
Mechanism of Notch Signaling Pathway in Malignant Progression of Glioblastoma and Targeted Therapy

Abstract
:1. Introduction
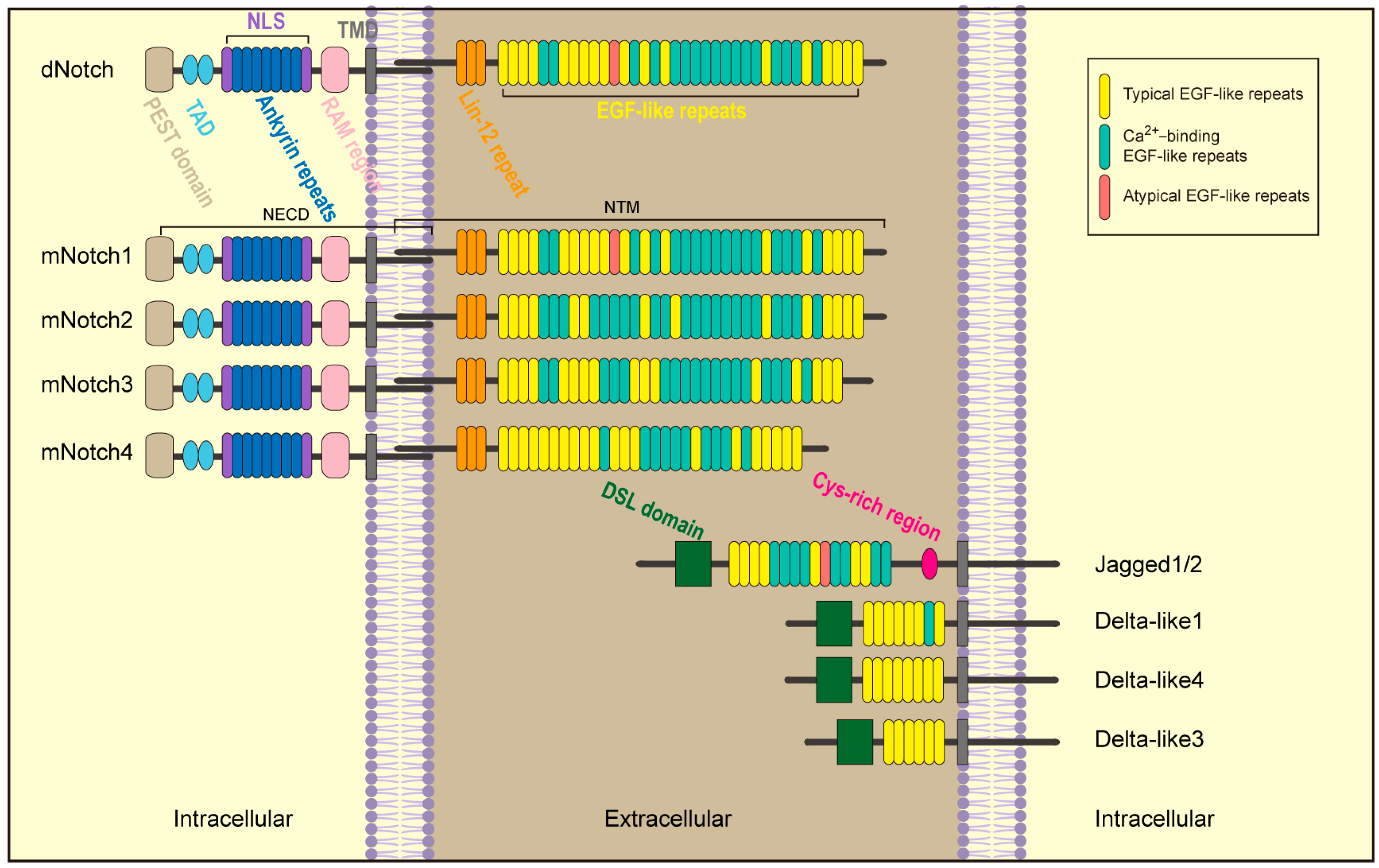
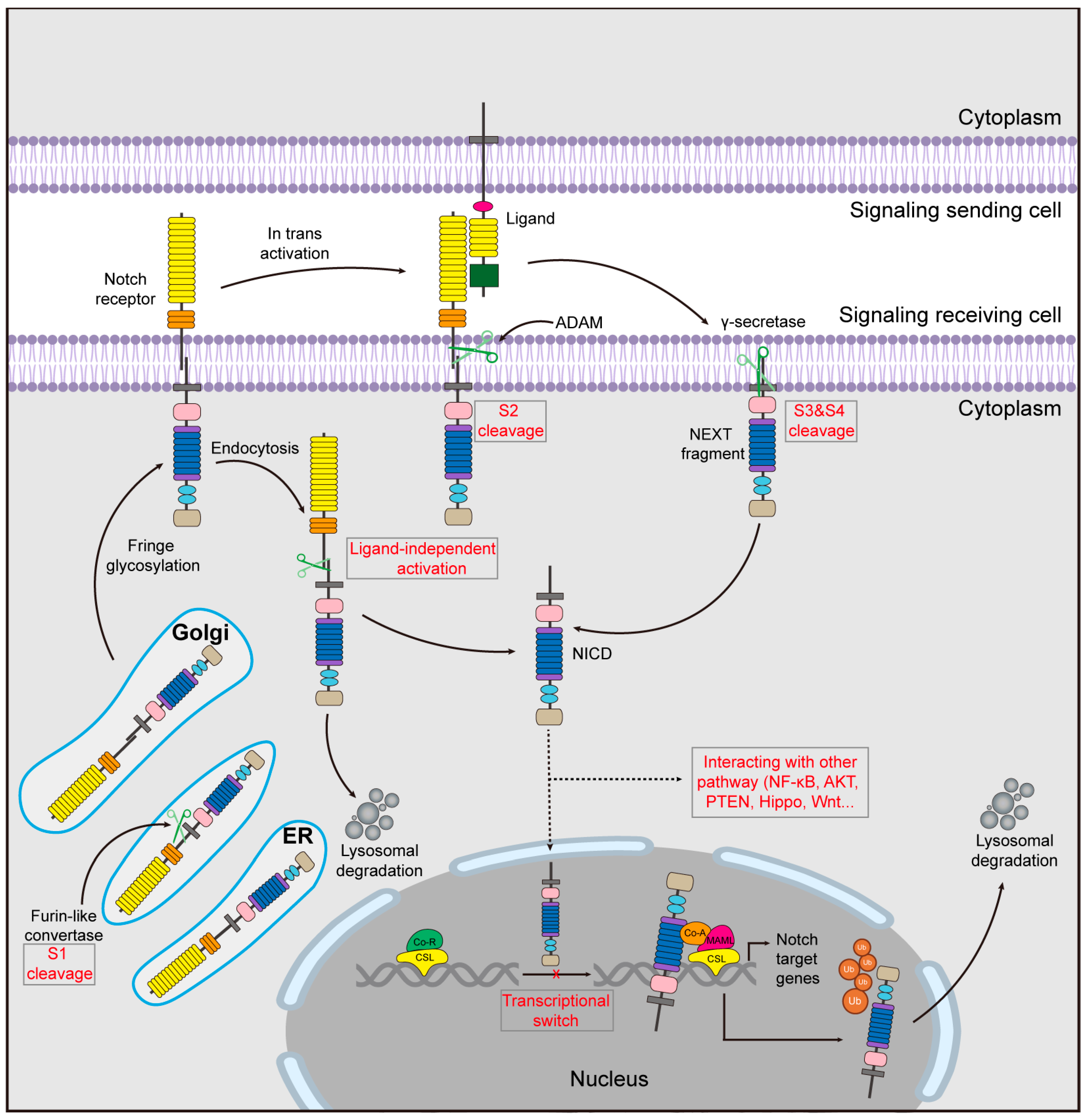
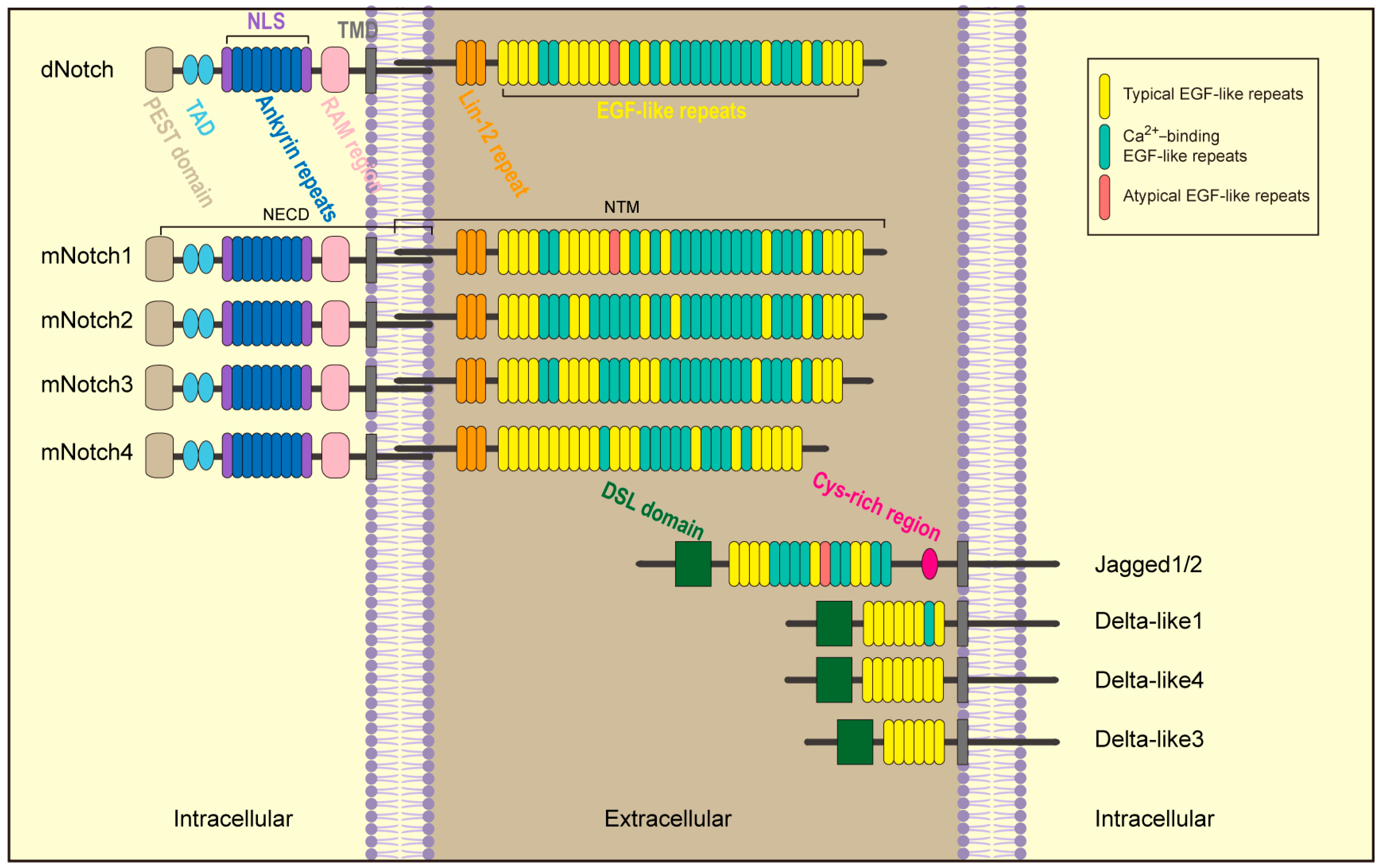
2. Notch
3. Notch Is Involved in Development and Progression of Glioblastoma
3.1. Proliferation and Growth
3.2. Migration and Invasion
3.3. Apoptosis
3.4. Regulation of Stemness
4. Status of Targeted Notch Therapy
4.1. Clinical Trials Targeting Notch in Cancer
4.2. Clinical Trials Targeting Notch in Glioblastoma
4.3. Targeting Notch in GBM
4.3.1. γ-Secretase Inhibitors in GBM
4.3.2. α-Secretase Inhibitors in GBM

{kind=link}
{kind=link}
| Class | Molecules | Testing Index | Biological Effects |
|---|---|---|---|
| γ-Secretase inhibitor (GSI) | DAPT(GSI-IX) [158] | Notch1; DLL1; VEGF | Improving sensitivity to radiotherapy; inducing cell differentiation. |
| LLNle [163] | Notch1; Notch2; Hey2; Myc; Akt | Inducing cell apoptosis. | |
| L-685,458 [164] | Hes1 | Inhibiting neurosphere formation. | |
| RO4929097 [154] | Notch2; Notch4 | Inhibiting neurosphere formation; reducing drug resistance. | |
| MRK003 [167] | Akt | Reducing drug resistance; inducing cell apoptosis. | |
| GSI-XVII [170] | Inducing cell apoptosis. | ||
| GSI-I [166] | Improving sensitivity to radiotherapy. | ||
| GSI-X [77] | Notch1 | Inducing cell apoptosis; suppressing cell invasion. | |
| α-Secretase inhibitor (ASI) | INCB3619 [65] | Notch1; p21; p53 | Inhibiting cell proliferation. |
| Other molecular inhibitor | Arsenic trioxide (ATO) [75] | Notch1; Hes1 | Improving sensitivity to radiotherapy. |
| AG1478 [172] | Notch1; EGFR | Inducing cell apoptosis. | |
| Retinoic acid [173] | Hes2; Hey1; Hey2 | Inducing neural differentiation; inhibiting cell proliferation. | |
| N-acetylcysteine [174] | Notch2; Hes1; Hey2 | Inhibiting cell proliferation; suppressing cell invasion and migration. | |
| dnMAML [175] | Hes1; Hey-L | Inhibiting cell proliferation; inducing cell apoptosis. | |
| Honokiol [176] | Notch3; Hes1 | Promoting the pharmacodynamics of TMZ. | |
| Bacoside A [108] | Notch1; Hes1 | Inducing cell apoptosis. | |
| Hypocretin-1 [109] | Notch1 | Inducing cell apoptosis. | |
| Resveratrol [110] | Notch1; p53 | Inducing cell apoptosis. |
4.3.3. Other Molecule Inhibitors
4.4. Notch and Glioblsatoma Resistance
5. Summary and Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Peng, W.; Shi, S.; Zhong, J.; Liang, H.; Hou, J.; Hu, X.; Wang, F.; Zhang, J.; Geng, S.; Sun, X.; et al. CBX3 accelerates the malignant progression of glioblastoma multiforme by stabilizing EGFR expression. Oncogene 2022, 41, 3051–3063. [Google Scholar] [CrossRef] [PubMed]
- Cadieux, B.; Ching, T.T.; VandenBerg, S.R.; Costello, J.F. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res. 2006, 66, 8469–8476. [Google Scholar] [CrossRef] [PubMed]
- McBain, C.; Lawrie, T.A.; Rogozińska, E.; Kernohan, A.; Robinson, T.; Jefferies, S. Treatment options for progression or recurrence of glioblastoma: A network meta-analysis. Cochrane Database Syst. Rev. 2021, 5, Cd013579. [Google Scholar] [CrossRef] [PubMed]
- Barrette, A.M.; Ronk, H.; Joshi, T.; Mussa, Z.; Mehrotra, M.; Bouras, A.; Nudelman, G.; Jesu Raj, J.G.; Bozec, D.; Lam, W.; et al. Anti-invasive efficacy and survival benefit of the YAP-TEAD inhibitor verteporfin in preclinical glioblastoma models. Neuro-oncology 2022, 24, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Witthayanuwat, S.; Pesee, M.; Supaadirek, C.; Supakalin, N.; Thamronganantasakul, K.; Krusun, S. Survival Analysis of Glioblastoma Multiforme. Asian Pac. J. Cancer Prev. APJCP 2018, 19, 2613–2617. [Google Scholar] [CrossRef] [PubMed]
- Quinones, A.; Le, A. The Multifaceted Glioblastoma: From Genomic Alterations to Metabolic Adaptations. Adv. Exp. Med. Biol. 2021, 1311, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Yong, R.L.; Paddison, P.; Zhu, J. Comparison of glioblastoma (GBM) molecular classification methods. Semin. Cancer Biol. 2018, 53, 201–211. [Google Scholar] [CrossRef] [PubMed]
- de La Coste, A.; Freitas, A.A. Notch signaling: Distinct ligands induce specific signals during lymphocyte development and maturation. Immunol. Lett. 2006, 102, 1–9. [Google Scholar] [CrossRef]
- Guruharsha, K.G.; Kankel, M.W.; Artavanis-Tsakonas, S. The Notch signalling system: Recent insights into the complexity of a conserved pathway. Nat. Rev. Genet. 2012, 13, 654–666. [Google Scholar] [CrossRef]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef]
- Koch, U.; Lehal, R.; Radtke, F. Stem cells living with a Notch. Development 2013, 140, 689–704. [Google Scholar] [CrossRef] [PubMed]
- Siebel, C.; Lendahl, U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef]
- Greenwald, I.; Kovall, R. Notch signaling: Genetics and structure. WormBook Online Rev. C. elegans Biol. 2013, 1–28. [Google Scholar] [CrossRef]
- Lino, M.M.; Merlo, A.; Boulay, J.L. Notch signaling in glioblastoma: A developmental drug target? BMC Med. 2010, 8, 72. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.K.; Stathias, V.; Maloof, M.E.; Vidovic, D.; Winterbottom, E.F.; Capobianco, A.J.; Clarke, J.; Schurer, S.; Robbins, D.J.; Ayad, N.G. Epigenetic pathways and glioblastoma treatment: Insights from signaling cascades. J. Cell. Biochem. 2015, 116, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Ramar, V.; Guo, S.; Hudson, B.; Liu, M. Progress in Glioma Stem Cell Research. Cancers 2023, 16, 102. [Google Scholar] [CrossRef]
- Sharifzad, F.; Ghavami, S.; Verdi, J.; Mardpour, S.; Mollapour Sisakht, M.; Azizi, Z.; Taghikhani, A.; Łos, M.J.; Fakharian, E.; Ebrahimi, M.; et al. Glioblastoma cancer stem cell biology: Potential theranostic targets. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2019, 42, 35–45. [Google Scholar] [CrossRef]
- Guichet, P.O.; Guelfi, S.; Teigell, M.; Hoppe, L.; Bakalara, N.; Bauchet, L.; Duffau, H.; Lamszus, K.; Rothhut, B.; Hugnot, J.P. Notch1 stimulation induces a vascularization switch with pericyte-like cell differentiation of glioblastoma stem cells. Stem Cells 2015, 33, 21–34. [Google Scholar] [CrossRef]
