
Cell Senescence in Heterotopic Ossification



Abstract
1. Fibrodysplasia Ossificans Progressiva and Acquired Forms of Heterotopic Ossification
2. Lesion Formation in Fibrodysplasia Ossificans Progressiva and Non-Hereditary or Acquired Forms of Heterotopic Ossification
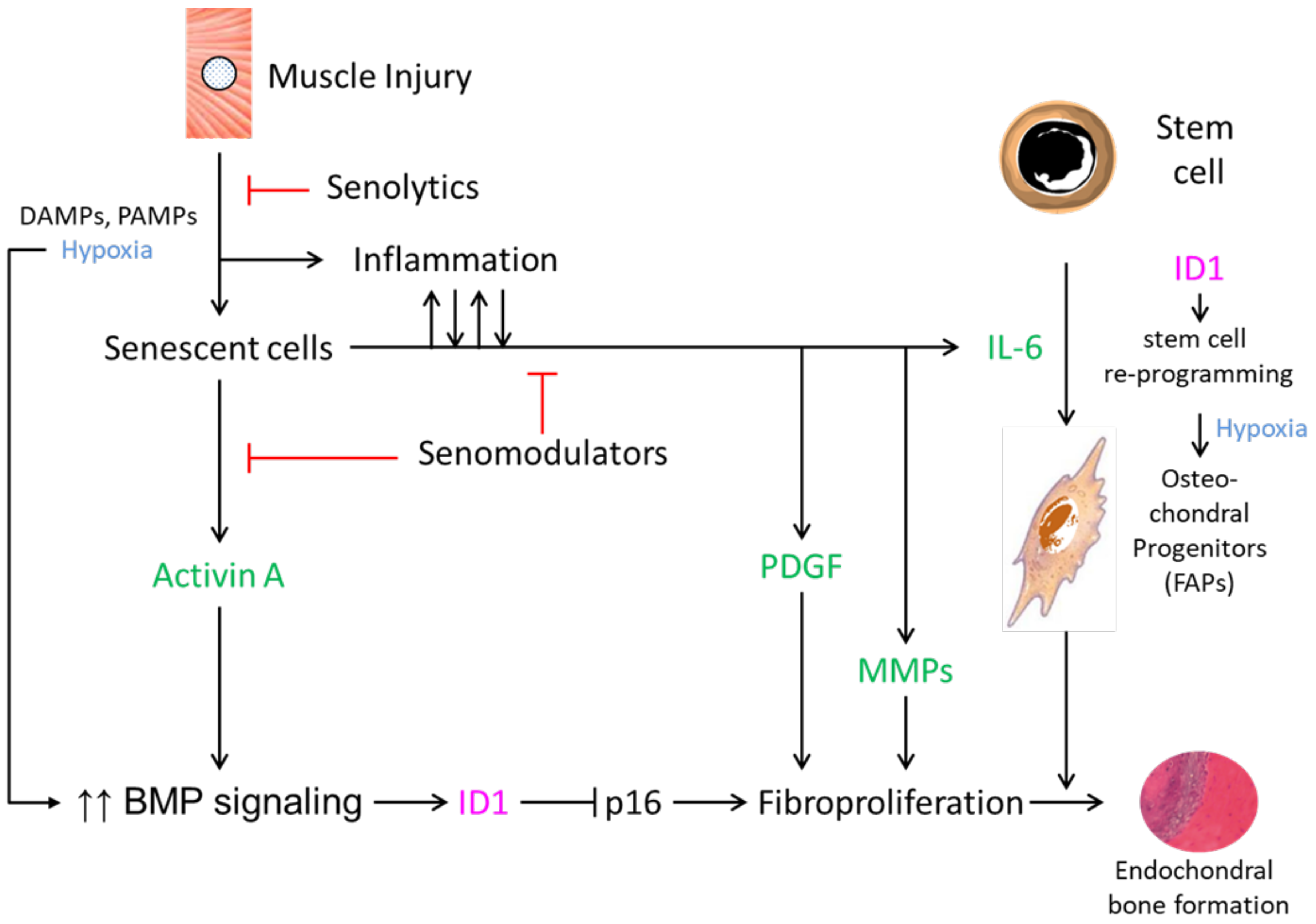
3. Possible Roles of Cellular Senescence in Heterotopic Ossification
4. Cellular Senescence and Urist’s Fundamental Concepts of HO Formation
5. Potential Use of Senotherapeutic Drugs in Disorders of Heterotopic Ossification
6. Challenges in the Clinical Use of Senotherapeutic Agents for Disorders of Heterotopic Ossification
7. Summary and Future Directions
Funding
Conflicts of Interest
References
- Pignolo, R.J.; Baujat, G.; Brown, M.A.; De Cunto, C.; Hsiao, E.C.; Keen, R.; Al Mukaddam, M.; Le Quan Sang, K.H.; Wilson, A.; Marino, R.; et al. The natural history of fibrodysplasia ossificans progressiva: A prospective, global 36-month study. Genet. Med. 2022, 24, 2422–2433. [Google Scholar] [CrossRef] [PubMed]
- Pignolo, R.J.; Shore, E.M.; Kaplan, F.S. Fibrodysplasia ossificans progressiva: Clinical and genetic aspects. Orphanet J. Rare Dis. 2011, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Pignolo, R.J.; Shore, E.M.; Kaplan, F.S. Fibrodysplasia ossificans progressiva: Diagnosis, management, and therapeutic horizons. Pediatr. Endocrinol. Rev. 2013, 10, 437–448. [Google Scholar] [PubMed]
- Pignolo, R.J.; Wang, H.; Kaplan, F.S. Fibrodysplasia Ossificans Progressiva (FOP): A Segmental Progeroid Syndrome. Front. Endocrinol. 2019, 10, 908. [Google Scholar] [CrossRef] [PubMed]
- Baujat, G.; Choquet, R.; Bouee, S.; Jeanbat, V.; Courouve, L.; Ruel, A.; Michot, C.; Le Quan Sang, K.H.; Lapidus, D.; Messiaen, C.; et al. Prevalence of fibrodysplasia ossificans progressiva (FOP) in France: An estimate based on a record linkage of two national databases. Orphanet J. Rare Dis. 2017, 12, 123. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.M.; Evans, D.A. Genetic aspects of fibrodysplasia ossificans progressiva. J. Med. Genet. 1982, 19, 35–39. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Hsiao, E.C.; Baujat, G.; Lapidus, D.; Sherman, A.; Kaplan, F.S. Prevalence of fibrodysplasia ossificans progressiva (FOP) in the United States: Estimate from three treatment centers and a patient organization. Orphanet J. Rare Dis. 2021, 16, 350. [Google Scholar] [CrossRef]
- Barruet, E.; Morales, B.M.; Cain, C.J.; Ton, A.N.; Wentworth, K.L.; Chan, T.V.; Moody, T.A.; Haks, M.C.; Ottenhoff, T.H.; Hellman, J.; et al. NF-kappaB/MAPK activation underlies ACVR1-mediated inflammation in human heterotopic ossification. JCI Insight 2018, 3, e122958. [Google Scholar] [CrossRef]
