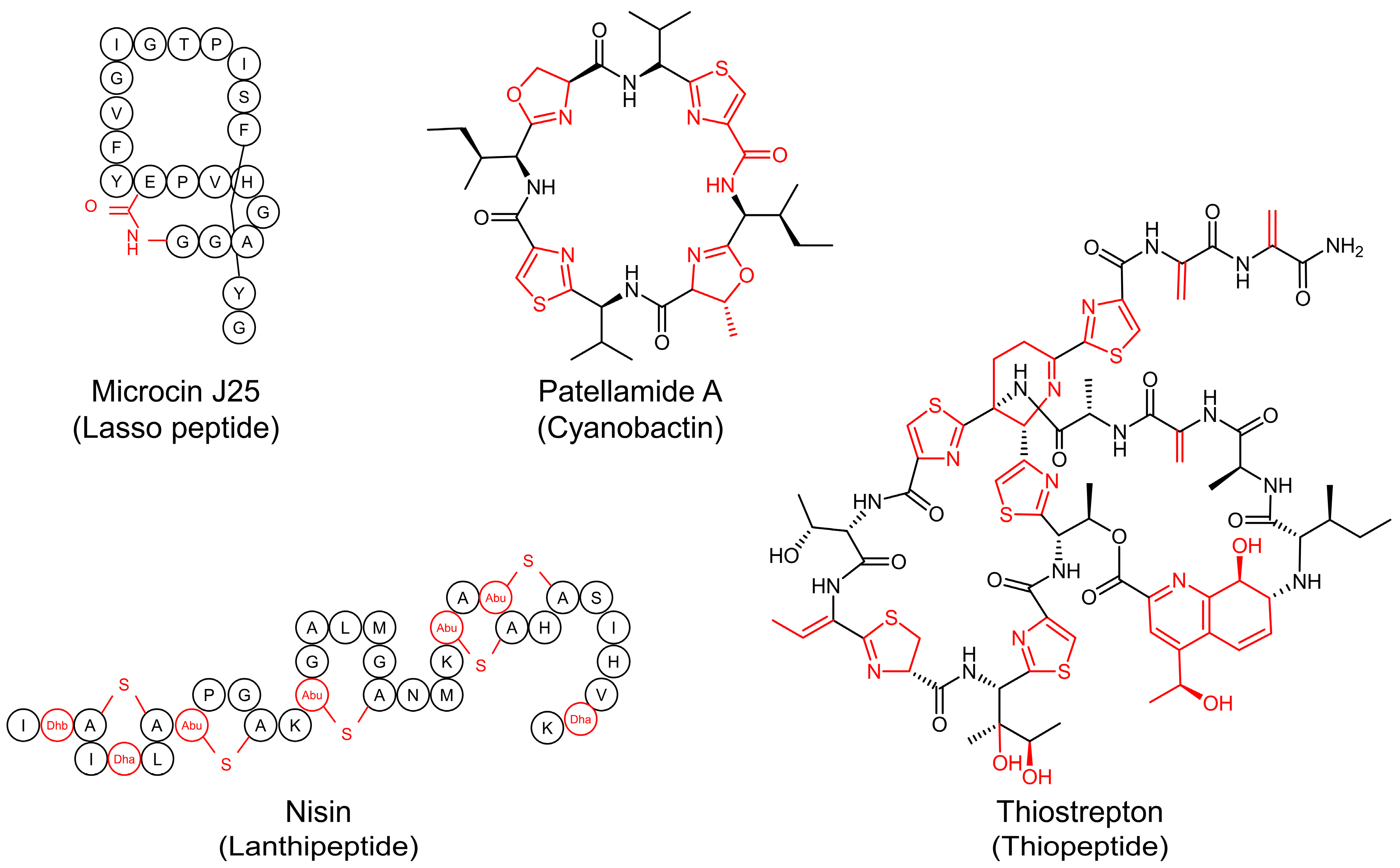
1. Introduction
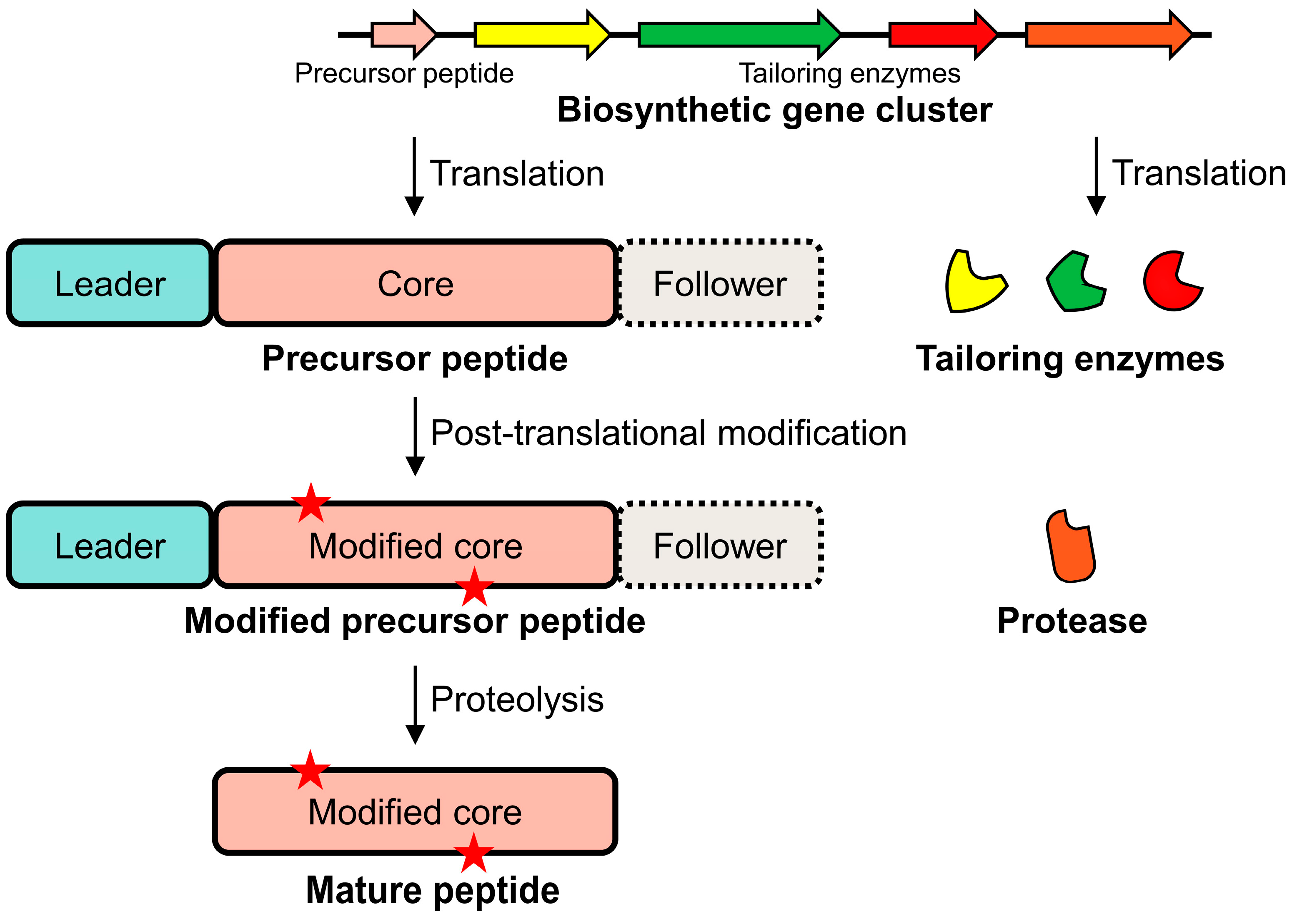
2. Biosynthetic Pathways of RiPPs
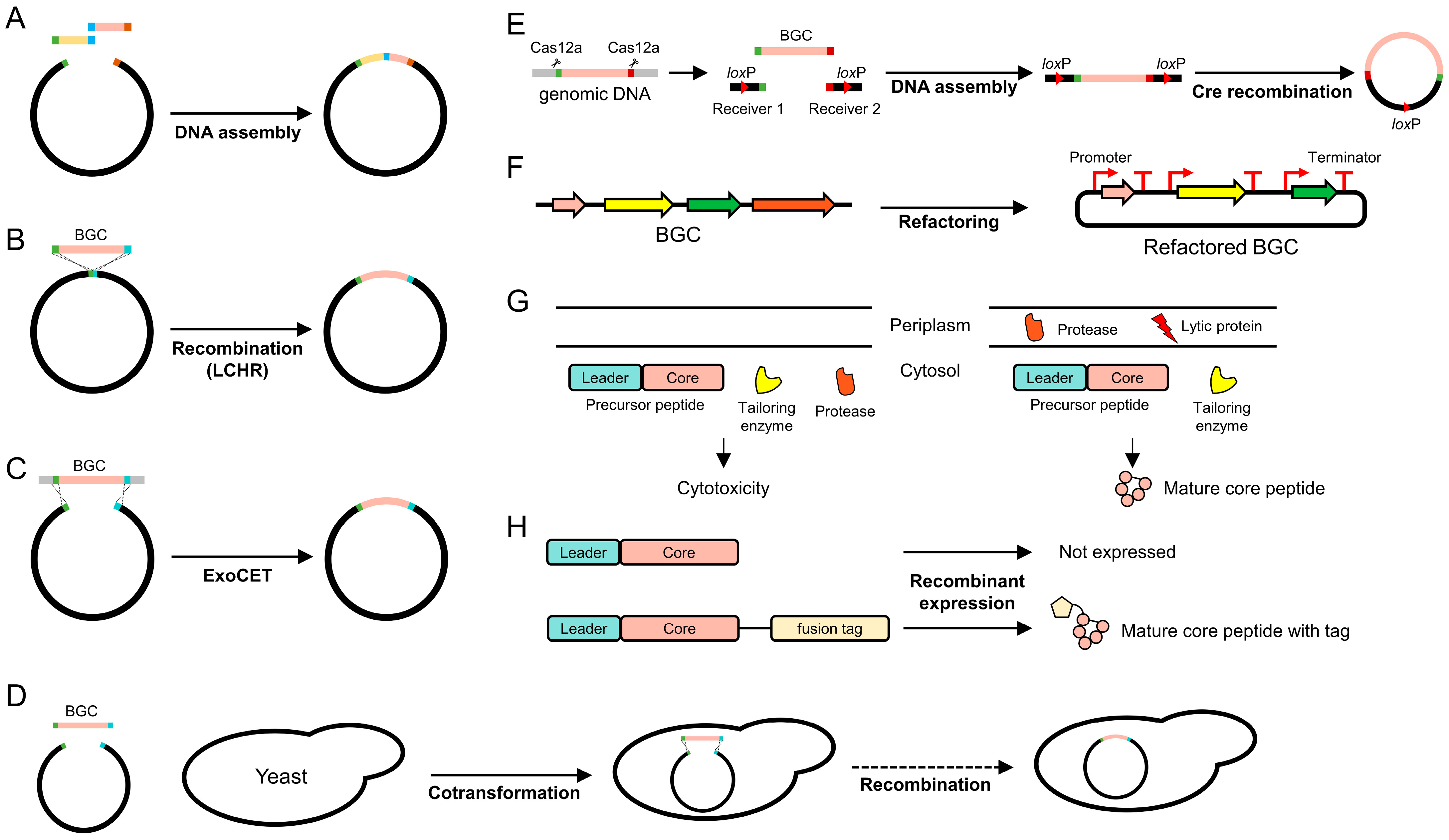
3. Advances in Synthetic Biology for RiPP Expression and Production
3.1. Genetic Manipulation for Heterologous Expression
3.2. Refactoring
3.3. Compartmentation
3.4. Fusion Tags
3.5. Plasmid Copy Number
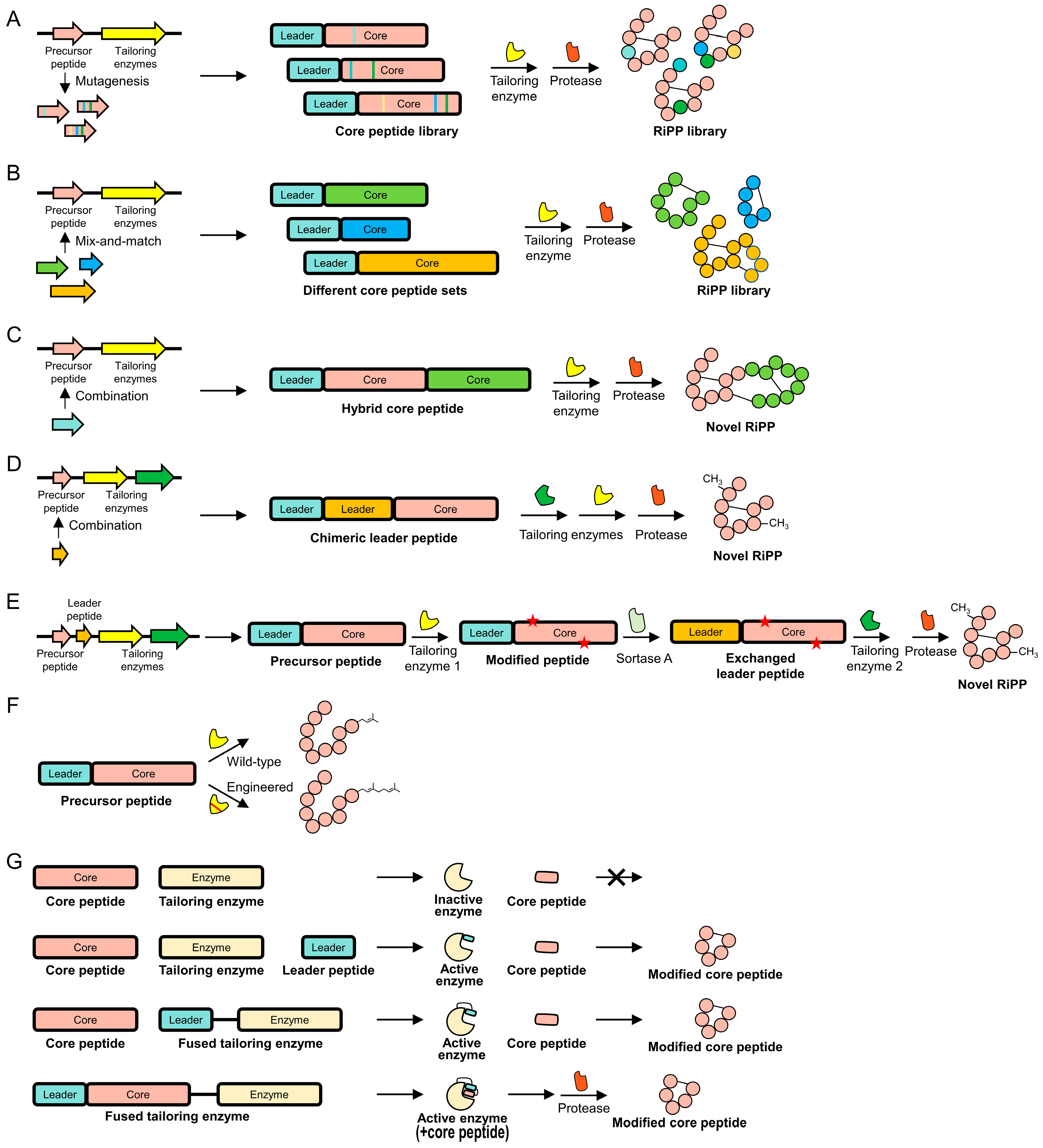
4. Strategies and Innovations in RiPP Engineering
4.1. Core Peptides
4.2. Leader Peptides
4.3. Tailoring Enzymes
4.4. Combinatorial Approach
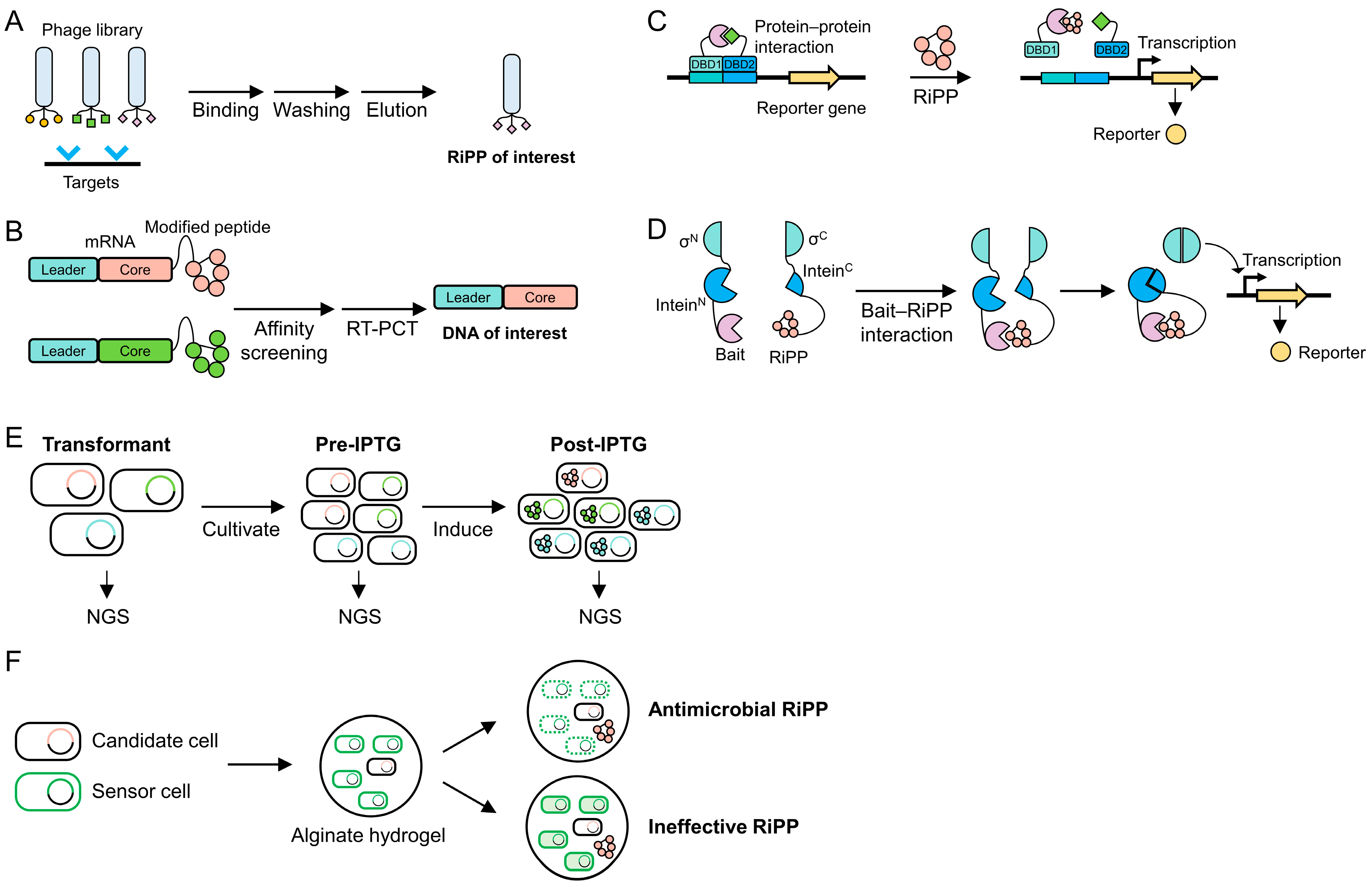
5. High-Throughput Screening Methods
5.1. Surface Display
5.2. mRNA Display
5.3. Two-Hybrid System
5.4. Intein-Based Genetic Circuit
5.5. Next-Generation Sequencing
5.6. Zone of Inhibition Assay
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products: Overview and Recommendations for a Universal Nomenclature. Nat. Prod. Rep. 2012, 30, 108–160. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Walsh, C.T. Assembly-Line Enzymology for Polyketide and Nonribosomal Peptide Antibiotics: Logic, Machinery, and Mechanisms. Chem. Rev. 2006, 106, 3468–3496. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.A.; Mitchell, D.A. RiPP Antibiotics: Biosynthesis and Engineering Potential. Curr. Opin. Microbiol. 2018, 45, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Jaarsma, A.H.; Kuipers, O.P. Antiviral Activities and Applications of Ribosomally Synthesized and Post-Translationally Modified Peptides (RiPPs). Cell. Mol. Life Sci. 2021, 78, 3921–3940. [Google Scholar] [CrossRef] [PubMed]
- Frattaruolo, L.; Lacret, R.; Cappello, A.R.; Truman, A.W. A Genomics-Based Approach Identifies a Thioviridamide-Like Compound with Selective Anticancer Activity. ACS Chem. Biol. 2017, 12, 2815–2822. [Google Scholar] [CrossRef]
- Pfeiffer, I.P.-M.; Schröder, M.-P.; Mordhorst, S. Opportunities and Challenges of RiPP-Based Therapeutics. Nat. Prod. Rep. 2024; Online ahead of print. [Google Scholar] [CrossRef]
- Zhou, D.; Wang, X.; Anjago, W.M.; Li, J.; Li, W.; Li, M.; Jiu, M.; Zhang, Q.; Zhang, J.; Deng, S.; et al. Borrelidin-Producing and Root-Colonizing Streptomyces Rochei is a Potent Biopesticide for Two Soil-Borne Oomycete-Caused Plant Diseases. Biol. Control 2024, 188, 105411. [Google Scholar] [CrossRef]
- Gharsallaoui, A.; Oulahal, N.; Joly, C.; Degraeve, P. Nisin as a Food Preservative: Part 1: Physicochemical Properties, Antimicrobial Activity, and Main Uses. Crit. Rev. Food Sci. Nutr. 2016, 56, 1262–1274. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, V. Insights into Post-Translational Modification Enzymes from RiPPs: A Toolkit for Applications in Peptide Synthesis. Biotechnol. Adv. 2022, 56, 107908. [Google Scholar] [CrossRef]
- Fu, Y.; Xu, Y.; Ruijne, F.; Kuipers, O.P. Engineering Lanthipeptides by Introducing a Large Variety of RiPP Modifications to Obtain New-to-Nature Bioactive Peptides. FEMS Microbiol. Rev. 2023, 47, fuad017. [Google Scholar] [CrossRef]
- Eslami, S.M.; van der Donk, W.A. Proteases Involved in Leader Peptide Removal during RiPP Biosynthesis. ACS Bio Med Chem Au 2024, 4, 20–36. [Google Scholar] [CrossRef]
- Si, T.; Tian, Q.; Min, Y.; Zhang, L.; Sweedler, J.V.; van der Donk, W.A.; Zhao, H. Rapid Screening of Lanthipeptide Analogs via In-Colony Removal of Leader Peptides in Escherichia coli. J. Am. Chem. Soc. 2018, 140, 11884–11888. [Google Scholar] [CrossRef]
