
Thrombopoietin, the Primary Regulator of Platelet Production: From Mythos to Logos, a Thirty-Year Journey

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Cloning of Thrombopoietin
3. The Interaction between Academia and Industry
4. Biochemical Characterization of Thrombopoietin and Its Receptor
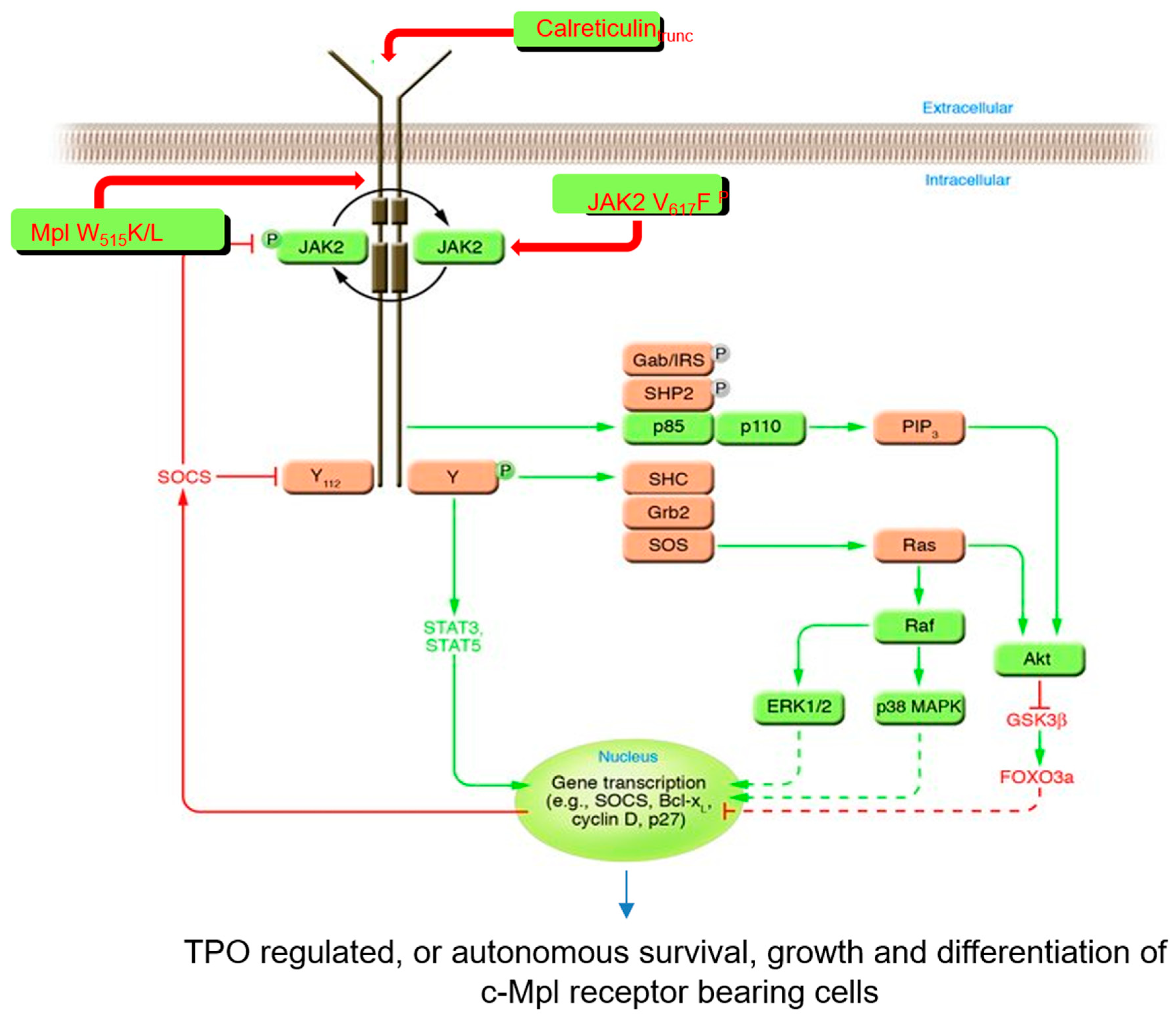
5. Thrombopoietin Signaling
6. The Relationship of Thrombopoietin and Other Hematopoietic Growth Factors
7. The Biological Activities of Thrombopoietin
8. The Thrombopoietin Receptor Is Involved in Myeloproliferative Neoplasms
9. A Mechanism by Which c-Mpl Participates in Hematopoietic Neoplasms
10. The Clinical Uses of Thrombopoietin and Thrombopoietin Receptor Agonists
Funding
Acknowledgments
Conflicts of Interest
References
- Carnot, P.; Deflandre, C. Sur l’activite hematopoietique des serum au cours de la regeneration du sang. Acad. Sci. M 1906, 3, 384. [Google Scholar]
- Pluznik, D.H.; Sachs, L. The cloning of normal mast cells in tissue culture. J. Cell. Comp. Physiol. 1965, 66, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Bradley, T.R.; Metcalf, D. The growth of mouse bone marrow cells in vitro. Aust. J. Exp. Biol. Med. Sci. 1966, 44, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Kung, S.K.; Goldwasser, E. Purification of human erythropoietin. J. Biol. Chem. 1977, 252, 5558–5564. [Google Scholar] [CrossRef]
- Goodwin, R.G.; Lupton, S.; Schmierer, A.; Hjerrild, K.J.; Jerzy, R.; Clevenger, W.; Gillis, S.; Cosman, D.; Namen, A.E. Human interleukin 7: Molecular cloning and growth factor activity on human and murine B-lineage cells. Proc. Natl. Acad. Sci. USA 1989, 86, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Souza, L.M.; Boone, T.C.; Gabrilove, J.; Lai, P.H.; Zsebo, K.M.; Murdock, D.C.; Chazin, V.R.; Bruszewski, J.; Lu, H.; Chen, K.K.; et al. Recombinant human granulocyte colony-stimulating factor: Effects on normal and leukemic myeloid cells. Science 1986, 232, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Ladner, M.B.; Martin, G.A.; Noble, J.A.; Wittman, V.P.; Warren, M.K.; McGrogan, M.; Stanley, E.R. cDNA cloning and exoression of murine macrophage colony-stimulating factor from L929 cells. Proc. Natl. Acad. Sci. USA 1988, 85, 6706–6710. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, T.; Konishi, M.; Mizuta, T.; Noma, T.; Honjo, T. Molecular cloning and structure of the human interleukin-5 gene. J. Biol. Chem. 1987, 262, 16580–16584. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.L.; Metcalf, D.; Nicola, N.A.; Johnson, G.R. Purification of a multipotential colony-stimulating factor from pokeweed mitogen-stimulated mouse spleen cell conditioned medium. J. Biol. Chem. 1985, 260, 6579–6587. [Google Scholar] [CrossRef]
- Martin, F.H.; Suggs, S.V.; Langley, K.E.; Lu, H.S.; Ting, J.; Okino, K.H.; Morris, C.F.; McNiece, I.K.; Jacobsen, F.W.; Mendiaz, E.A.; et al. Primary structure and functional expression of rat and human stem cell factor DNAs. Cell 1990, 63, 203–211. [Google Scholar] [CrossRef]