- Hori, K.; Sen, A.; Artavanis-Tsakonas, S. Notch signaling at a glance. J. Cell Sci. 2013, 126, 2135–2140. [Google Scholar] [CrossRef]
- Wang, M.M. Notch signaling and Notch signaling modifiers. Int. J. Biochem. Cell Biol. 2011, 43, 1550–1562. [Google Scholar] [CrossRef]
- Chillakuri, C.R.; Sheppard, D.; Lea, S.M.; Handford, P.A. Notch receptor-ligand binding and activation: Insights from molecular studies. Semin. Cell Dev. Biol. 2012, 23, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Fiúza, U.M.; Arias, A.M. Cell and molecular biology of Notch. J. Endocrinol. 2007, 194, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Sprinzak, D.; Blacklow, S.C. Biophysics of Notch Signaling. Annu. Rev. Biophys. 2021, 50, 157–189. [Google Scholar] [CrossRef] [PubMed]
- Cordle, J.; Johnson, S.; Tay, J.Z.; Roversi, P.; Wilkin, M.B.; de Madrid, B.H.; Shimizu, H.; Jensen, S.; Whiteman, P.; Jin, B.; et al. A conserved face of the Jagged/Serrate DSL domain is involved in Notch trans-activation and cis-inhibition. Nat. Struct. Mol. Biol. 2008, 15, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Chiba, S.; Saito, T.; Kumano, K.; Hirai, H. Physical interaction of Delta1, Jagged1, and Jagged2 with Notch1 and Notch3 receptors. Biochem. Biophys. Res. Commun. 2000, 276, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Logeat, F.; Bessia, C.; Brou, C.; LeBail, O.; Jarriault, S.; Seidah, N.G.; Israël, A. The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc. Natl. Acad. Sci. USA 1998, 95, 8108–8112. [Google Scholar] [CrossRef] [PubMed]
- Lovendahl, K.N.; Blacklow, S.C.; Gordon, W.R. The Molecular Mechanism of Notch Activation. Adv. Exp. Med. Biol. 2018, 1066, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Nauman, M.; Stanley, P. Glycans that regulate Notch signaling in the intestine. Biochem. Soc. Trans. 2022, 50, 689–701. [Google Scholar] [CrossRef]
- Fortini, M.E. Gamma-secretase-mediated proteolysis in cell-surface-receptor signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 673–684. [Google Scholar] [CrossRef]
- Zanotti, S.; Canalis, E. Notch Signaling and the Skeleton. Endocr. Rev. 2016, 37, 223–253. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Wallberg, A.E.; Pedersen, K.; Lendahl, U.; Roeder, R.G. p300 and PCAF act cooperatively to mediate transcriptional activation from chromatin templates by notch intracellular domains in vitro. Mol. Cell. Biol. 2002, 22, 7812–7819. [Google Scholar] [CrossRef] [PubMed]
- Kurooka, H.; Honjo, T. Functional interaction between the mouse notch1 intracellular region and histone acetyltransferases PCAF and GCN5. J. Biol. Chem. 2000, 275, 17211–17220. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Griffin, J.D. Modulation of Notch signaling by mastermind-like (MAML) transcriptional co-activators and their involvement in tumorigenesis. Semin. Cancer Biol. 2004, 14, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Jarriault, S.; Brou, C.; Logeat, F.; Schroeter, E.H.; Kopan, R.; Israel, A. Signalling downstream of activated mammalian Notch. Nature 1995, 377, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Iso, T.; Sartorelli, V.; Chung, G.; Shichinohe, T.; Kedes, L.; Hamamori, Y. HERP, a new primary target of Notch regulated by ligand binding. Mol. Cell. Biol. 2001, 21, 6071–6079. [Google Scholar] [CrossRef]
- Capaccione, K.M.; Pine, S.R. The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis 2013, 34, 1420–1430. [Google Scholar] [CrossRef]
- Stockhausen, M.T.; Kristoffersen, K.; Poulsen, H.S. Notch signaling and brain tumors. Adv. Exp. Med. Biol. 2012, 727, 289–304. [Google Scholar] [CrossRef]
- Yin, L.; Velazquez, O.C.; Liu, Z.J. Notch signaling: Emerging molecular targets for cancer therapy. Biochem. Pharmacol. 2010, 80, 690–701. [Google Scholar] [CrossRef]
- Borggrefe, T.; Oswald, F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell. Mol. Life Sci. CMLS 2009, 66, 1631–1646. [Google Scholar] [CrossRef]
- Chen, L.; Lu, H.; Peng, D.; Cao, L.L.; Ballout, F.; Srirmajayam, K.; Chen, Z.; Bhat, A.; Wang, T.C.; Capobianco, A.; et al. Activation of NOTCH signaling via DLL1 is mediated by APE1-redox-dependent NF-κB activation in oesophageal adenocarcinoma. Gut 2023, 72, 421–432. [Google Scholar] [CrossRef]
- Liu, J.; Shen, J.X.; Wen, X.F.; Guo, Y.X.; Zhang, G.J. Targeting Notch degradation system provides promise for breast cancer therapeutics. Crit. Rev. Oncol./Hematol. 2016, 104, 21–29. [Google Scholar] [CrossRef]
- Kopan, R. Notch signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011213. [Google Scholar] [CrossRef]
- Andersen, P.; Uosaki, H.; Shenje, L.T.; Kwon, C. Non-canonical Notch signaling: Emerging role and mechanism. Trends Cell Biol. 2012, 22, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Wang, Y.; Lee, J.T.; Huang, Z.; Wu, D.; Xu, A.; Lam, K.S. Adropin is a brain membrane-bound protein regulating physical activity via the NB-3/Notch signaling pathway in mice. J. Biol. Chem. 2014, 289, 25976–25986. [Google Scholar] [CrossRef] [PubMed]
- Ballester-López, C.; Conlon, T.M.; Ertüz, Z.; Greiffo, F.R.; Irmler, M.; Verleden, S.E.; Beckers, J.; Fernandez, I.E.; Eickelberg, O.; Yildirim, A. The Notch ligand DNER regulates macrophage IFNγ release in chronic obstructive pulmonary disease. EBioMedicine 2019, 43, 562–575. [Google Scholar] [CrossRef]
- Craft, C.S.; Broekelmann, T.J.; Mecham, R.P. Microfibril-associated glycoproteins MAGP-1 and MAGP-2 in disease. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 71–72, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavan, J.; Osborne, B.A. Notch and T Cell Function—A Complex Tale. Adv. Exp. Med. Biol. 2018, 1066, 339–354. [Google Scholar] [CrossRef]
- Sanalkumar, R.; Dhanesh, S.B.; James, J. Non-canonical activation of Notch signaling/target genes in vertebrates. Cell. Mol. Life Sci. CMLS 2010, 67, 2957–2968. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, L.; Zhao, S.; Zhao, X.Y.; Min, P.X.; Ma, Y.D.; Wang, Y.Y.; Chen, Y.; Tang, S.J.; Zhang, Y.J.; et al. Non-canonical Notch Signaling Regulates Actin Remodeling in Cell Migration by Activating PI3K/AKT/Cdc42 Pathway. Front. Pharmacol. 2019, 10, 370. [Google Scholar] [CrossRef] [PubMed]