- Duan, H.; Zhang, X.; Figeys, D. An Emerging Field: Post-Translational Modification in Microbiome. Proteomics 2023, 23, 2100389. [Google Scholar] [CrossRef]
- Wang, G. Post-Translational Modifications of Natural Antimicrobial Peptides and Strategies for Peptide Engineering. Curr. Biotechnol. 2012, 1, 72–79. [Google Scholar] [CrossRef]
- Gualillo, O.; Lago, F.; Casanueva, F.F.; Dieguez, C. One Ancestor, Several Peptides: Post-Translational Modifications of Preproghrelin Generate Several Peptides with Antithetical Effects. Mol. Cell. Endocrinol. 2006, 256, 1–8. [Google Scholar] [CrossRef]
- Sun, Z.; Xia, Z.; Bi, F.; Liu, J.-N. Expression and Purification of Human Urodilatin by Small Ubiquitin-Related Modifier Fusion in Escherichia coli. Appl. Microbiol. Biotechnol. 2008, 78, 495–502. [Google Scholar] [CrossRef]
- Li, J.F.; Zhang, J.; Zhang, Z.; Ma, H.W.; Zhang, J.X.; Zhang, S.Q. Production of Bioactive Human Beta-Defensin-4 in Escherichia coli Using SUMO Fusion Partner. Protein J. 2010, 29, 314–319. [Google Scholar] [CrossRef]
- Glassey, E.; King, A.M.; Anderson, D.A.; Zhang, Z.; Voigt, C.A. Functional Expression of Diverse Post-Translational Peptide-Modifying Enzymes in Escherichia coli under Uniform Expression and Purification Conditions. PLoS ONE 2022, 17, e0266488. [Google Scholar] [CrossRef]
- Vermeulen, R.R.; Van Staden, A.D.P.; Dicks, L. Heterologous Expression of the Class IIa Bacteriocins, Plantaricin 423 and Mundticin ST4SA, in Escherichia coli Using Green Fluorescent Protein as a Fusion Partner. Front. Microbiol. 2020, 11, 535667. [Google Scholar] [CrossRef]
- Van Zyl, W.F.; Van Staden, A.D.; Dicks, L.M.T.; Trindade, M. Use of the mCherry Fluorescent Protein to Optimize the Expression of Class I Lanthipeptides in Escherichia coli. Microb. Cell Factories 2023, 22, 149. [Google Scholar] [CrossRef]
- Guo, E.; Fu, L.; Fang, X.; Xie, W.; Li, K.; Zhang, Z.; Hong, Z.; Si, T. Robotic Construction and Screening of Lanthipeptide Variant Libraries in Escherichia coli. ACS Synth. Biol. 2022, 11, 3900–3911. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Dommaraju, S.R.; Huang, C.; Cui, H.; Pan, Y.; Nesic, M.; Zhu, L.; Sarlah, D.; Mitchell, D.A.; Zhao, H. Genome Mining Unveils a Class of Ribosomal Peptides with Two Amino Termini. Nat. Commun. 2023, 14, 1624. [Google Scholar] [CrossRef] [PubMed]
- Santos-Aberturas, J.; Chandra, G.; Frattaruolo, L.; Lacret, R.; Pham, T.H.; Vior, N.M.; Eyles, T.H.; Truman, A.W. Uncovering the Unexplored Diversity of Thioamidated Ribosomal Peptides in Actinobacteria Using the RiPPER Genome Mining Tool. Nucleic Acids Res. 2019, 47, 4624–4637. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.N.; et al. antiSMASH 7.0: New and Improved Predictions for Detection, Regulation, Chemical Structures and Visualisation. Nucleic Acids Res. 2023, 51, W46–W50. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, M.A.; Johnston, C.W.; Gunabalasingam, M.; Merwin, N.J.; Kieliszek, A.M.; MacLellan, R.J.; Li, H.; Ranieri, M.R.M.; Webster, A.L.H.; Cao, M.P.T.; et al. Comprehensive Prediction of Secondary Metabolite Structure and Biological Activity from Microbial Genome Sequences. Nat. Commun. 2020, 11, 6058. [Google Scholar] [CrossRef] [PubMed]