- Gough, N.M.; Gough, J.; Metcalf, D.; Kelso, A.; Grail, D.; Nicola, N.A.; Burgess, A.W.; Dunn, A.R. Molecular cloning of cDNA encoding a murine haematopoietic growth regulator, granulocyte-macrophage colony stimulating factor. Nature 1984, 309, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, E.; Cserhati, I.; Tanos, B. Demonstration and some properties of human thrombopoietin in thrombocythemic sera. Acta Haematol. 1958, 20, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Bartley, T.D.; Bogenberger, J.; Hunt, P.; Li, Y.-S.; Lu, H.-S.; Martin, F.; Chang, M.-S.; Samal, B.; Nichol, J.L.; Swift, S.; et al. Identification and cloning of a megakaryocyte growth and development factor that is a ligand for the cytokine receptor Mpl. Cell 1994, 77, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Sohma, Y.; Akahori, H.; Seki, N.; Hori, T.; Ogami, K.; Kato, T.; Shimada, Y.; Kawamura, K.; Miyazaki, H. Molecular cloning and chromosomal localization of the human thrombopoietin gene. FEBS Lett. 1994, 353, 57–61. [Google Scholar] [CrossRef]
- Wendling, F.; Varlet, P.; Charon, M.; Tambourin, P. A retrovirus complex inducing an acute myeloproliferative leukemia disorder in mice. Virology 1986, 149, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Vigon, I.; Mornon, J.P.; Cocault, L.; Mitjavila, I.T.; Tambourn, P.; Gisselbrecht, S.; Souyri, M. Molecular cloning and characterization of MPL, the human homolog of the v-mpl oncogene: Identification of a member of the hematopoietic growth factor receptor superfamily. Proc. Nati. Acad. Sci. USA 1992, 89, 5640–5644. [Google Scholar] [CrossRef]
- Martin, P.; Papayannopoulou, T. HEL cells: A new human erythroleukemia cell line with spontaneous and induced globin expression. Science 1982, 216, 1233–1235. [Google Scholar] [CrossRef]
- Lok, S.; Kaushansky, K.; Holly, R.D.; Kuijper, J.L.; Lofton-Day, C.E.; Oort, P.J.; Grant, F.J.; Heipel, M.D.; Burkhead, S.K.; Kramer, J.M.; et al. Cloning and expression of murine thrombopoietin cDNA and stimulation of platelet production in vivo. Nature 1994, 369, 565–568. [Google Scholar] [CrossRef]
- Kaushansky, K.; Lok, S.; Holly, R.D.; Broudy, V.C.; Lin, N.; Bailey, M.C.; Forstrom, J.W.; Buddle, M.; Oort, P.J.; Hagen, F.S.; et al. Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature 1994, 369, 568–571. [Google Scholar] [CrossRef]
- de Sauvage, F.J.; Hass, P.E.; Spencer, S.D.; Malloy, B.E.; Gurney, A.L.; Spencer, S.A.; Darbonne, W.C.; Henzel, W.J.; Wong, S.C.; Kuang, W.-J.; et al. Stimulation of megakaryocytopoiesis and thrombopoiesis by the c-Mpl ligand. Nature 1994, 369, 533–538. [Google Scholar] [CrossRef]
- Bazan, J.F. Unraveling the structure of IL-2. Science 1992, 257, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Brandhuber, B.J.; Boone, T.; Kenney, W.C.; McKay, D.B. Three dimensional structure of interleukin 2. Science 1987, 238, 1707–1709. [Google Scholar] [CrossRef] [PubMed]
