

Brain-Derived Neurotrophic Factor, Nociception, and Pain


Abstract
:1. Introduction
1.1. A Brief Overview of BDNF Functions in the Normal and Pathologic Nervous System and the Cellular Processing of BDNF
1.2. Pain as a Complex and Multifaceted Sensory Experience

2. Role of BDNF in Nociception and Pain: Insights and Mechanisms
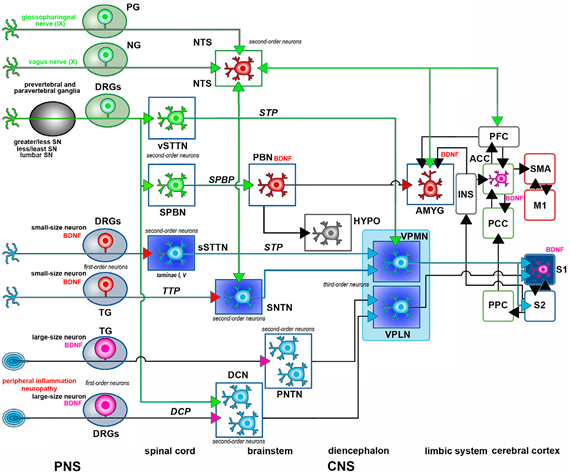
2.1. BDNF in Pain Pathways Neurons
2.1.1. Primary Sensory Neurons
2.1.2. Second-Order Neurons
2.1.3. Higher-Order Neurons
Brainstem
Amygdala
Cerebral Cortex

{kind=link}
{kind=link}
{kind=link}
| Location | Type of Neurons | Function | Ref |
|---|---|---|---|
| DRGs | Small- to medium-size PSNs | Nociception | [54,55,56] |
| DRGs | Small- to medium-size PSNs | Inflammatory nociception | [78] |
| JG (X) | Small- to medium-size PSNs | Nociception | [40] |
| TG (V) | Small- to medium-size PSNs | Nociception | [64,65] |
| DRGs | Large-size PSNs | Nociception (after injury) | [59] |
| SNTN | SSNs | Localization study | [33] |
| CN | SSNs | Localization study | [33] |
| PBN | Projection neurons | Nociception | [33] |
| S1 | Cortical neurons | Inflammatory/neuropathic pain | [74] |
| Au1 | Cortical neurons | Chronic pain | [76] |
| ACC | Cortical neurons | Inflammatory/neuropathic pain | [74,75] |
| Location | Type of Neurons | Function | Ref |
|---|---|---|---|
| DRGs | Small- to medium-size PSNs | Nociception | [79] |
| NG (X) | Small- to medium-size PSNs | Chemoafferent neurons | [40,69] |
| PG (IX) | Small- to medium-size PSNs | Chemoafferent neurons | [40,69] |
| NTS | Small- to medium-size SSNs | Modulation of cardiovascular afferents | [33,70] |
| AMY | Projection neurons to PBN | Hyperalgesia | [73] |
2.2. Neuronal Mechanisms
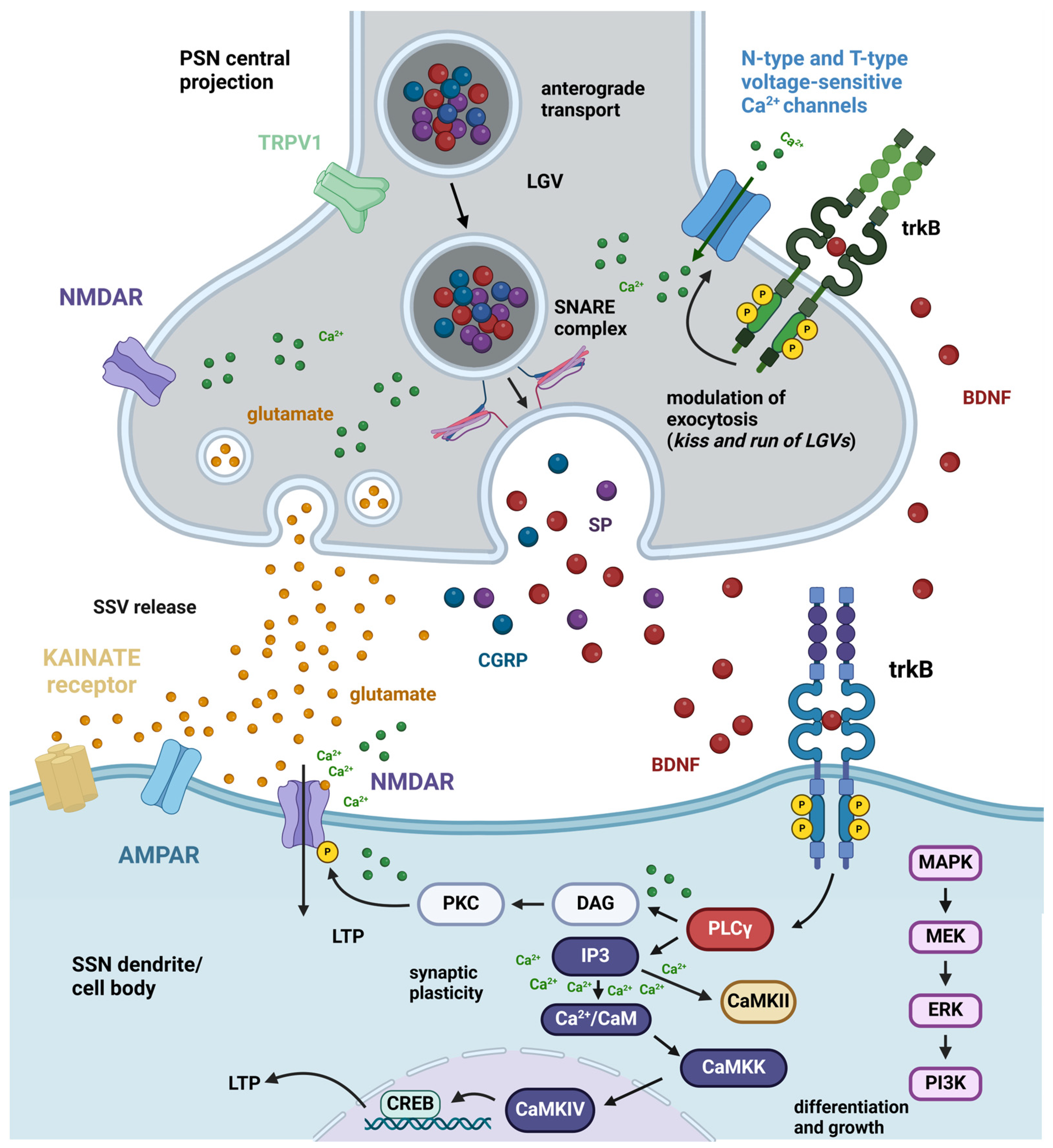
2.2.1. Ex Vivo Studies on Nociceptive/Pain Modulation by BDNF
2.2.2. In Vivo Studies on Inflammatory Pain Modulation by BDNF and proBDNF
BDNF
proBDNF
2.2.3. In Vivo Studies on Neuropathic Pain Modulation by BDNF
- DRG/Spinal cord
Trigeminal System
2.3. BDNF in Glia and Other Cells of Relevance in Nociceptive/Pain Pathways—Localization and Mechanisms
2.3.1. Glial Cells
- Schwann Cells
- Microglia
- Astrocytes
- Oligodendroglia
2.3.2. Immune Cells
2.4. BDNF Polymorphisms Related to Pain Signaling
3. BDNF and Neuronal Sensitization
3.1. Peripheral Sensitization
3.2. Central Sensitization
3.3. Hyperalgesic Priming in the Spinal Cord Dorsal Horn
4. Implications for Pain Management
5. Clinical Trials