- Tietz, J.I.; Schwalen, C.J.; Patel, P.S.; Maxson, T.; Blair, P.M.; Tai, H.-C.; Zakai, U.I.; Mitchell, D.A. A New Genome-Mining Tool Redefines the Lasso Peptide Biosynthetic Landscape. Nat. Chem. Biol. 2017, 13, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, A.M.; Medema, M.H.; van Wezel, G.P. Omics-Based Strategies to Discover Novel Classes of RiPP Natural Products. Curr. Opin. Biotechnol. 2021, 69, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.H.; Truman, A.W. Genome Mining Strategies for Ribosomally Synthesised and Post-Translationally Modified Peptides. Comput. Struct. Biotechnol. J. 2020, 18, 1838–1851. [Google Scholar] [CrossRef] [PubMed]
- Smanski, M.J.; Zhou, H.; Claesen, J.; Shen, B.; Fischbach, M.A.; Voigt, C.A. Synthetic Biology to Access and Expand Nature’s Chemical Diversity. Nat. Rev. Microbiol. 2016, 14, 135–149. [Google Scholar] [CrossRef]
- Thibodeaux, C.J. The Conformationally Dynamic Structural Biology of Lanthipeptide Biosynthesis. Curr. Opin. Struct. Biol. 2023, 81, 102644. [Google Scholar] [CrossRef]
- Lohans, C.T.; Vederas, J.C. Structural Characterization of Thioether-Bridged Bacteriocins. J. Antibiot. 2014, 67, 23–30. [Google Scholar] [CrossRef]
- Enghiad, B.; Huang, C.; Guo, F.; Jiang, G.; Wang, B.; Tabatabaei, S.K.; Martin, T.A.; Zhao, H. Cas12a-Assisted Precise Targeted Cloning Using in Vivo Cre-Lox Recombination. Nat. Commun. 2021, 12, 1171. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, Z.; Ke, J.; Engel, Y.; Shi, Y.-M.; Robinson, D.; Bingol, K.; Zhang, Z.; Bowen, B.; Louie, K.; et al. CRAGE Enables Rapid Activation of Biosynthetic Gene Clusters in Undomesticated Bacteria. Nat. Microbiol. 2019, 4, 2498–2510. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.; Jia, R.; Yin, J.; Li, A.; Xia, L.; Yin, Y.; Müller, R.; Fu, J.; Stewart, A.F.; et al. ExoCET: Exonuclease in Vitro Assembly Combined with RecET Recombination for Highly Efficient Direct DNA Cloning from Complex Genomes. Nucleic Acids Res. 2018, 46, e28. [Google Scholar] [CrossRef] [PubMed]
- Wuisan, Z.G.; Kresna, I.D.M.; Böhringer, N.; Lewis, K.; Schäberle, T.F. Optimization of Heterologous Darobactin A Expression and Identification of the Minimal Biosynthetic Gene Cluster. Metab. Eng. 2021, 66, 123–136. [Google Scholar] [CrossRef]
- King, A.M.; Zhang, Z.; Glassey, E.; Siuti, P.; Clardy, J.; Voigt, C.A. Systematic Mining of the Human Microbiome Identifies Antimicrobial Peptides with Diverse Activity Spectra. Nat. Microbiol. 2023, 8, 2420–2434. [Google Scholar] [CrossRef] [PubMed]
- Ayikpoe, R.S.; Shi, C.; Battiste, A.J.; Eslami, S.M.; Ramesh, S.; Simon, M.A.; Bothwell, I.R.; Lee, H.; Rice, A.J.; Ren, H.; et al. A Scalable Platform to Discover Antimicrobials of Ribosomal Origin. Nat. Commun. 2022, 13, 6135. [Google Scholar] [CrossRef]
- Cao, L.; Do, T.; Zhu, A.; Duan, J.; Alam, N.; Link, A.J. Genome Mining and Discovery of Imiditides, a Family of RiPPs with a Class-Defining Aspartimide Modification. J. Am. Chem. Soc. 2023, 145, 18834–18845. [Google Scholar] [CrossRef] [PubMed]
- Bowler, M.M.; Glavatskikh, M.; Pecot, C.V.; Kireev, D.; Bowers, A.A. Enzymatic Macrolactamization of mRNA Display Libraries for Inhibitor Selection. ACS Chem. Biol. 2023, 18, 166–175. [Google Scholar] [CrossRef]