- Pignolo, R.J.; McCarrick-Walmsley, R.; Wang, H.; Qiu, S.; Hunter, J.; Barr, S.; He, K.; Zhang, H.; Kaplan, F.S. Plasma-Soluble Biomarkers for Fibrodysplasia Ossificans Progressiva (FOP) Reflect Acute and Chronic Inflammatory States. J. Bone Miner. Res. 2022, 37, 475–483. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Bedford-Gay, C.; Liljesthrom, M.; Durbin-Johnson, B.P.; Shore, E.M.; Rocke, D.M.; Kaplan, F.S. The Natural History of Flare-Ups in Fibrodysplasia Ossificans Progressiva (FOP): A Comprehensive Global Assessment. J. Bone Miner. Res. 2016, 31, 650–656. [Google Scholar] [CrossRef]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Xu, M.; Seemann, P.; Connor, J.M.; Glaser, D.L.; Carroll, L.; Delai, P.; Fastnacht-Urban, E.; Forman, S.J.; Gillessen-Kaesbach, G.; et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum. Mutat. 2009, 30, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Lounev, V.; Groppe, J.C.; Brewer, N.; Wentworth, K.L.; Smith, V.; Xu, M.; Schomburg, L.; Bhargava, P.; Al Mukaddam, M.; Hsiao, E.C.; et al. MMP-9 deficiency confers resilience in Fibrodysplasia Ossificans Progressiva in a man and mice. J. Bone Miner. Res. 2024, zjae029. [Google Scholar] [CrossRef]
- Garland, D.E.; Blum, C.E.; Waters, R.L. Periarticular heterotopic ossification in head-injured adults. Incidence and location. J. Bone Jt. Surg. 1980, 62, 1143–1146. [Google Scholar] [CrossRef]
- Neal, B.; Gray, H.; MacMahon, S.; Dunn, L. Incidence of heterotopic bone formation after major hip surgery. ANZ J. Surg. 2002, 72, 808–821. [Google Scholar] [CrossRef] [PubMed]
- Mohler, E.R., 3rd; Gannon, F.; Reynolds, C.; Zimmerman, R.; Keane, M.G.; Kaplan, F.S. Bone formation and inflammation in cardiac valves. Circulation 2001, 103, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.D.; Morales, D.V.; Welsh, C.H.; McDermott, M.T.; Schwarz, M.I. Calcium deposition with or without bone formation in the lung. Am. J. Respir. Crit. Care Med. 2002, 165, 1654–1669. [Google Scholar] [CrossRef]
- Kraft, C.T.; Agarwal, S.; Ranganathan, K.; Wong, V.W.; Loder, S.; Li, J.; Delano, M.J.; Levi, B. Trauma-induced heterotopic bone formation and the role of the immune system: A review. J. Trauma Acute Care Surg. 2016, 80, 156–165. [Google Scholar] [CrossRef]
- Moraes, J.R.; Moraes, F.R. Effect of a persistent inflammatory process on experimental heterotopic ossification. The influence of macrophages. Braz. J. Med. Biol. Res. 1993, 26, 53–66. [Google Scholar]
- Brennan, T.A.; Lindborg, C.M.; Bergbauer, C.R.; Wang, H.; Kaplan, F.S.; Pignolo, R.J. Mast cell inhibition as a therapeutic approach in fibrodysplasia ossificans progressiva (FOP). Bone 2018, 109, 259–266. [Google Scholar] [CrossRef]
- Convente, M.R.; Chakkalakal, S.A.; Yang, E.; Caron, R.J.; Zhang, D.; Kambayashi, T.; Kaplan, F.S.; Shore, E.M. Depletion of Mast Cells and Macrophages Impairs Heterotopic Ossification in an Acvr1(R206H) Mouse Model of Fibrodysplasia Ossificans Progressiva. J. Bone Miner. Res. 2018, 33, 269–282. [Google Scholar] [CrossRef]
- Egan, K.P.; Duque, G.; Keenan, M.A.; Pignolo, R.J. Circulating osteogentic precursor cells in non-hereditary heterotopic ossification. Bone 2018, 109, 61–64. [Google Scholar] [CrossRef]