- Feese, M.D.; Tamada, T.; Kato, Y.; Maeda, Y.; Hirose, M.; Matsukura, Y.; Shigematsu, H.; Muto, T.; Matsumoto, A.; Watarai, H.; et al. Structure of the receptor-binding domain of human thrombopoietin determined by complexation with a neutralizing antibody fragment. Proc. Natl. Acad. Sci. USA 2004, 101, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, R.; Hirose, M.; Kato, Y.; Feese, M.D.; Tamada, T.; Shigematsu, H.; Watarai, H.; Maeda, Y.; Tahara, T.; Kato, T.; et al. 2002 Crystallization of the functional domain of human thrombopoietin using an antigen-binding fragment derived from neutralizing monoclonal antibody. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 856–858. [Google Scholar] [CrossRef] [PubMed]
- Waters, M.J. The growth hormone receptor. Growth Horm. IGF Res. 2016, 28, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Uzé, G.; Mogensen, K.E.; Gresser, I.; Tovey, M.G.; Grütter, M.; Meyer, F. Biological activities and receptor binding of two human recombinant interferons and their hybrids. J. Gen. Virol. 1986, 67, 1633–1643. [Google Scholar] [CrossRef]
- Kaushansky, K.; Shoemaker, S.G.; Alfaro, S.; Brown, C. The hematopoietic activity of granulocyte-macrophage colony-stimulating factor is dependent upon two distinct regions of the molecule: A functional analysis based upon the activities of hybrid growth factors. Proc. Natl. Acad. Sci. USA 1989, 86, 1213–1217. [Google Scholar] [CrossRef] [PubMed]
- Hercus, T.R.; Cambareri, B.; Dottore, M.; Woodcock, J.; Bagley, C.J.; Vadas, M.A.; Shannon, M.F.; Lopez, A.F. Identification of residues in the first and fourth helices of human granulocyte-macrophage colony-stimulating factor involved in biologic activity and in binding to the alpha- and beta-chains of its receptor. Blood 1994, 83, 3500–3508. [Google Scholar] [CrossRef] [PubMed]
- Middleton, S.A.; Johnson, D.L.; Jin, R.; McMahon, F.J.; Collins, A.; Tullai, J.; Gruninger, R.H.; Jolliffe, L.K.; Mulcahy, L.S. Identification of a critical ligand binding determinant of the human erythropoietin receptor. Evidence for common ligand binding motifs in the cytokine receptor family. J. Biol. Chem. 1996, 271, 14045–14054. [Google Scholar] [CrossRef]
- Tsutsumi, N.; Masoumi, Z.; James, S.C.; Tucker, J.A.; Winkelmann, H.; Grey, W.; Picton, L.K.; Moss, L.; Wilson, S.C.; Caveney, N.A.; et al. Structure of the thrombopoietin-MPL receptor complex is a blueprint for biasing hematopoiesis. Cell 2023, 186, 4189–4203. [Google Scholar] [CrossRef]
- Hansen, G.; Hercus, T.R.; McClure, B.J.; Stomski, F.C.; Dottore, M.; Powell, J.; Ramshaw, H.; Woodcock, J.M.; Xu, Y.; Guthridge, M.; et al. The structure of the GM-CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell 2008, 134, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Ihle, J.N. Cytokine receptor signalling. Nature 1995, 377, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Wilks, A.F.; Harpur, A.G. Cytokine signal transduction and the JAK family of protein tyrosine kinases. Bioessays 1994, 16, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Remy, I.; Wilson, I.A.; Michnick, S.W. Erythropoietin receptor activation by a ligand-induced conformation change. Science 1999, 283, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Drachman, J.; Griffin, J.D.; Kaushansky, K. Stimulation of tyrosine kinase activity by MPL-ligand (thrombopoietin). J. Biol. Chem. 1995, 270, 4979–4982. [Google Scholar] [CrossRef] [PubMed]