- Sarkar, S.; Gu, W.; Schmidt, E.W. Applying Promiscuous RiPP Enzymes to Peptide Backbone N-Methylation Chemistry. ACS Chem. Biol. 2022, 17, 2165–2178. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, Y.; Zhai, G.; Fu, S.; Xia, Y.; Hu, B.; Cai, X.; Zhang, Y.; Li, Y.; Deng, Z.; et al. A Cell-Free Platform Based on Nisin Biosynthesis for Discovering Novel Lanthipeptides and Guiding Their Overproduction In Vivo. Adv. Sci. 2020, 7, 2001616. [Google Scholar] [CrossRef]
- Kunakom, S.; Eustáquio, A.S. Heterologous Production of Lasso Peptide Capistruin in a Burkholderia Host. ACS Synth. Biol. 2020, 9, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Eastman, K.A.S.; Kincannon, W.M.; Bandarian, V. Leveraging Substrate Promiscuity of a Radical S-Adenosyl-L-Methionine RiPP Maturase toward Intramolecular Peptide Cross-Linking Applications. ACS Cent. Sci. 2022, 8, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Kuipers, O.P. Nisin- and Ripcin-Derived Hybrid Lanthipeptides Display Selective Antimicrobial Activity against Staphylococcus Aureus. ACS Synth. Biol. 2021, 10, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Stoffels, K.; Broos, J.; Kuipers, O.P. Engineering Hybrid Lantibiotics Yields the Highly Stable and Bacteriocidal Peptide Cerocin V. Microbiol. Res. 2024, 282, 127640. [Google Scholar] [CrossRef]
- Franz, L.; Koehnke, J. Leader Peptide Exchange to Produce Hybrid, New-to-Nature Ribosomal Natural Products. Chem. Commun. 2021, 57, 6372–6375. [Google Scholar] [CrossRef]
- Estrada, P.; Morita, M.; Hao, Y.; Schmidt, E.W.; Nair, S.K. A Single Amino Acid Switch Alters the Isoprene Donor Specificity in Ribosomally Synthesized and Post-Translationally Modified Peptide Prenyltransferases. J. Am. Chem. Soc. 2018, 140, 8124–8127. [Google Scholar] [CrossRef] [PubMed]
- Lacerna, N.I.; Cong, Y.; Schmidt, E.W. An Autocatalytic Peptide Cyclase Improves Fidelity and Yield of Circular Peptides In Vivo and In Vitro. ACS Synth. Biol. 2024, 13, 394–401. [Google Scholar] [CrossRef]
- Fleming, S.R.; Himes, P.M.; Ghodge, S.V.; Goto, Y.; Suga, H.; Bowers, A.A. Exploring the Post-Translational Enzymology of PaaA by mRNA Display. J. Am. Chem. Soc. 2020, 142, 5024–5028. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.A.; Chang, J.S.; Onaka, H.; Goto, Y.; Suga, H. Accurate Models of Substrate Preferences of Post-Translational Modification Enzymes from a Combination of mRNA Display and Deep Learning. ACS Cent. Sci. 2022, 8, 814–824. [Google Scholar] [CrossRef]
- King, A.M.; Anderson, D.A.; Glassey, E.; Segall-Shapiro, T.H.; Zhang, Z.; Niquille, D.L.; Embree, A.C.; Pratt, K.; Williams, T.L.; Gordon, D.B.; et al. Selection for Constrained Peptides That Bind to a Single Target Protein. Nat. Commun. 2021, 12, 6343. [Google Scholar] [CrossRef]
- Thokkadam, A.; Do, T.; Ran, X.; Brynildsen, M.P.; Yang, Z.J.; Link, A.J. High-Throughput Screen Reveals the Structure–Activity Relationship of the Antimicrobial Lasso Peptide Ubonodin. ACS Cent. Sci. 2023, 9, 540–550. [Google Scholar] [CrossRef]
- Yang, X.; Lennard, K.R.; He, C.; Walker, M.C.; Ball, A.T.; Doigneaux, C.; Tavassoli, A.; van der Donk, W.A. A Lanthipeptide Library Used to Identify a Protein–Protein Interaction Inhibitor. Nat. Chem. Biol. 2018, 14, 375–380. [Google Scholar] [CrossRef]