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| 1NMP | 1-(1, 1-dimethyl ethyl)-3-(1-naphthalenyl methyl)-1H-pyrazole [3,4-d] pyrimidine-4-amine |
| ACC | anterior cingulate cortex |
| ALS | amyotrophic lateral sclerosis |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor |
| AMYG | amygdala |
| APC | adenomatous polyposis coli tumor suppressor protein |
| aPKC | atypical isoform of PKC |
| Au1 | primary auditory area of the cerebral cortex |
| BBB | blood-brain barrier |
| BDNF | brain-derived neurotrophic factor |
| B-MEPS | brief measure of emotional preoperative stress |
| [Ca++]i | intracellular calcium concentration |
| CAMK | calcium/calmodulin-stimulated protein kinase |
| CaMKII | calcium/calmodulin-dependent protein kinase II |
| CaMKIV | calcium/calmodulin-dependent protein kinase IV |
| CCI | chronic constriction injury |
| CCL2 | chemokine (C-C motif) ligand 2 |
| CCR2 | C-C chemokine receptor type 2 |
| CFA | complete Freund adjuvant |
| CGRP | calcitonin gene-related peptide |
| C-LTMRs | C- low-threshold mechanoreceptors |
| CN | cuneiform nucleus |
| CNS | central nervous system |
| CPM | conditional pain modulation |
| COX2 | cyclooxygenase-2 |
| CREB | cAMP response element-binding protein |
| CSF-1 | colony-stimulating factor 1 |
| CTTH | chronic tension-type headache |
| DAG | diacylglycerol |
| D-APV | D-2-amino-5-phosphonovaleric acid |
| DCP | dorsal column pathway |
| DREAM | downstream regulatory element antagonist modulator |
| DRGs | dorsal root ganglia |
| EAAT3 | excitatory amino acid transporter 3 |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| GABA | γ-amino-butyric acid |
| GABAAR | γ-amino-butyric acid receptor A |
| GDNF | glial-derived neurotrophic factor |
| GFAP | glial fibrillary acidic protein |
| GLUN2B-NMDA | NMDA receptor GluN2B |
| GlyR | glycine receptor |
| HYPO | hypothalamus |
| IAN | inferior alveolar nerve |
| Iba1 | ionized calcium-binding adaptor molecule 1 |
| IL-10 | interleukin-10 |
| IL-1β | interleukin-1β |
| IL-6 | interleukin-6 |
| INS | insular cortex |
| IP3 | inositol 1,4,5-trisphosphate |
| JG | jugular ganglion |
| KCC2 | K-Cl co-transporter 2 |
| LGV | large granular vesicle |
| LTD | long-term depression |
| LTP | long-term potentiation |
| MAO-B | type-B monoamine oxidase |
| MAPK | mitogen-activated protein kinase |
| mEPSC | miniature excitatory postsynaptic currents |
| mIPSC | miniature inhibitory postsynaptic currents |
| mTORC1 | mammalian target of rapamycin complex 1 |