- Kaushansky, A.; Kaushansky, K. Systems biology of megakaryocytes. Adv. Exp. Med. Biol. 2014, 844, 59–84. [Google Scholar] [PubMed]
- Kaushansky, K. The molecular mechanisms that control thrombopoiesis. J. Clin. Investig. 2005, 115, 3339–3347. [Google Scholar] [CrossRef] [PubMed]
- Linden, H.M.; Kaushansky, K. The glycan domain of thrombopoietin (TPO) acts in trans to enhance secretion of the hormone and other cytokines. J. Biol. Chem. 2002, 277, 35240–35247. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.S.; Hokom, M.M.; Chen, J.L.; Skrine, J.; Faust, J.; Nichol, J.; Hunt, P. The role of megakaryocyte growth and development factor in terminal stages of thrombopoiesis. Br. J. Haematol. 1996, 95, 227–233. [Google Scholar] [CrossRef]
- Kaushansky, K.; Broudy, V.C.; Grossmann, A.; Humes, J.; Lin, N.; Ren, H.-P.; Bailey, M.C.; Papayannopoulou Th Forstrom, J.W.; Sprugel, K.H. Thrombopoietin expands erythroid progenitors, increases red cell production, and enhances erythroid recovery after myelosuppressive therapy. J. Clin. Investig. 1995, 96, 1683–1687. [Google Scholar] [CrossRef]
- Sitnicka, E.; Lin, N.; Priestley, G.V.; Fox, N.; Broudy, V.C.; Wolf, N.S.; Kaushansky, K. The effect of thrombopoietin on the proliferation and differentiation of murine hematopoietic stem cells. Blood 1996, 87, 4998–5005. [Google Scholar] [CrossRef]
- Ku, H.; Yonemura, Y.; Kaushansky, K.; Ogawa, M. Thrombopoietin, the ligand for the Mpl receptor, synergizes with steel factor and other early-acting cytokines in supporting proliferation of primitive hematopoietic progenitors of mice. Blood 1996, 87, 4544–4551. [Google Scholar] [CrossRef] [PubMed]
- Zeigler, F.C.; de Sauvage, F.; Widmer, H.R.; Keller, G.A.; Donahue, C.; Schreiber, R.D.; Malloy, B.; Hass, P.; Eaton, D.; Matthews, W. In vitro megakaryocytopoietic and thrombopoietic activity of c-mpl ligand (TPO) on purified murine hematopoietic stem cells. Blood 1994, 84, 4045–4052. [Google Scholar] [CrossRef] [PubMed]
- Solar, G.P.; Kerr, W.G.; Zeigler, F.C.; Hess, D.; Donahue, C.; de Sauvage, F.J.; Eaton, D.L. Role of c-mpl in early hematopoiesis. Blood 1998, 92, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Ballmaier, M.; Germeshausen, M.; Krukemeier, S.; Welte, K. Thrombopoietin is essential for the maintenance of normal hematopoiesis in humans: Development of aplastic anemia in patients with congenital amegakaryocytic thrombocytopenia. Ann. N. Y. Acad. Sci. 2003, 996, 17–25. [Google Scholar] [CrossRef]
- Germeshausen, M.; Ballmaier, M. CAMT-MPL: Congenital amegakaryocytic thrombocytopenia caused by MPL mutations-heterogeneity of a monogenic disorder—A comprehensive analysis of 56 patients. Haematologica 2021, 106, 2439–2448. [Google Scholar] [CrossRef]