- Wongchana, W.; Kongkavitoon, P.; Tangtanatakul, P.; Sittplangkoon, C.; Butta, P.; Chawalitpong, S.; Pattarakankul, T.; Osborne, B.A.; Palaga, T. Notch signaling regulates the responses of lipopolysaccharide-stimulated macrophages in the presence of immune complexes. PLoS ONE 2018, 13, e0198609. [Google Scholar] [CrossRef] [PubMed]
- Konishi, J.; Yi, F.; Chen, X.; Vo, H.; Carbone, D.P.; Dang, T.P. Notch3 cooperates with the EGFR pathway to modulate apoptosis through the induction of bim. Oncogene 2010, 29, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, H.; Chen, Y.; Qiao, G.; Jiang, W.; Ni, P.; Liu, X.; Ma, L. miR-598 inhibits metastasis in colorectal cancer by suppressing JAG1/Notch2 pathway stimulating EMT. Exp. Cell Res. 2017, 352, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef] [PubMed]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. 2017, 12, 245–275. [Google Scholar] [CrossRef]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef]
- Purow, B.W.; Haque, R.M.; Noel, M.W.; Su, Q.; Burdick, M.J.; Lee, J.; Sundaresan, T.; Pastorino, S.; Park, J.K.; Mikolaenko, I.; et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005, 65, 2353–2363. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cui, Y.; Gao, G.; Zhao, Z.; Zhang, H.; Wang, X. Notch1 is an independent prognostic factor for patients with glioma. J. Surg. Oncol. 2011, 103, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, C.; Meng, Q.; Li, S.; Sun, X.; Bo, Y.; Yao, W. siRNA targeting Notch-1 decreases glioma stem cell proliferation and tumor growth. Mol. Biol. Rep. 2012, 39, 2497–2503. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kesari, S.; Rooney, C.; Strack, P.R.; Chen, J.; Shen, H.; Wu, L.; Griffin, J.D. Inhibition of notch signaling blocks growth of glioblastoma cell lines and tumor neurospheres. Genes Cancer 2010, 1, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Floyd, D.H.; Kefas, B.; Seleverstov, O.; Mykhaylyk, O.; Dominguez, C.; Comeau, L.; Plank, C.; Purow, B. Alpha-secretase inhibition reduces human glioblastoma stem cell growth in vitro and in vivo by inhibiting Notch. Neuro-oncology 2012, 14, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Moutal, A.; Honnorat, J.; Massoma, P.; Désormeaux, P.; Bertrand, C.; Malleval, C.; Watrin, C.; Chounlamountri, N.; Mayeur, M.E.; Besançon, R.; et al. CRMP5 Controls Glioblastoma Cell Proliferation and Survival through Notch-Dependent Signaling. Cancer Res. 2015, 75, 3519–3528. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Lin, X.; Yang, X.; Dong, H.; Yue, X.; Andrade, K.C.; Guo, Z.; Yang, J.; Wu, L.; Zhu, X.; et al. Downregulation of RND3/RhoE in glioblastoma patients promotes tumorigenesis through augmentation of notch transcriptional complex activity. Cancer Med. 2015, 4, 1404–1416. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guessous, F.; Zhang, Y.; Dipierro, C.; Kefas, B.; Johnson, E.; Marcinkiewicz, L.; Jiang, J.; Yang, Y.; Schmittgen, T.D.; et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009, 69, 7569–7576. [Google Scholar] [CrossRef] [PubMed]
- Panza, S.; Russo, U.; Giordano, F.; Leggio, A.; Barone, I.; Bonofiglio, D.; Gelsomino, L.; Malivindi, R.; Conforti, F.L.; Naimo, G.D.; et al. Leptin and Notch Signaling Cooperate in Sustaining Glioblastoma Multiforme Progression. Biomolecules 2020, 10, 886. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Q.; Geng, R.; Liu, H.; Yuan, F.; Xu, Y.; Qi, Y.; Jiang, H.; Chen, Q.; Liu, B. Notch intracellular domain regulates glioblastoma proliferation through the Notch1 signaling pathway. Oncol. Lett. 2021, 21, 303. [Google Scholar] [CrossRef]
- Wu, J.; Wang, N.; Yang, Y.; Jiang, G.; Zhan, H.; Li, F. LINC01152 upregulates MAML2 expression to modulate the progression of glioblastoma multiforme via Notch signaling pathway. Cell Death Dis. 2021, 12, 115. [Google Scholar] [CrossRef] [PubMed]
- Li, J.L.; Sainson, R.C.; Shi, W.; Leek, R.; Harrington, L.S.; Preusser, M.; Biswas, S.; Turley, H.; Heikamp, E.; Hainfellner, J.A.; et al. Delta-like 4 Notch ligand regulates tumor angiogenesis, improves tumor vascular function, and promotes tumor growth in vivo. Cancer Res. 2007, 67, 11244–11253. [Google Scholar] [CrossRef]
- Lim, K.J.; Rajan, K.; Eberhart, C.G. Effects of Zeng Sheng Ping/ACAPHA on malignant brain tumor growth and Notch signaling. Anticancer Res. 2012, 32, 2689–2696. [Google Scholar]
- Wong, H.A.; Fatimy, R.E.; Onodera, C.; Wei, Z.; Yi, M.; Mohan, A.; Gowrisankaran, S.; Karmali, P.; Marcusson, E.; Wakimoto, H.; et al. The Cancer Genome Atlas Analysis Predicts MicroRNA for Targeting Cancer Growth and Vascularization in Glioblastoma. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 1234–1247. [Google Scholar] [CrossRef]
- Wu, J.; Ji, Z.; Liu, H.; Liu, Y.; Han, D.; Shi, C.; Shi, C.; Wang, C.; Yang, G.; Chen, X.; et al. Arsenic trioxide depletes cancer stem-like cells and inhibits repopulation of neurosphere derived from glioblastoma by downregulation of Notch pathway. Toxicol. Lett. 2013, 220, 61–69. [Google Scholar] [CrossRef]
- Chigurupati, S.; Venkataraman, R.; Barrera, D.; Naganathan, A.; Madan, M.; Paul, L.; Pattisapu, J.V.; Kyriazis, G.A.; Sugaya, K.; Bushnev, S.; et al. Receptor channel TRPC6 is a key mediator of Notch-driven glioblastoma growth and invasiveness. Cancer Res. 2010, 70, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Cenciarelli, C.; Marei, H.E.; Zonfrillo, M.; Casalbore, P.; Felsani, A.; Giannetti, S.; Trevisi, G.; Althani, A.; Mangiola, A. The interference of Notch1 target Hes1 affects cell growth, differentiation and invasiveness of glioblastoma stem cells through modulation of multiple oncogenic targets. Oncotarget 2017, 8, 17873–17886. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.C.; Lin, Y.C.; Liu, C.H.; Chung, H.C.; Wang, Y.T.; Lin, Y.W.; Ma, H.I.; Tu, P.H.; Lawler, S.E.; Chen, R.H. USP11 regulates PML stability to control Notch-induced malignancy in brain tumours. Nat. Commun. 2014, 5, 3214. [Google Scholar] [CrossRef]