- Egan, K.P.; Kim, J.H.; Mohler, E.R., 3rd; Pignolo, R.J. Role for circulating osteogenic precursor cells in aortic valvular disease. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2965–2971. [Google Scholar] [CrossRef][Green Version]
- Foley, K.L.; Hebela, N.; Keenan, M.A.; Pignolo, R.J. Histopathology of periarticular non-hereditary heterotopic ossification. Bone 2018, 109, 65–70. [Google Scholar] [CrossRef]
- Gannon, F.H.; Glaser, D.; Caron, R.; Thompson, L.D.; Shore, E.M.; Kaplan, F.S. Mast cell involvement in fibrodysplasia ossificans progressiva. Hum. Pathol. 2001, 32, 842–848. [Google Scholar] [CrossRef]
- Gannon, F.H.; Kaplan, F.S.; Olmsted, E.; Finkel, G.C.; Zasloff, M.A.; Shore, E. Bone morphogenetic protein 2/4 in early fibromatous lesions of fibrodysplasia ossificans progressiva. Hum. Pathol. 1997, 28, 339–343. [Google Scholar] [CrossRef]
- Gannon, F.H.; Valentine, B.A.; Shore, E.M.; Zasloff, M.A.; Kaplan, F.S. Acute lymphocytic infiltration in an extremely early lesion of fibrodysplasia ossificans progressiva. Clin. Orthop. Relat. Res. 1998, 346, 19–25. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Tabas, J.A.; Gannon, F.H.; Finkel, G.; Hahn, G.V.; Zasloff, M.A. The histopathology of fibrodysplasia ossificans progressiva. An endochondral process. J. Bone Jt. Surg. 1993, 75, 220–230. [Google Scholar] [CrossRef]
- Wang, H.; Lindborg, C.; Lounev, V.; Kim, J.H.; McCarrick-Walmsley, R.; Xu, M.; Mangiavini, L.; Groppe, J.C.; Shore, E.M.; Schipani, E.; et al. Cellular Hypoxia Promotes Heterotopic Ossification by Amplifying BMP Signaling. J. Bone Miner. Res. 2016, 31, 1652–1665. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Q.; Kaplan, F.S.; Pignolo, R.J. Clearance of Senescent Cells from Injured Muscle Abrogates Heterotopic Ossification in Mouse Models of Fibrodysplasia Ossificans Progressiva. J. Bone Miner. Res. 2022, 37, 95–107. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Passos, J.F.; Khosla, S.; Tchkonia, T.; Kirkland, J.L. Reducing Senescent Cell Burden in Aging and Disease. Trends Mol. Med. 2020, 26, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Chiche, A.; Le Roux, I.; von Joest, M.; Sakai, H.; Aguin, S.B.; Cazin, C.; Salam, R.; Fiette, L.; Alegria, O.; Flamant, P.; et al. Injury-Induced Senescence Enables In Vivo Reprogramming in Skeletal Muscle. Cell Stem Cell 2017, 20, 407–414.e404. [Google Scholar] [CrossRef] [PubMed]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marion, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Munoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef]
- Wang, H.; Shore, E.M.; Pignolo, R.J.; Kaplan, F.S. Activin A amplifies dysregulated BMP signaling and induces chondro-osseous differentiation of primary connective tissue progenitor cells in patients with fibrodysplasia ossificans progressiva (FOP). Bone 2018, 109, 218–224. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Al Mukaddam, M.; Pignolo, R.J. Acute unilateral hip pain in fibrodysplasia ossificans progressiva (FOP). Bone 2018, 109, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Urist, M.R. Bone: Formation by autoinduction. Science 1965, 150, 893–899. [Google Scholar] [CrossRef]