- Li, Y.; Hetet, G.; Kiladjian, J.J.; Gardin, C.; Grandchamp, B.; Briere, J. Proto-oncogene c-mpl is involved in spontaneous megakaryocytopoiesis in myeloproliferative disorders. Br. J. Haematol. 1996, 92, 60–66. [Google Scholar] [CrossRef]
- Prchal, J.F.; Axelrad, A.A. Letter: Bone-marrow responses in polycythemia vera. N. Engl. J. Med. 1974, 290, 1382. [Google Scholar]
- Casadevall, N.; Vainchenker, W.; Lacombe, C.; Vinci, G.; Chapman, J.; Breton-Gorius, J.; Varet, B. Erythroid progenitors in polycythemia vera: Demonstration of their hypersensitivity to erythropoietin using serum free cultures. Blood 1982, 59, 447–451. [Google Scholar] [CrossRef]
- Kimura, H.; Ishibashi, T.; Sato, T.; Matsuda, S.; Uchida, T.; Kariyone, S. Megakaryocytic colony formation (CFU-Meg) in essential thrombocythemia: Quantitative and qualitative abnormalities of bone marrow CFU-Meg. Am. J. Hematol. 1987, 24, 23–30. [Google Scholar]
- Kaushansky, K. The role of the MPL receptor in myeloproliferative disorders. Leukemia 1998, 12, S47–S50. [Google Scholar]
- Kaushansky, K. Etiology of the myeloproliferative disorders: The role of thrombopoietin. Sem. Hematol. 2003, 40, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Cocault, L.; Bouscary, D.; Le Bousse Kerdiles, C.; Clay, D.; Picard, F.; Gisselbrecht, S.; Souyri, M. Ectopic expression of murine TPO receptor (c-mpl) in mice is pathogenic and induces erythroblastic proliferation. Blood 1996, 88, 1656–1665. [Google Scholar] [CrossRef]
- Vigon, I.; Dreyfus, F.; Melle, J.; Viguie, F.; Ribrag, V.; Cocault, L.; Souyri, M.; Gisselbrecht, S. Expression of the c-mpl proto-oncogene in human hematologic malignancies. Blood 1993, 82, 877–883. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Plo, I.; Marty, C.; Varghese, L.N.; Constantinescu, S.N. The role of the thrombopoietin receptor MPL in myeloproliferative neoplasms: Recent findings and potential therapeutic applications. Expert Rev. Hematol. 2019, 12, 437–448. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Tiedt, R.; Hao-Shen, H.; Sobas, M.A.; Looser, R.; Dirnhofer, S.; Schwaller, J.; Skoda, R.C. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood 2008, 111, 3931–3940. [Google Scholar] [CrossRef]
- Sangkhae, V.; Etheridge, S.E.; Kaushansky, K.; Hitchcock, I.S. The thrombopoietin receptor, c-MPL, is critical for development of JAK2V617F-positive MPNs. Blood 2014, 124, 3956–3963. [Google Scholar] [CrossRef]
- Araki, M.; Yang, Y.; Masubuchi, N.; Hironaka, Y.; Takei, H.; Morishita, S.; Mizukami, Y.; Kan, S.; Shirane, S.; Edahiro, Y.; et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 2016, 127, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.I.; Marty, C.; Gryshkova, V.; Defour, J.P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 2016, 127, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Spivak, J.L.; Moliterno, A. The thrombopoietin receptor, MPL, is a therapeutic target of opportunity in the MPNs. Front. Oncol. 2021, 11, 641613. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef] [PubMed]