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Reineke, E.L.; Liu, Y.; Kao, H.Y. Promyelocytic leukemia protein controls cell migration in response to hydrogen peroxide and insulin-like growth factor-1. J. Biol. Chem. 2010, 285, 9485–9492. [Google Scholar] [CrossRef]
- Sivasankaran, B.; Degen, M.; Ghaffari, A.; Hegi, M.E.; Hamou, M.F.; Ionescu, M.C.; Zweifel, C.; Tolnay, M.; Wasner, M.; Mergenthaler, S.; et al. Tenascin-C is a novel RBPJkappa-induced target gene for Notch signaling in gliomas. Cancer Res. 2009, 69, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Poulikakos, P.I.; Irie, H.Y.; Parekh, S.; Reddy, E.P. CDK4: A master regulator of the cell cycle and its role in cancer. Genes Cancer 2022, 13, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.; D’Amico, M.; Montalto, F.I.; Malivindi, R.; Chimento, A.; Conforti, F.L.; Pezzi, V.; Panno, M.L.; Andò, S.; De Amicis, F. Cdk4 Regulates Glioblastoma Cell Invasion and Stemness and Is Target of a Notch Inhibitor Plus Resveratrol Combined Treatment. Int. J. Mol. Sci. 2023, 24, 10094. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Maciaczyk, D.; Picard, D.; Zhao, L.; Koch, K.; Herrera-Rios, D.; Li, G.; Marquardt, V.; Pauck, D.; Hoerbelt, T.; Zhang, W.; et al. CBF1 is clinically prognostic and serves as a target to block cellular invasion and chemoresistance of EMT-like glioblastoma cells. Br. J. Cancer 2017, 117, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Raghu, H.; Gondi, C.S.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Specific knockdown of uPA/uPAR attenuates invasion in glioblastoma cells and xenografts by inhibition of cleavage and trafficking of Notch -1 receptor. Mol. Cancer 2011, 10, 130. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wei, L.; Hu, B.; Zhang, J.; Wei, J.; Qian, Z.; Zou, D. RBM8A Promotes Glioblastoma Growth and Invasion Through the Notch/STAT3 Pathway. Front. Oncol. 2021, 11, 736941. [Google Scholar] [CrossRef]
- Wei, L.; Zou, C.; Chen, L.; Lin, Y.; Liang, L.; Hu, B.; Mao, Y.; Zou, D. Molecular Insights and Prognosis Associated With RBM8A in Glioblastoma. Front. Mol. Biosci. 2022, 9, 876603. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Pan, M.; Jiang, Q.; Hu, B.; Zhao, J.; Zou, C.; Chen, L.; Tang, C.; Zou, D. Eukaryotic initiation factor 4 A-3 promotes glioblastoma growth and invasion through the Notch1-dependent pathway. BMC Cancer 2023, 23, 550. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, T.; Zhang, J.; Mao, Q.; Li, S.; Xiong, W.; Qiu, Y.; Xie, Q.; Ge, J. Notch1 promotes glioma cell migration and invasion by stimulating β-catenin and NF-κB signaling via AKT activation. Cancer Sci. 2012, 103, 181–190. [Google Scholar] [CrossRef]
- Hai, L.; Zhang, C.; Li, T.; Zhou, X.; Liu, B.; Li, S.; Zhu, M.; Lin, Y.; Yu, S.; Zhang, K.; et al. Notch1 is a prognostic factor that is distinctly activated in the classical and proneural subtype of glioblastoma and that promotes glioma cell survival via the NF-κB(p65) pathway. Cell Death Dis. 2018, 9, 158. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Zhou, X.; Li, T.; Liu, P.; Hai, L.; Tong, L.; Ma, H.; Tao, Z.; Xie, Y.; Zhang, C.; et al. Notch1 signaling pathway promotes invasion, self-renewal and growth of glioma initiating cells via modulating chemokine system CXCL12/CXCR4. J. Exp. Clin. Cancer Res. CR 2019, 38, 339. [Google Scholar] [CrossRef]
- Friedrich, T.; Ferrante, F.; Pioger, L.; Nist, A.; Stiewe, T.; Andrau, J.C.; Bartkuhn, M.; Giaimo, B.D.; Borggrefe, T. Notch-dependent and -independent functions of transcription factor RBPJ. Nucleic Acids Res. 2022, 50, 7925–7937. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Tanaka, S.; Jiapaer, S.; Sabit, H.; Tamai, S.; Kinoshita, M.; Nakada, M. RBPJ contributes to the malignancy of glioblastoma and induction of proneural-mesenchymal transition via IL-6-STAT3 pathway. Cancer Sci. 2020, 111, 4166–4176. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Sarkar, F.H. The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr. Drug Targets 2010, 11, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Noseda, M.; McLean, G.; Niessen, K.; Chang, L.; Pollet, I.; Montpetit, R.; Shahidi, R.; Dorovini-Zis, K.; Li, L.; Beckstead, B.; et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ. Res. 2004, 94, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Niessen, K.; Fu, Y.; Chang, L.; Hoodless, P.A.; McFadden, D.; Karsan, A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J. Cell Biol. 2008, 182, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Chen, Q.; Liu, B.; Wang, L.; Zhang, S.; Ji, B. Knockdown of Rab21 inhibits proliferation and induces apoptosis in human glioma cells. Cell. Mol. Biol. Lett. 2017, 22, 30. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Bergsland, E.; Aggarwal, R.; Aparicio, A.; Beltran, H.; Crabtree, J.S.; Hann, C.L.; Ibrahim, T.; Byers, L.A.; Sasano, H.; et al. DLL3 as an Emerging Target for the Treatment of Neuroendocrine Neoplasms. Oncologist 2022, 27, 940–951. [Google Scholar] [CrossRef]
- Hu, B.; Nandhu, M.S.; Sim, H.; Agudelo-Garcia, P.A.; Saldivar, J.C.; Dolan, C.E.; Mora, M.E.; Nuovo, G.J.; Cole, S.E.; Viapiano, M.S. Fibulin-3 promotes glioma growth and resistance through a novel paracrine regulation of Notch signaling. Cancer Res. 2012, 72, 3873–3885. [Google Scholar] [CrossRef]
- Xing, Z.Y.; Sun, L.G.; Guo, W.J. Elevated expression of Notch-1 and EGFR induced apoptosis in glioblastoma multiforme patients. Clin. Neurol. Neurosurg. 2015, 131, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Kefas, B.; Comeau, L.; Erdle, N.; Montgomery, E.; Amos, S.; Purow, B. Pyruvate kinase M2 is a target of the tumor-suppressive microRNA-326 and regulates the survival of glioma cells. Neuro-oncology 2010, 12, 1102–1112. [Google Scholar] [CrossRef]