- Wang, H.; Behrens, E.M.; Pignolo, R.J.; Kaplan, F.S. ECSIT links TLR and BMP signaling in FOP connective tissue progenitor cells. Bone 2018, 109, 201–209. [Google Scholar] [CrossRef]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Alessi Wolken, D.M.; Idone, V.; Hatsell, S.J.; Yu, P.B.; Economides, A.N. The obligatory role of Activin A in the formation of heterotopic bone in Fibrodysplasia Ossificans Progressiva. Bone 2018, 109, 210–217. [Google Scholar] [CrossRef]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef]
- Hino, K.; Horigome, K.; Nishio, M.; Komura, S.; Nagata, S.; Zhao, C.; Jin, Y.; Kawakami, K.; Yamada, Y.; Ohta, A.; et al. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J. Clin. Investig. 2017, 127, 3339–3352. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef]
- Lees-Shepard, J.B.; Yamamoto, M.; Biswas, A.A.; Stoessel, S.J.; Nicholas, S.E.; Cogswell, C.A.; Devarakonda, P.M.; Schneider, M.J., Jr.; Cummins, S.M.; Legendre, N.P.; et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 2018, 9, 471. [Google Scholar] [CrossRef]
- Almalki, S.G.; Agrawal, D.K. Effects of matrix metalloproteinases on the fate of mesenchymal stem cells. Stem Cell Res. Ther. 2016, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- Ben David, D.; Reznick, A.Z.; Srouji, S.; Livne, E. Exposure to pro-inflammatory cytokines upregulates MMP-9 synthesis by mesenchymal stem cells-derived osteoprogenitors. Histochem. Cell Biol. 2008, 129, 589–597. [Google Scholar] [CrossRef]
- Hayashi, Y.; Hsiao, E.C.; Sami, S.; Lancero, M.; Schlieve, C.R.; Nguyen, T.; Yano, K.; Nagahashi, A.; Ikeya, M.; Matsumoto, Y.; et al. BMP-SMAD-ID promotes reprogramming to pluripotency by inhibiting p16/INK4A-dependent senescence. Proc. Natl. Acad. Sci. USA 2016, 113, 13057–13062. [Google Scholar] [CrossRef]
- Matsuo, K.; Lepinski, A.; Chavez, R.D.; Barruet, E.; Pereira, A.; Moody, T.A.; Ton, A.N.; Sharma, A.; Hellman, J.; Tomoda, K.; et al. ACVR1(R206H) extends inflammatory responses in human induced pluripotent stem cell-derived macrophages. Bone 2021, 153, 116129. [Google Scholar] [CrossRef]
- Agarwal, S.; Loder, S.; Cholok, D.; Li, J.; Breuler, C.; Drake, J.; Brownley, C.; Peterson, J.; Li, S.; Levi, B. Surgical Excision of Heterotopic Ossification Leads to Re-Emergence of Mesenchymal Stem Cell Populations Responsible for Recurrence. Stem Cells Transl. Med. 2017, 6, 799–806. [Google Scholar] [CrossRef]
- Mueller, A.A.; van Velthoven, C.T.; Fukumoto, K.D.; Cheung, T.H.; Rando, T.A. Intronic polyadenylation of PDGFRalpha in resident stem cells attenuates muscle fibrosis. Nature 2016, 540, 276–279. [Google Scholar] [CrossRef]
- Wosczyna, M.N.; Biswas, A.A.; Cogswell, C.A.; Goldhamer, D.J. Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J. Bone Miner. Res. 2012, 27, 1004–1017. [Google Scholar] [CrossRef]