- Luque Paz, D.; Kralovics, R.; Skoda, R.C. Genetic basis and molecular profiling in myeloproliferative neoplasms. Blood 2023, 141, 1909–1921. [Google Scholar] [CrossRef]
- Li, J.; Yang, C.; Yea, X. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood 2001, 98, 3241–3248. [Google Scholar] [CrossRef] [PubMed]
- Basser, R.; O’Flaherty, E.; Green, M.; Edmonds, M.; Nichols, J. Development of pancytopenia with neutralizing antibodies to thrombopoietin after multicycle chemotherapy supported by megakaryocyte growth and development factor. Blood 2002, 99, 2599–2602. [Google Scholar] [CrossRef] [PubMed]
- Molineux, G.; Newland, A. Development of romiplostim for the treatment of patients with chronic immune thrombocytopenia: From bench to bedside. Br. J. Haematol. 2010, 150, 9–20. [Google Scholar] [CrossRef]
- Kuter, D.J.; Bussel, J.B.; Lyons, R.M.; Pullarkat, V.; Gernsheimer, T.B.; Senecal, F.M.; Aledort, L.M.; George, J.N.; Kessler, C.M.; Sanz, M.A.; et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: A double-blind randomised controlled trial. Lancet 2008, 371, 395–403. [Google Scholar] [CrossRef]
- Marshall, A.L.; Scarpone, R.; De Greef, M.; Bird, R.; Kuter, D.J. Remissions after long-term use of romiplostim for immune thrombocytopenia. Haematologica 2016, 101, e476–e478. [Google Scholar] [CrossRef]
- Cuker, A.; Despotovic, J.M.; Grace, R.F.; Kruse, C.; Lambert, M.P.; Liebman, H.A.; Lyons, R.M.; McCrae, K.R.; Pullarkat, V.; Wasser, J.S.; et al. Tapering thrombopoietin receptor agonists in primary immune thrombocytopenia: Expert consensus based on the RAND/UCLA modified Delphi panel method. Res. Pract. Thromb. Haemost. 2020, 5, 69–80. [Google Scholar] [CrossRef]
- Jenkins, J.M.; Williams, D.; Deng, Y.; Uhl, J.; Kitchen, V.; Collins, D.; Erickson-Miller, C.L. Phase 1 clinical study of eltrombopag, an oral, nonpeptide thrombopoietin receptor agonist. Blood 2007, 109, 4739–4741. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.N.; Bussel, J.B.; Cheng, G.; Meyer, O.; Bailey, C.K.; Arning, M.; Brainsky, A. EXTEND Study Group Safety and efficacy of eltrombopag for treatment of chronic immune thrombocytopenia: Results of the long-term, open-label EXTEND study. Blood 2013, 121, 537–545. [Google Scholar] [CrossRef]
- Townsley, D.M.; Scheinberg, P.; Winkler, T.; Desmond, R.; Dumitriu, B.; Rios, O.; Weinstein, B.; Valdez, J.; Lotter, J.; Feng, X.; et al. Eltrombopag Added to Standard Immunosuppression for Aplastic Anemia. N. Engl. J. Med. 2017, 376, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M.; McCafferty, E.H.; Blair, H.A. Lusutrombopag: A review in thrombocytopenia in patients with chronic liver disease prior to a scheduled procedure. Drugs 2019, 79, 1689–1695. [Google Scholar] [CrossRef] [PubMed]