- Wu, Z.; Wu, Y.; Tian, Y.; Sun, X.; Liu, J.; Ren, H.; Liang, C.; Song, L.; Hu, H.; Wang, L.; et al. Differential effects of miR-34c-3p and miR-34c-5p on the proliferation, apoptosis and invasion of glioma cells. Oncol. Lett. 2013, 6, 1447–1452. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Li, J.; Xu, T.; Zhou, D.D.; Zhang, L.; Wang, X. MicroRNA-145 induces apoptosis of glioma cells by targeting BNIP3 and Notch signaling. Oncotarget 2017, 8, 61510–61527. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Li, R.J.; Wang, H.D. γ-secretase inhibitor DAPT sensitizes t-AUCB-induced apoptosis of human glioblastoma cells in vitro via blocking the p38 MAPK/MAPKAPK2/Hsp27 pathway. Acta Pharmacol. Sin. 2014, 35, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Bessette, B.; Durand, K.; Giraud, S.; Bégaud, G.; Mathonnet, M.; Lalloué, F. Decrease in Fas-induced apoptosis by the γ-secretase inhibitor is dependent on p75(NTR) in a glioblastoma cell line. Exp. Ther. Med. 2012, 3, 873–877. [Google Scholar] [CrossRef]
- Ding, D.; Lim, K.S.; Eberhart, C.G. Arsenic trioxide inhibits Hedgehog, Notch and stem cell properties in glioblastoma neurospheres. Acta Neuropathol. Commun. 2014, 2, 31. [Google Scholar] [CrossRef] [PubMed]
- Aithal, M.G.S.; Rajeswari, N. Bacoside A Induced Sub-G0 Arrest and Early Apoptosis in Human Glioblastoma Cell Line U-87 MG through Notch Signaling Pathway. Brain Tumor Res. Treat. 2019, 7, 25–32. [Google Scholar] [CrossRef]
- Huan, R.; Yue, J.; Lan, J.; Wang, J.; Cheng, Y.; Zhang, J.; Tan, Y. Hypocretin-1 suppresses malignant progression of glioblastoma cells through Notch1 signaling pathway. Brain Res. Bull. 2023, 196, 46–58. [Google Scholar] [CrossRef]
- Lin, H.; Xiong, W.; Zhang, X.; Liu, B.; Zhang, W.; Zhang, Y.; Cheng, J.; Huang, H. Notch-1 activation-dependent p53 restoration contributes to resveratrol-induced apoptosis in glioblastoma cells. Oncol. Rep. 2011, 26, 925–930. [Google Scholar] [CrossRef]
- Dianat-Moghadam, H.; Sharifi, M.; Salehi, R.; Keshavarz, M.; Shahgolzari, M.; Amoozgar, Z. Engaging stemness improves cancer immunotherapy. Cancer Lett. 2023, 554, 216007. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Zhao, Y.; Chen, W.; Peng, W.; Wang, Y.; Xiong, S.; Aruna; Li, Y.; Yang, Y.; Chen, S.; et al. The stromal-tumor amplifying STC1-Notch1 feedforward signal promotes the stemness of hepatocellular carcinoma. J. Transl. Med. 2023, 21, 236. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Guo, A.A.; King, P.; Guo, S.; Saafir, T.; Jiang, Y.; Liu, M. TRPM7 Induces Tumorigenesis and Stemness Through Notch Activation in Glioma. Front. Pharmacol. 2020, 11, 590723. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bar-Lev, L.; Sharife, H.; Grunewald, M.; Mogilevsky, M.; Licht, T.; Goveia, J.; Taverna, F.; Paldor, I.; Carmeliet, P.; et al. Identification of vascular cues contributing to cancer cell stemness and function. Angiogenesis 2022, 25, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, M.; Xing, F.; Wang, M.; Wang, B.; Qian, D. Human cytomegalovirus infection promotes the stemness of U251 glioma cells. J. Med. Virol. 2017, 89, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Linder, B.; Wehle, A.; Hehlgans, S.; Bonn, F.; Dikic, I.; Rödel, F.; Seifert, V.; Kögel, D. Arsenic Trioxide and (-)-Gossypol Synergistically Target Glioma Stem-Like Cells via Inhibition of Hedgehog and Notch Signaling. Cancers 2019, 11, 350. [Google Scholar] [CrossRef] [PubMed]
- Tchorz, J.S.; Tome, M.; Cloëtta, D.; Sivasankaran, B.; Grzmil, M.; Huber, R.M.; Rutz-Schatzmann, F.; Kirchhoff, F.; Schaeren-Wiemers, N.; Gassmann, M.; et al. Constitutive Notch2 signaling in neural stem cells promotes tumorigenic features and astroglial lineage entry. Cell Death Dis. 2012, 3, e325. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, L.; Zhou, Y.; Dong, L.; Ma, W.; Lv, L.; Zhang, J.; Wang, X. Glioblastoma Stem Cell-Derived Exosomes Enhance Stemness and Tumorigenicity of Glioma Cells by Transferring Notch1 Protein. Cell. Mol. Neurobiol. 2020, 40, 767–784. [Google Scholar] [CrossRef] [PubMed]
- Ylivinkka, I.; Sihto, H.; Tynninen, O.; Hu, Y.; Laakso, A.; Kivisaari, R.; Laakkonen, P.; Keski-Oja, J.; Hyytiäinen, M. Motility of glioblastoma cells is driven by netrin-1 induced gain of stemness. J. Exp. Clin. Cancer Res. CR 2017, 36, 9. [Google Scholar] [CrossRef]
- Smith, A.M.; Gibbons, H.M.; Oldfield, R.L.; Bergin, P.M.; Mee, E.W.; Curtis, M.A.; Faull, R.L.; Dragunow, M. M-CSF increases proliferation and phagocytosis while modulating receptor and transcription factor expression in adult human microglia. J. Neuroinflammation 2013, 10, 85. [Google Scholar] [CrossRef]
- Chockalingam, S.; Ghosh, S.S. Amelioration of cancer stem cells in macrophage colony stimulating factor-expressing U87MG-human glioblastoma upon 5-fluorouracil therapy. PLoS ONE 2013, 8, e83877. [Google Scholar] [CrossRef]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhang, P.; Chen, S.; Chen, Z.; Qiu, Y.; Peng, P.; Huang, W.; Cheng, F.; Zhang, Y.; Li, H.; et al. FAM129A promotes self-renewal and maintains invasive status via stabilizing the Notch intracellular domain in glioma stem cells. Neuro-oncology 2023, 25, 1788–1801. [Google Scholar] [CrossRef] [PubMed]
- Rajakulendran, N.; Rowland, K.J.; Selvadurai, H.J.; Ahmadi, M.; Park, N.I.; Naumenko, S.; Dolma, S.; Ward, R.J.; So, M.; Lee, L.; et al. Wnt and Notch signaling govern self-renewal and differentiation in a subset of human glioblastoma stem cells. Genes Dev. 2019, 33, 498–510. [Google Scholar] [CrossRef]
- Shen, F.; Song, C.; Liu, Y.; Zhang, J.; Wei Song, S. IGFBP2 promotes neural stem cell maintenance and proliferation differentially associated with glioblastoma subtypes. Brain Res. 2019, 1704, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, X.; Chen, A.T.; Gao, X.; Himes, B.T.; Zhang, H.; Chen, Z.; Wang, J.; Sheu, W.C.; Deng, G.; et al. ZNF117 regulates glioblastoma stem cell differentiation towards oligodendroglial lineage. Nat. Commun. 2022, 13, 2196. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, L.; Zhang, R.; Yi, Y.; Ma, Y.; Yan, K.; Jiang, X.; Wang, X. ADAM17 regulates self-renewal and differentiation of U87 glioblastoma stem cells. Neurosci. Lett. 2013, 537, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Xia, S.; Lal, B.; Eberhart, C.G.; Quinones-Hinojosa, A.; Maciaczyk, J.; Matsui, W.; Dimeco, F.; Piccirillo, S.M.; Vescovi, A.L.; et al. DNER, an epigenetically modulated gene, regulates glioblastoma-derived neurosphere cell differentiation and tumor propagation. Stem Cells 2009, 27, 1473–1486. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; He, J.; Liu, Y.; Byun, J.; Vivekanandan, A.; Pennathur, S.; Fan, X.; Lubman, D.M. Dose-dependent proteomic analysis of glioblastoma cancer stem cells upon treatment with γ-secretase inhibitor. Proteomics 2011, 11, 4529–4540. [Google Scholar] [CrossRef]