- Schmitt, S.; Montalbán-López, M.; Peterhoff, D.; Deng, J.; Wagner, R.; Held, M.; Kuipers, O.P.; Panke, S. Analysis of Modular Bioengineered Antimicrobial Lanthipeptides at Nanoliter Scale. Nat. Chem. Biol. 2019, 15, 437–443. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, Y.; Vélasquez, J.E.; van der Donk, W.A. Evolution of Lanthipeptide Synthetases. Proc. Natl. Acad. Sci. USA 2012, 109, 18361–18366. [Google Scholar] [CrossRef]
- McBrayer, D.N.; Gantman, B.K.; Tal-Gan, Y. N-Methylation of Amino Acids in Gelatinase Biosynthesis-Activating Pheromone Identifies Key Site for Stability Enhancement with Retention of the Enterococcus Faecalis Fsr Quorum Sensing Circuit Response. ACS Infect. Dis. 2019, 5, 1035–1041. [Google Scholar] [CrossRef]
- Su, Y.; Han, M.; Meng, X.; Feng, Y.; Luo, S.; Yu, C.; Zheng, G.; Zhu, S. Discovery and Characterization of a Novel C-Terminal Peptide Carboxyl Methyltransferase in a Lassomycin-like Lasso Peptide Biosynthetic Pathway. Appl. Microbiol. Biotechnol. 2019, 103, 2649–2664. [Google Scholar] [CrossRef]
- Acedo, J.Z.; Bothwell, I.R.; An, L.; Trouth, A.; Frazier, C.; van der Donk, W.A. O-Methyltransferase-Mediated Incorporation of a β-Amino Acid in Lanthipeptides. J. Am. Chem. Soc. 2019, 141, 16790–16801. [Google Scholar] [CrossRef]
- Gavrish, E.; Sit, C.S.; Cao, S.; Kandror, O.; Spoering, A.; Peoples, A.; Ling, L.; Fetterman, A.; Hughes, D.; Bissell, A.; et al. Lassomycin, a Ribosomally Synthesized Cyclic Peptide, Kills Mycobacterium Tuberculosis by Targeting the ATP-Dependent Protease ClpC1P1P2. Chem. Biol. 2014, 21, 509–518. [Google Scholar] [CrossRef]
- Maget-Dana, R.; Ptak, M. Interactions of Surfactin with Membrane Models. Biophys. J. 1995, 68, 1937–1943. [Google Scholar] [CrossRef] [PubMed]
- He, B.-B.; Cheng, Z.; Zhong, Z.; Gao, Y.; Liu, H.; Li, Y.-X. Expanded Sequence Space of Radical S-Adenosylmethionine-Dependent Enzymes Involved in Post-Translational Macrocyclization**. Angew. Chem. Int. Ed. 2022, 61, e202212447. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; van der Donk, W.A. Biosynthesis of 3-Thia-α-Amino Acids on a Carrier Peptide. Proc. Natl. Acad. Sci. USA 2022, 119, e2205285119. [Google Scholar] [CrossRef]
- Guo, L.; Stoffels, K.; Broos, J.; Kuipers, O.P. Altering Specificity and Enhancing Stability of the Antimicrobial Peptides Nisin and Rombocin through Dehydrated Amino Acid Residue Engineering. Peptides 2024, 174, 171152. [Google Scholar] [CrossRef]
- Rutledge, P.J.; Challis, G.L. Discovery of Microbial Natural Products by Activation of Silent Biosynthetic Gene Clusters. Nat. Rev. Microbiol. 2015, 13, 509–523. [Google Scholar] [CrossRef]
- Xue, Y.; Braslavsky, I.; Quake, S.R. Temperature Effect on Polymerase Fidelity. J. Biol. Chem. 2021, 297, 101270. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic Assembly of DNA Molecules up to Several Hundred Kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Casini, A.; Storch, M.; Baldwin, G.S.; Ellis, T. Bricks and Blueprints: Methods and Standards for DNA Assembly. Nat. Rev. Mol. Cell Biol. 2015, 16, 568–576. [Google Scholar] [CrossRef]
- Li, L.; Zhao, Y.; Ruan, L.; Yang, S.; Ge, M.; Jiang, W.; Lu, Y. A Stepwise Increase in Pristinamycin II Biosynthesis by Streptomyces Pristinaespiralis through Combinatorial Metabolic Engineering. Metab. Eng. 2015, 29, 12–25. [Google Scholar] [CrossRef]
- Sharan, S.K.; Thomason, L.C.; Kuznetsov, S.G.; Court, D.L. Recombineering: A Homologous Recombination-Based Method of Genetic Engineering. Nat. Protoc. 2009, 4, 206–223. [Google Scholar] [CrossRef]