- Terrault, N.; Chen, Y.C.; Izumi, N.; Kayali, Z.; Mitrut, P.; Tak, W.Y.; Allen, L.F.; Hassanein, T. Avatrombopag before procedures reduces need for platelet transfusion in patients with chronic liver disease and thrombocytopenia. Gastroenterology 2018, 155, 705–718. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Jiang, D.; Gernsheimer, T.; Liebman, H.; Lee, S.; Wojdyla, M.; Vredenburg, M.; Cuker, A. Adults with immune thrombocytopenia who switched to avatrombopag following prior treatment with eltrombopag or romiplostim: A multicentre US study. Br. J. Haematol. 2022, 197, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hu, Q.; Yang, C.; Chen, M.; Han, B. Comparison of Eltrombopag and avatrombopag in the treatment of refractory/relapsed aplastic anemia: A single-center retrospective study in China. Ther. Adv. Hematol. 2023, 14, 20406207231191310. [Google Scholar] [CrossRef]
- Kuter, D.J. Treatment of chemotherapy-induced thrombocytopenia in patients with non-hematologic malignancies. Haematologica 2022, 107, 1243–1263. [Google Scholar] [CrossRef]
- Al-Samkari, H. Thrombopoietin receptor agonists for chemotherapy-induced thrombocytopenia: A new solution for an old problem. Am. Soc. Hematol. Educ. Program 2022, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Goshua, G.; Sinha, P.; Kunst, N.; Pischel, L.; Lee, A.I.; Cuker, A. Cost-effectiveness of second-line therapies in adults with chronic immune thrombocytopenia. Am. J. Hematol. 2023, 98, 122–130. [Google Scholar] [CrossRef]
- Tremblay, G.; Dolph, M.; Roy, A.N.; Said, Q.; Forsythe, A. The Cost-effectiveness of Eltrombopag for the Treatment of Immune Thrombocytopenia in the United States. Clin. Ther. 2020, 42, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Mahévas, M.; Fain, O.; Ebbo, M.; Roudot-Thoraval, F.; Limal, N.; Khellaf, M.; Schleinitz, N.; Bierling, P.; Languille, L.; Godeau, B.; et al. The temporary use of thrombopoietin-receptor agonists may induce a prolonged remission in adult chronic immune thrombocytopenia. Results of a French observational study. Br. J. Hematol. 2014, 165, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Barlassina, A.; González-López, T.J.; Cooper, N.; Zaja, F. European Delphi panel to build consensus on tapering and discontinuing thrombopoietin receptor agonists in immune thrombocytopenia. Platelets 2023, 34, 2170999. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaushansky, K. Thrombopoietin, the Primary Regulator of Platelet Production: From Mythos to Logos, a Thirty-Year Journey. Biomolecules 2024, 14, 489. https://doi.org/10.3390/biom14040489
Kaushansky K. Thrombopoietin, the Primary Regulator of Platelet Production: From Mythos to Logos, a Thirty-Year Journey. Biomolecules. 2024; 14(4):489. https://doi.org/10.3390/biom14040489
Chicago/Turabian StyleKaushansky, Kenneth. 2024. "Thrombopoietin, the Primary Regulator of Platelet Production: From Mythos to Logos, a Thirty-Year Journey" Biomolecules 14, no. 4: 489. https://doi.org/10.3390/biom14040489
APA StyleKaushansky, K. (2024). Thrombopoietin, the Primary Regulator of Platelet Production: From Mythos to Logos, a Thirty-Year Journey. Biomolecules, 14(4), 489. https://doi.org/10.3390/biom14040489