- Ulasov, I.V.; Mijanovic, O.; Savchuk, S.; Gonzalez-Buendia, E.; Sonabend, A.; Xiao, T.; Timashev, P.; Lesniak, M.S. TMZ regulates GBM stemness via MMP14-DLL4-Notch3 pathway. Int. J. Cancer 2020, 146, 2218–2228. [Google Scholar] [CrossRef]
- Baisiwala, S.; Hall, R.R., III; Saathoff, M.R.; Shireman, J.M.; Park, C.; Budhiraja, S.; Goel, C.; Warnke, L.; Hardiman, C.; Wang, J.Y.; et al. LNX1 Modulates Notch1 Signaling to Promote Expansion of the Glioma Stem Cell Population during Temozolomide Therapy in Glioblastoma. Cancers 2020, 12, 3505. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, H.; Glotzbach, A.; Kolbe, K.; Leonhardt, G.; Loosse, C.; Müller, T. Small things matter: Implications of APP intracellular domain AICD nuclear signaling in the progression and pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2017, 156, 189–213. [Google Scholar] [CrossRef] [PubMed]
- DeAngelo, D.J.; Stone, R.M.; Silverman, L.B.; Stock, W.; Attar, E.C.; Fearen, I.; Dallob, A.; Matthews, C.Z.; Stone, J.R.; Freedman, S.J.; et al. A phase I clinical trial of the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. J. Clin. Oncol. 2006, 24 (Suppl. 18), 6585. [Google Scholar] [CrossRef]
- Wong, G.T.; Manfra, D.; Poulet, F.M.; Zhang, Q.; Josien, H.; Bara, T.; Engstrom, L.; Pinzon-Ortiz, M.; Fine, J.S.; Lee, H.J.; et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem. 2004, 279, 12876–12882. [Google Scholar] [CrossRef] [PubMed]
- van Es, J.H.; van Gijn, M.E.; Riccio, O.; van den Born, M.; Vooijs, M.; Begthel, H.; Cozijnsen, M.; Robine, S.; Winton, D.J.; Radtke, F.; et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005, 435, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Messersmith, W.A.; Shapiro, G.I.; Cleary, J.M.; Jimeno, A.; Dasari, A.; Huang, B.; Shaik, M.N.; Cesari, R.; Zheng, X.; Reynolds, J.M.; et al. A Phase I, dose-finding study in patients with advanced solid malignancies of the oral γ-secretase inhibitor PF-03084014. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 60–67. [Google Scholar] [CrossRef]
- Papayannidis, C.; DeAngelo, D.J.; Stock, W.; Huang, B.; Shaik, M.N.; Cesari, R.; Zheng, X.; Reynolds, J.M.; English, P.A.; Ozeck, M.; et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 2015, 5, e350. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Messersmith, W.A.; Mikulski, S.M.; Papadopoulos, K.P.; Kwak, E.L.; Gibbon, D.G.; Patnaik, A.; Falchook, G.S.; Dasari, A.; Shapiro, G.I.; et al. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 2348–2353. [Google Scholar] [CrossRef]
- Knoechel, B.; Bhatt, A.; Pan, L.; Pedamallu, C.S.; Severson, E.; Gutierrez, A.; Dorfman, D.M.; Kuo, F.C.; Kluk, M.; Kung, A.L.; et al. Complete hematologic response of early T-cell progenitor acute lymphoblastic leukemia to the γ-secretase inhibitor BMS-906024: Genetic and epigenetic findings in an outlier case. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000539. [Google Scholar] [CrossRef]
- Villalobos, V.M.; Hall, F.; Jimeno, A.; Gore, L.; Kern, K.; Cesari, R.; Huang, B.; Schowinsky, J.T.; Blatchford, P.J.; Hoffner, B.; et al. Long-Term Follow-Up of Desmoid Fibromatosis Treated with PF-03084014, an Oral Gamma Secretase Inhibitor. Ann. Surg. Oncol. 2018, 25, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; O’Sullivan Coyne, G.; Do, K.T.; Turkbey, B.; Meltzer, P.S.; Polley, E.; Choyke, P.L.; Meehan, R.; Vilimas, R.; Horneffer, Y.; et al. Clinical Activity of the γ-Secretase Inhibitor PF-03084014 in Adults With Desmoid Tumors (Aggressive Fibromatosis). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Lehal, R.; Zaric, J.; Vigolo, M.; Urech, C.; Frismantas, V.; Zangger, N.; Cao, L.; Berger, A.; Chicote, I.; Loubéry, S.; et al. Pharmacological disruption of the Notch transcription factor complex. Proc. Natl. Acad. Sci. USA 2020, 117, 16292–16301. [Google Scholar] [CrossRef] [PubMed]
- Stoeck, A.; Lejnine, S.; Truong, A.; Pan, L.; Wang, H.; Zang, C.; Yuan, J.; Ware, C.; MacLean, J.; Garrett-Engele, P.W.; et al. Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer Discov. 2014, 4, 1154–1167. [Google Scholar] [CrossRef] [PubMed]
- Yan, M. Therapeutic promise and challenges of targeting DLL4/NOTCH1. Vasc. Cell 2011, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, E.G.; LoRusso, P.; Strother, R.M.; Diamond, J.R.; Younger, A.; Messersmith, W.A.; Adriaens, L.; Liu, L.; Kao, R.J.; DiCioccio, A.T.; et al. A Phase I First-in-Human Study of Enoticumab (REGN421), a Fully Human Delta-like Ligand 4 (Dll4) Monoclonal Antibody in Patients with Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 2695–2703. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.; Zhang, G.; Wu, Y.; Stawicki, S.; Liang, W.C.; Chanthery, Y.; Kowalski, J.; Watts, R.J.; Callahan, C.; Kasman, I.; et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 2006, 444, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.I.; Bendell, J.C.; Bullock, A.; LoConte, N.K.; Hatoum, H.; Ritch, P.; Hool, H.; Leach, J.W.; Sanchez, J.; Sohal, D.P.S.; et al. A randomized phase II trial of nab-paclitaxel and gemcitabine with tarextumab or placebo in patients with untreated metastatic pancreatic cancer. Cancer Med. 2019, 8, 5148–5157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, Y.; Li, X.; Yuan, X.; Chu, Q. Targeting the Notch signaling pathway and the Notch ligand, DLL3, in small cell lung cancer. Biomed. Pharmacother. 2023, 159, 114248. [Google Scholar] [CrossRef]
- Blackhall, F.; Jao, K.; Greillier, L.; Cho, B.C.; Penkov, K.; Reguart, N.; Majem, M.; Nackaerts, K.; Syrigos, K.; Hansen, K.; et al. Efficacy and Safety of Rovalpituzumab Tesirine Compared With Topotecan as Second-Line Therapy in DLL3-High SCLC: Results From the Phase 3 TAHOE Study. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2021, 16, 1547–1558. [Google Scholar] [CrossRef]