- Fu, J.; Bian, X.; Hu, S.; Wang, H.; Huang, F.; Seibert, P.M.; Plaza, A.; Xia, L.; Müller, R.; Stewart, A.F.; et al. Full-Length RecE Enhances Linear-Linear Homologous Recombination and Facilitates Direct Cloning for Bioprospecting. Nat. Biotechnol. 2012, 30, 440–446. [Google Scholar] [CrossRef]
- Kouprina, N.; Larionov, V. Transformation-Associated Recombination (TAR) Cloning for Genomics Studies and Synthetic Biology. Chromosoma 2016, 125, 621–632. [Google Scholar] [CrossRef]
- Nagy, A. Cre Recombinase: The Universal Reagent for Genome Tailoring. Genesis 2000, 26, 99–109. [Google Scholar] [CrossRef]
- Kuipers, A.; Wierenga, J.; Rink, R.; Kluskens, L.D.; Driessen, A.J.M.; Kuipers, O.P.; Moll, G.N. Sec-Mediated Transport of Posttranslationally Dehydrated Peptides in Lactococcus Lactis. Appl. Environ. Microbiol. 2006, 72, 7626–7633. [Google Scholar] [CrossRef]
- Jiménez, J.J.; Diep, D.B.; Borrero, J.; Gútiez, L.; Arbulu, S.; Nes, I.F.; Herranz, C.; Cintas, L.M.; Hernández, P.E. Cloning Strategies for Heterologous Expression of the Bacteriocin Enterocin A by Lactobacillus Sakei Lb790, Lb. Plantarum NC8 and Lb. Casei CECT475. Microb. Cell Factories 2015, 14, 166. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.; Zhang, D.; Li, X.; Yu, H.; Shen, Z. Overexpression of Specific Proton Motive Force-Dependent Transporters Facilitate the Export of Surfactin in Bacillus Subtilis. J. Ind. Microbiol. Biotechnol. 2015, 42, 93–103. [Google Scholar] [CrossRef]
- Kuipers, A.; de Boef, E.; Rink, R.; Fekken, S.; Kluskens, L.D.; Driessen, A.J.M.; Leenhouts, K.; Kuipers, O.P.; Moll, G.N. NisT, the Transporter of the Lantibiotic Nisin, Can Transport Fully Modified, Dehydrated, and Unmodified Prenisin and Fusions of the Leader Peptide with Non-Lantibiotic Peptides. J. Biol. Chem. 2004, 279, 22176–22182. [Google Scholar] [CrossRef]
- Huo, L.; Rachid, S.; Stadler, M.; Wenzel, S.C.; Müller, R. Synthetic Biotechnology to Study and Engineer Ribosomal Bottromycin Biosynthesis. Chem. Biol. 2012, 19, 1278–1287. [Google Scholar] [CrossRef]
- Havarstein, L.S.; Diep, D.B.; Nes, I.F. A Family of Bacteriocin ABC Transporters Carry out Proteolytic Processing of Their Substrates Concomitant with Export. Mol. Microbiol. 1995, 16, 229–240. [Google Scholar] [CrossRef]
- Hegemann, J.D.; Jeanne Dit Fouque, K.; Santos-Fernandez, M.; Fernandez-Lima, F. A Bifunctional Leader Peptidase/ABC Transporter Protein Is Involved in the Maturation of the Lasso Peptide Cochonodin I from Streptococcus Suis. J. Nat. Prod. 2021, 84, 2683–2691. [Google Scholar] [CrossRef]
- Rücker, E.; Schneider, G.; Steinhäuser, K.; Löwer, R.; Hauber, J.; Stauber, R.H. Rapid Evaluation and Optimization of Recombinant Protein Production Using GFP Tagging. Protein Expr. Purif. 2001, 21, 220–223. [Google Scholar] [CrossRef]
- Fernandez, H.N.; Kretsch, A.M.; Kunakom, S.; Kadjo, A.E.; Mitchell, D.A.; Eustáquio, A.S. High-Yield Lasso Peptide Production in a Burkholderia Bacterial Host by Plasmid Copy Number Engineering. ACS Synth. Biol. 2024, 13, 337–350. [Google Scholar] [CrossRef]
- Nguyen, N.A.; Cong, Y.; Hurrell, R.C.; Arias, N.; Garg, N.; Puri, A.W.; Schmidt, E.W.; Agarwal, V. A Silent Biosynthetic Gene Cluster from a Methanotrophic Bacterium Potentiates Discovery of a Substrate Promiscuous Proteusin Cyclodehydratase. ACS Chem. Biol. 2022, 17, 1577–1585. [Google Scholar] [CrossRef]