- Davies, O.G.; Grover, L.M.; Lewis, M.P.; Liu, Y. PDGF is a potent initiator of bone formation in a tissue engineered model of pathological ossification. J. Tissue Eng. Regen. Med. 2018, 12, e355–e367. [Google Scholar] [CrossRef]
- Davis, E.L.; Sonnet, C.; Lazard, Z.W.; Henslee, G.; Gugala, Z.; Salisbury, E.A.; Strecker, E.V.; Davis, T.A.; Forsberg, J.A.; Davis, A.R.; et al. Location-dependent heterotopic ossification in the rat model: The role of activated matrix metalloproteinase 9. J. Orthop. Res. 2016, 34, 1894–1904. [Google Scholar] [CrossRef]
- Rodenberg, E.; Azhdarinia, A.; Lazard, Z.W.; Hall, M.; Kwon, S.K.; Wilganowski, N.; Salisbury, E.A.; Merched-Sauvage, M.; Olmsted-Davis, E.A.; Sevick-Muraca, E.M.; et al. Matrix metalloproteinase-9 is a diagnostic marker of heterotopic ossification in a murine model. Tissue Eng. Part A 2011, 17, 2487–2496. [Google Scholar] [CrossRef]
- Shi, W.Z.; Ju, J.Y.; Xiao, H.J.; Xue, F.; Wu, J.; Pan, M.M.; Ni, W.F. Dynamics of MMP-9, MMP-2 and TIMP-1 in a rat model of brain injury combined with traumatic heterotopic ossification. Mol. Med. Rep. 2017, 15, 2129–2135. [Google Scholar] [CrossRef]
- Vu, T.H.; Shipley, J.M.; Bergers, G.; Berger, J.E.; Helms, J.A.; Hanahan, D.; Shapiro, S.D.; Senior, R.M.; Werb, Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 1998, 93, 411–422. [Google Scholar] [CrossRef]
- Hari, P.; Millar, F.R.; Tarrats, N.; Birch, J.; Quintanilla, A.; Rink, C.J.; Fernandez-Duran, I.; Muir, M.; Finch, A.J.; Brunton, V.G.; et al. The innate immune sensor Toll-like receptor 2 controls the senescence-associated secretory phenotype. Sci. Adv. 2019, 5, eaaw0254. [Google Scholar] [CrossRef]
- Xu, M.; Tchkonia, T.; Kirkland, J.L. Perspective: Targeting the JAK/STAT pathway to fight age-related dysfunction. Pharmacol. Res. 2016, 111, 152–154. [Google Scholar] [CrossRef]
- Kasembeli, M.M.; Bharadwaj, U.; Robinson, P.; Tweardy, D.J. Contribution of STAT3 to Inflammatory and Fibrotic Diseases and Prospects for its Targeting for Treatment. Int. J. Mol. Sci. 2018, 19, 2299. [Google Scholar] [CrossRef]
- Madaro, L.; Passafaro, M.; Sala, D.; Etxaniz, U.; Lugarini, F.; Proietti, D.; Alfonsi, M.V.; Nicoletti, C.; Gatto, S.; De Bardi, M.; et al. Denervation-activated STAT3-IL-6 signalling in fibro-adipogenic progenitors promotes myofibres atrophy and fibrosis. Nat. Cell Biol. 2018, 20, 917–927. [Google Scholar] [CrossRef]
- Otero-Albiol, D.; Carnero, A. Cellular senescence or stemness: Hypoxia flips the coin. J. Exp. Clin. Cancer Res. 2021, 40, 243. [Google Scholar] [CrossRef]
- van Vliet, T.; Varela-Eirin, M.; Wang, B.; Borghesan, M.; Brandenburg, S.M.; Franzin, R.; Evangelou, K.; Seelen, M.; Gorgoulis, V.; Demaria, M. Physiological hypoxia restrains the senescence-associated secretory phenotype via AMPK-mediated mTOR suppression. Mol. Cell 2021, 81, 2041–2052.e2046. [Google Scholar] [CrossRef]
- Wang, H.; Kaplan, F.S.; Pignolo, R.J. The HIF-1alpha and mTOR Pathways Amplify Heterotopic Ossification. Biomolecules 2024, 14, 147. [Google Scholar] [CrossRef]