- Rudin, C.M.; Pietanza, M.C.; Bauer, T.M.; Ready, N.; Morgensztern, D.; Glisson, B.S.; Byers, L.A.; Johnson, M.L.; Burris, H.A., 3rd; Robert, F.; et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: A first-in-human, first-in-class, open-label, phase 1 study. Lancet. Oncol. 2017, 18, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Morgensztern, D.; Besse, B.; Greillier, L.; Santana-Davila, R.; Ready, N.; Hann, C.L.; Glisson, B.S.; Farago, A.F.; Dowlati, A.; Rudin, C.M.; et al. Efficacy and Safety of Rovalpituzumab Tesirine in Third-Line and Beyond Patients with DLL3-Expressing, Relapsed/Refractory Small-Cell Lung Cancer: Results From the Phase II TRINITY Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 6958–6966. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.L.; Zvirbule, Z.; Laktionov, K.; Helland, A.; Cho, B.C.; Gutierrez, V.; Colinet, B.; Lena, H.; Wolf, M.; Gottfried, M.; et al. Rovalpituzumab Tesirine as a Maintenance Therapy After First-Line Platinum-Based Chemotherapy in Patients With Extensive-Stage-SCLC: Results From the Phase 3 MERU Study. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2021, 16, 1570–1581. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Shimizu, F.; Hovinga, K.; Beal, K.; Karimi, S.; Droms, L.; Peck, K.K.; Gutin, P.; Iorgulescu, J.B.; Kaley, T.; et al. Molecular and Clinical Effects of Notch Inhibition in Glioma Patients: A Phase 0/I Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4786–4796. [Google Scholar] [CrossRef] [PubMed]
- Peereboom, D.M.; Ye, X.; Mikkelsen, T.; Lesser, G.J.; Lieberman, F.S.; Robins, H.I.; Ahluwalia, M.S.; Sloan, A.E.; Grossman, S.A. A Phase II and Pharmacodynamic Trial of RO4929097 for Patients With Recurrent/Progressive Glioblastoma. Neurosurgery 2021, 88, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Pan, E.; Supko, J.G.; Kaley, T.J.; Butowski, N.A.; Cloughesy, T.; Jung, J.; Desideri, S.; Grossman, S.; Ye, X.; Park, D.M. Phase I study of RO4929097 with bevacizumab in patients with recurrent malignant glioma. J. Neuro-Oncol. 2016, 130, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Cao, Y.; Yue, Z.; Liu, J. Gamma-secretase inhibitor DAPT suppresses glioblastoma growth via uncoupling of tumor vessel density from vessel function. Clin. Exp. Med. 2013, 13, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Kristoffersen, K.; Villingshøj, M.; Poulsen, H.S.; Stockhausen, M.T. Level of Notch activation determines the effect on growth and stem cell-like features in glioblastoma multiforme neurosphere cultures. Cancer Biol. Ther. 2013, 14, 625–637. [Google Scholar] [CrossRef]
- Kristoffersen, K.; Nedergaard, M.K.; Villingshøj, M.; Borup, R.; Broholm, H.; Kjær, A.; Poulsen, H.S.; Stockhausen, M.T. Inhibition of Notch signaling alters the phenotype of orthotopic tumors formed from glioblastoma multiforme neurosphere cells but does not hamper intracranial tumor growth regardless of endogene Notch pathway signature. Cancer Biol. Ther. 2014, 15, 862–877. [Google Scholar] [CrossRef]
- Lu, S.; Lyu, Z.; Wang, Z.; Kou, Y.; Liu, C.; Li, S.; Hu, M.; Zhu, H.; Wang, W.; Zhang, C.; et al. Lipin 1 deficiency causes adult-onset myasthenia with motor neuron dysfunction in humans and neuromuscular junction defects in zebrafish. Theranostics 2021, 11, 2788–2805. [Google Scholar] [CrossRef]
- Tulip, I.J.; Kim, S.O.; Kim, E.J.; Kim, J.; Lee, J.Y.; Kim, H.; Kim, S.C. Combined inhibition of STAT and Notch signalling effectively suppresses tumourigenesis by inducing apoptosis and inhibiting proliferation, migration and invasion in glioblastoma cells. Anim. Cells Syst. 2021, 25, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Staberg, M.; Michaelsen, S.R.; Olsen, L.S.; Nedergaard, M.K.; Villingshøj, M.; Stockhausen, M.T.; Hamerlik, P.; Poulsen, H.S. Combined EGFR- and notch inhibition display additive inhibitory effect on glioblastoma cell viability and glioblastoma-induced endothelial cell sprouting in vitro. Cancer Cell Int. 2016, 16, 34. [Google Scholar] [CrossRef] [PubMed]
- Monticone, M.; Biollo, E.; Fabiano, A.; Fabbi, M.; Daga, A.; Romeo, F.; Maffei, M.; Melotti, A.; Giaretti, W.; Corte, G.; et al. z-Leucinyl-leucinyl-norleucinal induces apoptosis of human glioblastoma tumor-initiating cells by proteasome inhibition and mitotic arrest response. Mol. Cancer Res. MCR 2009, 7, 1822–1834. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.K.; Segarra, M.; Sierra Mde, L.; Sainson, R.C.; Tosato, G.; Harris, A.L. Regulation of CXCR4 by the Notch ligand delta-like 4 in endothelial cells. Cancer Res. 2008, 68, 1889–1895. [Google Scholar] [CrossRef]
- Giordano, F.; Montalto, F.I.; Panno, M.L.; Andò, S.; De Amicis, F. A Notch inhibitor plus Resveratrol induced blockade of autophagy drives glioblastoma cell death by promoting a switch to apoptosis. Am. J. Cancer Res. 2021, 11, 5933–5950. [Google Scholar] [PubMed]
- Lin, J.; Zhang, X.M.; Yang, J.C.; Ye, Y.B.; Luo, S.Q. γ-secretase inhibitor-I enhances radiosensitivity of glioblastoma cell lines by depleting CD133+ tumor cells. Arch. Med. Res. 2010, 41, 519–529. [Google Scholar] [CrossRef]
- Tanaka, S.; Nakada, M.; Yamada, D.; Nakano, I.; Todo, T.; Ino, Y.; Hoshii, T.; Tadokoro, Y.; Ohta, K.; Ali, M.A.; et al. Strong therapeutic potential of γ-secretase inhibitor MRK003 for CD44-high and CD133-low glioblastoma initiating cells. J. Neuro-Oncol. 2015, 121, 239–250. [Google Scholar] [CrossRef]
- Natsumeda, M.; Maitani, K.; Liu, Y.; Miyahara, H.; Kaur, H.; Chu, Q.; Zhang, H.; Kahlert, U.D.; Eberhart, C.G. Targeting Notch Signaling and Autophagy Increases Cytotoxicity in Glioblastoma Neurospheres. Brain Pathol. 2016, 26, 713–723. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Cheng, M.; Koch, K.; Marchionni, L.; Fan, X.; Raabe, E.H.; Maciaczyk, J.; Glunde, K.; Eberhart, C.G. Alterations in cellular metabolome after pharmacological inhibition of Notch in glioblastoma cells. Int. J. Cancer 2016, 138, 1246–1255. [Google Scholar] [CrossRef]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef]