- Precord, T.W.; Mahanta, N.; Mitchell, D.A. Reconstitution and Substrate Specificity of the Thioether-Forming Radical S-Adenosylmethionine Enzyme in Freyrasin Biosynthesis. ACS Chem. Biol. 2019, 14, 1981–1989. [Google Scholar] [CrossRef]
- Burkhart, B.J.; Kakkar, N.; Hudson, G.A.; van der Donk, W.A.; Mitchell, D.A. Chimeric Leader Peptides for the Generation of Non-Natural Hybrid RiPP Products. ACS Cent. Sci. 2017, 3, 629–638. [Google Scholar] [CrossRef]
- Jin, L.; Wu, X.; Xue, Y.; Jin, Y.; Wang, S.; Chen, Y. Mutagenesis of NosM Leader Peptide Reveals Important Elements in Nosiheptide Biosynthesis. Appl. Environ. Microbiol. 2017, 83, e02880-16. [Google Scholar] [CrossRef]
- Liu, F.; van Heel, A.J.; Kuipers, O.P. Leader- and Terminal Residue Requirements for Circularin A Biosynthesis Probed by Systematic Mutational Analyses. ACS Synth. Biol. 2023, 12, 852–862. [Google Scholar] [CrossRef]
- Levengood, M.R.; Patton, G.C.; van der Donk, W.A. The Leader Peptide Is Not Required for Post-Translational Modification by Lacticin 481 Synthetase. J. Am. Chem. Soc. 2007, 129, 10314–10315. [Google Scholar] [CrossRef]
- Oman, T.J.; Knerr, P.J.; Bindman, N.A.; Velásquez, J.E.; van der Donk, W.A. An Engineered Lantibiotic Synthetase That Does Not Require a Leader Peptide on Its Substrate. J. Am. Chem. Soc. 2012, 134, 6952–6955. [Google Scholar] [CrossRef]
- Patel, K.P.; Silsby, L.M.; Li, G.; Bruner, S.D. Structure-Based Engineering of Peptide Macrocyclases for the Chemoenzymatic Synthesis of Microviridins. J. Org. Chem. 2021, 86, 11212–11219. [Google Scholar] [CrossRef]
- Reyna-González, E.; Schmid, B.; Petras, D.; Süssmuth, R.D.; Dittmann, E. Leader Peptide-Free In Vitro Reconstitution of Microviridin Biosynthesis Enables Design of Synthetic Protease-Targeted Libraries. Angew. Chem. Int. Ed. 2016, 55, 9398–9401. [Google Scholar] [CrossRef]
- Koehnke, J.; Mann, G.; Bent, A.F.; Ludewig, H.; Shirran, S.; Botting, C.; Lebl, T.; Houssen, W.E.; Jaspars, M.; Naismith, J.H. Structural Analysis of Leader Peptide Binding Enables Leader-Free Cyanobactin Processing. Nat. Chem. Biol. 2015, 11, 558–563. [Google Scholar] [CrossRef]
- Bosma, T.; Kuipers, A.; Bulten, E.; de Vries, L.; Rink, R.; Moll, G.N. Bacterial Display and Screening of Posttranslationally Thioether-Stabilized Peptides. Appl. Environ. Microbiol. 2011, 77, 6794–6801. [Google Scholar] [CrossRef][Green Version]
- Wang, X.S.; Chen, P.-H.C.; Hampton, J.T.; Tharp, J.M.; Reed, C.A.; Das, S.K.; Wang, D.-S.; Hayatshahi, H.S.; Shen, Y.; Liu, J.; et al. A Genetically Encoded, Phage-Displayed Cyclic-Peptide Library. Angew. Chem. Int. Ed. 2019, 58, 15904–15909. [Google Scholar] [CrossRef]
- Urban, J.H.; Moosmeier, M.A.; Aumüller, T.; Thein, M.; Bosma, T.; Rink, R.; Groth, K.; Zulley, M.; Siegers, K.; Tissot, K.; et al. Phage Display and Selection of Lanthipeptides on the Carboxy-Terminus of the Gene-3 Minor Coat Protein. Nat. Commun. 2017, 8, 1500. [Google Scholar] [CrossRef]
- Hetrick, K.J.; Walker, M.C.; van der Donk, W.A. Development and Application of Yeast and Phage Display of Diverse Lanthipeptides. ACS Cent. Sci. 2018, 4, 458–467. [Google Scholar] [CrossRef]
- Gold, L. mRNA Display: Diversity Matters during in Vitro Selection. Proc. Natl. Acad. Sci. USA 2001, 98, 4825–4826. [Google Scholar] [CrossRef]