- Kim, E.C.; Kim, J.R. Senotherapeutics: Emerging strategy for healthy aging and age-related disease. BMB Rep. 2019, 52, 47–55. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. FEBS J. 2023, 290, 1362–1383. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Tchkonia, T.; Kirkland, J.L. Aging, Cell Senescence, and Chronic Disease: Emerging Therapeutic Strategies. JAMA 2018, 320, 1319–1320. [Google Scholar] [CrossRef]
- Lee, D.; Park, Y.H.; Lee, J.E.; Kim, H.S.; Min, K.Y.; Jo, M.G.; Kim, H.S.; Choi, W.S.; Kim, Y.M. Dasatinib Inhibits Lyn and Fyn Src-Family Kinases in Mast Cells to Suppress Type I Hypersensitivity in Mice. Biomol. Ther. 2020, 28, 456–464. [Google Scholar] [CrossRef]
- Moon, H.; Choi, H.H.; Lee, J.Y.; Moon, H.J.; Sim, S.S.; Kim, C.J. Quercetin inhalation inhibits the asthmatic responses by exposure to aerosolized-ovalbumin in conscious guinea-pigs. Arch. Pharmacal Res. 2008, 31, 771–778. [Google Scholar] [CrossRef]
- Nagai, K.; Takahashi, Y.; Mikami, I.; Fukusima, T.; Oike, H.; Kobori, M. The hydroxyflavone, fisetin, suppresses mast cell activation induced by interaction with activated T cell membranes. Br. J. Pharmacol. 2009, 158, 907–919. [Google Scholar] [CrossRef]
- Shishehbor, F.; Behroo, L.; Ghafouriyan Broujerdnia, M.; Namjoyan, F.; Latifi, S.M. Quercetin effectively quells peanut-induced anaphylactic reactions in the peanut sensitized rats. Iran. J. Allergy Asthma Immunol. 2010, 9, 27–34. [Google Scholar]
- Alexander, K.A.; Tseng, H.W.; Fleming, W.; Jose, B.; Salga, M.; Kulina, I.; Millard, S.M.; Pettit, A.R.; Genet, F.; Levesque, J.P. Inhibition of JAK1/2 Tyrosine Kinases Reduces Neurogenic Heterotopic Ossification After Spinal Cord Injury. Front. Immunol. 2019, 10, 377. [Google Scholar] [CrossRef]
- Lin, H.; Shi, F.; Jiang, S.; Wang, Y.; Zou, J.; Ying, Y.; Huang, D.; Luo, L.; Yan, X.; Luo, Z. Metformin attenuates trauma-induced heterotopic ossification via inhibition of Bone Morphogenetic Protein signalling. J. Cell. Mol. Med. 2020, 24, 14491–14501. [Google Scholar] [CrossRef]
- Chen, X.; Wang, S.; Xu, W.; Zhao, M.; Zhang, Y.; Xiao, H. Metformin Directly Binds to MMP-9 to Improve Plaque Stability. J. Cardiovasc. Dev. Dis. 2023, 10, 54. [Google Scholar] [CrossRef]
- Hwang, C.; Pagani, C.A.; Das, N.; Marini, S.; Huber, A.K.; Xie, L.; Jimenez, J.; Brydges, S.; Lim, W.K.; Nannuru, K.C.; et al. Activin A does not drive post-traumatic heterotopic ossification. Bone 2020, 138, 115473. [Google Scholar] [CrossRef]
- Li, J.; Sun, Z.; Luo, G.; Wang, S.; Cui, H.; Yao, Z.; Xiong, H.; He, Y.; Qian, Y.; Fan, C. Quercetin Attenuates Trauma-Induced Heterotopic Ossification by Tuning Immune Cell Infiltration and Related Inflammatory Insult. Front. Immunol. 2021, 12, 649285. [Google Scholar] [CrossRef]