- Gersey, Z.; Osiason, A.D.; Bloom, L.; Shah, S.; Thompson, J.W.; Bregy, A.; Agarwal, N.; Komotar, R.J. Therapeutic Targeting of the Notch Pathway in Glioblastoma Multiforme. World Neurosurg. 2019, 131, 252–263.e252. [Google Scholar] [CrossRef] [PubMed]
- Cenciarelli, C.; Marei, H.E.; Zonfrillo, M.; Pierimarchi, P.; Paldino, E.; Casalbore, P.; Felsani, A.; Vescovi, A.L.; Maira, G.; Mangiola, A. PDGF receptor alpha inhibition induces apoptosis in glioblastoma cancer stem cells refractory to anti-Notch and anti-EGFR treatment. Mol. Cancer 2014, 13, 247. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.; Wang, S.; Sang, Y.; Sun, P.; Lal, B.; Goodwin, C.R.; Guerrero-Cazares, H.; Quinones-Hinojosa, A.; Laterra, J.; Xia, S. Regulation of glioblastoma stem cells by retinoic acid: Role for Notch pathway inhibition. Oncogene 2011, 30, 3454–3467. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Liu, A.D.; Hou, G.Q.; Zhang, X.; Ren, K.; Chen, X.Z.; Li, S.S.C.; Wu, Y.S.; Cao, X. N-acetylcysteine decreases malignant characteristics of glioblastoma cells by inhibiting Notch2 signaling. J. Exp. Clin. Cancer Res. CR 2019, 38, 2. [Google Scholar] [CrossRef]
- Opačak-Bernardi, T.; Ryu, J.S.; Raucher, D. Effects of cell penetrating Notch inhibitory peptide conjugated to elastin-like polypeptide on glioblastoma cells. J. Drug Target. 2017, 25, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Patel, S.; Imran, M.; Maalik, A.; Arshad, M.U.; Saeed, F.; Mabkhot, Y.N.; Al-Showiman, S.S.; Ahmad, N.; Elsharkawy, E. Honokiol: An anticancer lignan. Biomed. Pharmacother. 2018, 107, 555–562. [Google Scholar] [CrossRef]
- Zhang, T.D.; Chen, G.Q.; Wang, Z.G.; Wang, Z.Y.; Chen, S.J.; Chen, Z. Arsenic trioxide, a therapeutic agent for APL. Oncogene 2001, 20, 7146–7153. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Cheng, L.; Shi, Y.; Ke, S.Q.; Huang, Z.; Fang, X.; Chu, C.W.; Xie, Q.; Bian, X.W.; Rich, J.N.; et al. Arsenic trioxide disrupts glioma stem cells via promoting PML degradation to inhibit tumor growth. Oncotarget 2015, 6, 37300–37315. [Google Scholar] [CrossRef]
- Carlsson, S.K.; Brothers, S.P.; Wahlestedt, C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol. Med. 2014, 6, 1359–1370. [Google Scholar] [CrossRef]
- Zheng, X.; Tang, Q.; Ren, L.; Liu, J.; Li, W.; Fu, W.; Wang, J.; Du, G. A narrative review of research progress on drug therapies for glioblastoma multiforme. Ann. Transl. Med. 2021, 9, 943. [Google Scholar] [CrossRef]
- Hiddingh, L.; Tannous, B.A.; Teng, J.; Tops, B.; Jeuken, J.; Hulleman, E.; Boots-Sprenger, S.H.; Vandertop, W.P.; Noske, D.P.; Kaspers, G.J.; et al. EFEMP1 induces γ-secretase/Notch-mediated temozolomide resistance in glioblastoma. Oncotarget 2014, 5, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.L.; Chang, H.H.; Chen, Y.C.; Chang, Y.C.; Chen, Y.; Tsai, W.C. Molecular Mechanisms of KDELC2 on Glioblastoma Tumorigenesis and Temozolomide Resistance. Biomedicines 2020, 8, 339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Guo, S.; Liang, C.; Lian, M. Long intergenic noncoding RNA 00021 promotes glioblastoma temozolomide resistance by epigenetically silencing p21 through Notch pathway. IUBMB Life 2020, 72, 1747–1756. [Google Scholar] [CrossRef]
- Alafate, W.; Xu, D.; Wu, W.; Xiang, J.; Ma, X.; Xie, W.; Bai, X.; Wang, M.; Wang, J. Loss of PLK2 induces acquired resistance to temozolomide in GBM via activation of notch signaling. J. Exp. Clin. Cancer Res. CR 2020, 39, 239. [Google Scholar] [CrossRef] [PubMed]
- Pustchi, S.E.; Avci, N.G.; Akay, Y.M.; Akay, M. Astrocytes Decreased the Sensitivity of Glioblastoma Cells to Temozolomide and Bay 11-7082. Int. J. Mol. Sci. 2020, 21, 7154. [Google Scholar] [CrossRef]
- Lai, I.C.; Shih, P.H.; Yao, C.J.; Yeh, C.T.; Wang-Peng, J.; Lui, T.N.; Chuang, S.E.; Hu, T.S.; Lai, T.Y.; Lai, G.M. Elimination of cancer stem-like cells and potentiation of temozolomide sensitivity by Honokiol in glioblastoma multiforme cells. PLoS ONE 2015, 10, e0114830. [Google Scholar] [CrossRef] [PubMed]
- Minniti, G.; Niyazi, M.; Alongi, F.; Navarria, P.; Belka, C. Current status and recent advances in reirradiation of glioblastoma. Radiat. Oncol. 2021, 16, 36. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.M.; Kim, J.Y.; Cho, H.J.; Lee, W.J.; Nguyen, D.; Kim, S.S.; Oh, Y.T.; Kim, H.J.; Jung, C.W.; Pinero, G.; et al. Tissue factor is a critical regulator of radiation therapy-induced glioblastoma remodeling. Cancer Cell 2023, 41, 1480–1497.e1489. [Google Scholar] [CrossRef] [PubMed]
- Yahyanejad, S.; King, H.; Iglesias, V.S.; Granton, P.V.; Barbeau, L.M.; van Hoof, S.J.; Groot, A.J.; Habets, R.; Prickaerts, J.; Chalmers, A.J.; et al. NOTCH blockade combined with radiation therapy and temozolomide prolongs survival of orthotopic glioblastoma. Oncotarget 2016, 7, 41251–41264. [Google Scholar] [CrossRef]
- Wu, W.; Klockow, J.L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H.E. Glioblastoma multiforme (GBM): An overview of current therapies and mechanisms of resistance. Pharmacol. Res. 2021, 171, 105780. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, B.; Cai, L.; Li, Y.; Huang, Y.; Zhou, Y.; Sun, X.; Yang, W.; Sun, T. G9a promotes immune suppression by targeting the Fbxw7/Notch pathway in glioma stem cells. CNS Neurosci. Ther. 2023, 29, 2508–2521. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Gu, S.; Chen, J.; Yuan, Z.; Liang, P.; Cui, H. Mechanism of Notch Signaling Pathway in Malignant Progression of Glioblastoma and Targeted Therapy. Biomolecules 2024, 14, 480. https://doi.org/10.3390/biom14040480
Wang S, Gu S, Chen J, Yuan Z, Liang P, Cui H. Mechanism of Notch Signaling Pathway in Malignant Progression of Glioblastoma and Targeted Therapy. Biomolecules. 2024; 14(4):480. https://doi.org/10.3390/biom14040480
Chicago/Turabian StyleWang, Shenghao, Sikuan Gu, Junfan Chen, Zhiqiang Yuan, Ping Liang, and Hongjuan Cui. 2024. "Mechanism of Notch Signaling Pathway in Malignant Progression of Glioblastoma and Targeted Therapy" Biomolecules 14, no. 4: 480. https://doi.org/10.3390/biom14040480