- Bratkovič, T. Progress in Phage Display: Evolution of the Technique and Its Applications. Cell. Mol. Life Sci. 2010, 67, 749–767. [Google Scholar] [CrossRef]
- Cwirla, S.E.; Peters, E.A.; Barrett, R.W.; Dower, W.J. Peptides on Phage: A Vast Library of Peptides for Identifying Ligands. Proc. Natl. Acad. Sci. USA 1990, 87, 6378–6382. [Google Scholar] [CrossRef]
- Zade, H.M.; Keshavarz, R.; Shekarabi, H.S.Z.; Bakhshinejad, B. Biased Selection of Propagation-Related TUPs from Phage Display Peptide Libraries. Amino Acids 2017, 49, 1293–1308. [Google Scholar] [CrossRef]
- Fields, S.; Sternglanz, R. The Two-Hybrid System: An Assay for Protein-Protein Interactions. Trends Genet. 1994, 10, 286–292. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RiPP Product | Class | Biological Activity | Ref. |
|---|---|---|---|
| Thiovarsolin | Thioamitides | Unidentified | [23] |
| Daptide | Daptide | Hemolytic activity | [22] |
| Imiditide | Imiditide | Unidentified | [38] |
| Mycetolassin | Lasso peptide | Unidentified | [42] |
| 7 RiPPs | Lanthipeptide, lasso peptide, LAP | Unidentified | [32] |
| 30 RiPPs | Lanthipeptide, lasso peptide, graspetide, glycocin, LAP, thioamitide | Antimicrobial activity against ESKAPE pathogens | [37] |
| 24 RiPPs | Lanthipeptide, lasso peptide | Antimicrobial activity against human pathogens | [36] |
| Octreotide analogs | Ranthipeptide | Unidentified | [43] |
| Hybrid RiPPs | Lanthipeptide | Antimicrobial activity against antibiotic-resistant MRSA strain | [44] |
| Hybrid RiPPs | Lanthipeptide | Antimicrobial activity against antibiotic-resistant MRSA strain | [45] |
| Hybrid RiPPs | Cyanobactin, microviridin | Unidentified | [46] |
| Prenylated lanthipeptides | Lanthipeptide | Unidentified | [47] |
| Cycle peptides | Cyanobactin | Unidentified | [48] |
| Cycle peptides | Cyanobactin | Unidentified | [40] |
| Pantocin A analogs | Pantocin | Unidentified | [49] |
| Lactazole analogs | Thiopeptide | Unidentified | [50] |
| Freyrasin analogs | Ranthipeptide | Binding to the SARS-CoV-2 Spike receptor | [51] |
| Ubonodin analogs | Lasso peptide | Antimicrobial activity against opportunistic human pathogens | [52] |
| Cycle peptides | Lasso peptide | Anticancer activity | [39] |
| XY3-3 | Lanthipeptide | Inhibition to HIV infection | [53] |
| Hybrid RiPPs | Lanthipeptide | Antimicrobial activity against pathogenic bacteria | [54] |
| Halα analogs | Lanthipeptide | Antimicrobial activity | [21] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, S.-W.; Won, H.-S. Advancements in the Application of Ribosomally Synthesized and Post-Translationally Modified Peptides (RiPPs). Biomolecules 2024, 14, 479. https://doi.org/10.3390/biom14040479
Han S-W, Won H-S. Advancements in the Application of Ribosomally Synthesized and Post-Translationally Modified Peptides (RiPPs). Biomolecules. 2024; 14(4):479. https://doi.org/10.3390/biom14040479
Chicago/Turabian StyleHan, Sang-Woo, and Hyung-Sik Won. 2024. "Advancements in the Application of Ribosomally Synthesized and Post-Translationally Modified Peptides (RiPPs)" Biomolecules 14, no. 4: 479. https://doi.org/10.3390/biom14040479
APA StyleHan, S.-W., & Won, H.-S. (2024). Advancements in the Application of Ribosomally Synthesized and Post-Translationally Modified Peptides (RiPPs). Biomolecules, 14(4), 479. https://doi.org/10.3390/biom14040479