- Peterson, J.R.; Eboda, O.N.; Brownley, R.C.; Cilwa, K.E.; Pratt, L.E.; De La Rosa, S.; Agarwal, S.; Buchman, S.R.; Cederna, P.S.; Morris, M.D.; et al. Effects of aging on osteogenic response and heterotopic ossification following burn injury in mice. Stem Cells Dev. 2015, 24, 205–213. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Al Mukaddam, M.; Baujat, G.; Brown, M.A.; De Cunto, C.; Hsiao, E.C.; Keen, R.; Le Quan Sang, K.H.; Grogan, D.R.; Marino, R.; et al. Study methodology and insights from the palovarotene clinical development program in fibrodysplasia ossificans progressiva. BMC Med. Res. Methodol. 2023, 23, 269. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Kaplan, F.S. Druggable targets, clinical trial design and proposed pharmacological management in fibrodysplasia ossificans progressiva. Expert Opin. Orphan Drugs 2020, 8, 101–109. [Google Scholar] [CrossRef]


{kind=link}
| Type | Class | Example(s) | Mechanism(s) of Action | Comments |
|---|---|---|---|---|
| Senolytics | Kinase inhibitors | Dasatinib (D) | SCAP trageting | Often used in combination (e.g., D + Q); mast cell stabilizer |
| HSP-90 inhibitors | Geldanamycin | Same as class | ||
| p53/p21 pathway modulators | FOXO4-DRI | Same as class | ||
| Turmeric derivatives | Curcumin and analogs | Downregulation of Nrf2 and NF-κB | ||
| Cardiac glycosides | Prosciliaridin A, Digoxin, Strophanthidin | Na+/K+ ATPase inhibition | May be limited by toxicities | |
| Galactose modified pro-drugs | SSK1, Nav-Gal | SA-β-galactosidase | ||
| PROTACs | PZ15227 | BCL-XL degadation | ||
| PPARα agonist | Fenofibrate | Same as class | ||
| Bioflavanoids | Fisetin, Quercetin (Q) | SCAP targeting (PI3K/AKT) | Mast cell stabilizer | |
| Antibiotics | Azithromycin | Autophagy and glycolysis inhibition | ||
| Senomorphics | NF-κB inhibitors | SR12343 | Same as class | |
| P38MAPK inhibitors | SB203580 | Same as class | ||
| JAK/STAT inhibitors | Ruxolitnib, tofacitinib | JAK 1/2 inhibitor | Infectious adverse events | |
| ATM inhibitors | KU-60019 | Same as class | ||
| Statins | Atorvastatin, Pravastatin | HMG-CoA reductase inhibitor | ||
| Rapalogs | Rapamycin | Regulation of mTOR, Nrf2, NF-κB | ||
| SIRT1 activator | Resveratrol | Same a s class | ||
| Biguanides | Metformin | Regulation of mTOR, Nrf2, NF-κB | Other pathways may also be involved | |
| Bioflavanoids | Apigenin | IRAK/NF-κB | ||
| Quercetin | Proteosome activator |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pignolo, R.J.; Kaplan, F.S.; Wang, H. Cell Senescence in Heterotopic Ossification. Biomolecules 2024, 14, 485. https://doi.org/10.3390/biom14040485
Pignolo RJ, Kaplan FS, Wang H. Cell Senescence in Heterotopic Ossification. Biomolecules. 2024; 14(4):485. https://doi.org/10.3390/biom14040485
Chicago/Turabian StylePignolo, Robert J., Frederick S. Kaplan, and Haitao Wang. 2024. "Cell Senescence in Heterotopic Ossification" Biomolecules 14, no. 4: 485. https://doi.org/10.3390/biom14040485
APA StylePignolo, R. J., Kaplan, F. S., & Wang, H. (2024). Cell Senescence in Heterotopic Ossification. Biomolecules, 14(4), 485. https://doi.org/10.3390/biom14040485