| NaV1.8 | voltage-gated sodium channel 1.8 |
| NF-κB | nuclear factor-kappa B |
| NG | nodose ganglion |
| NGF | nerve growth factor |
| NMDA | N-methyl-d-aspartate |
| NMDAR | N-methyl-d-aspartate receptor |
| NO | nitric oxide |
| NT-3 | neurotrophin 3 |
| NT-4/5 | neurotrophin-4/5 |
| NTS | nucleus tractus solitarius |
| P2RX4 | ATP-gated purinergic receptor 4 |
| p75NTR | low-affinity p75 pan neurotrophin receptor |
| PAC1R | PACAP type I receptor |
| PACAP | pituitary adenylate cyclase-activating peptide |
| PAG | periaqueductal gray |
| PAR2 | proteinase-activated receptor 2 |
| PBN | parabrachial nucleus |
| PFC | prefrontal cortex |
| PG | petrosal ganglion |
| PGE2 | prostaglandin E2 |
| PI3K | phosphoinositide 3-kinase |
| PKA | protein kinase A |
| PKC | protein kinase C |
| PKCε | protein kinase type C ε |
| PLC | phospholipase C |
| PLCγ | phospholipase C γ |
| PNS | peripheral nervous system |
| proBDNF | BDNF pro-protein |
| PSN | primary sensory neuron |
| rTMS | repetitive transcranial magnetic stimulation |
| RvD1 | resolvin D1 |
| S1 | first-order somatosensory area of the cerebral cortex |
| S2 | second-order somatosensory area of the cerebral cortex |
| SCI | spinal cord injury |
| sEPSC | spontaneous excitatory postsynaptic currents |
| SHP2 | Src homology-2 domain-containing protein tyrosine phosphatase-2 |
| sIPSC | spontaneous inhibitory postsynaptic currents |
| SNARE | soluble N-ethylmaleimide-sensitive-factor attachment protein receptor |
| SNL | spinal nerve ligation |
| SNTN | spinal nucleus of the trigeminal nerve |
| SorCS2 | sortilin-related VPS10 domain-containing receptor 2 |
| SPBP | spinoparabrachial pathway |
| SSN | somatic sensory neuron |
| STP | spinothalamic pathway |
| tDCS | transcranial direct current stimulation |
| TENS | transcutaneous electrical nerve stimulation |
| TET1 | ten-eleven translocation methylcytosine dioxygenase |
| TG | trigeminal ganglion |
| TGN | trans-Golgi network |
| TNFα | tumor necrosis α |
| trkB | tropomyosin receptor kinase B |
| trkB.T1 | truncated isoform 1 of trkB |
| TRP | transient receptor potential |
| TRPA1 | transient receptor potential cation channel, subfamily A, member 1 |
| TRPV1 | transient receptor potential vanilloid receptor 1 |
| TTP | trigeminothalamic pathway |
| VPLN | ventroposterior lateral nucleus of the thalamus |
| VPMN | ventroposterior medial nucleus of the thalamus |
| WDR | wide dynamic range |
| ZIP | Zrt, Irt-like proteins |
References
- Ateaque, S.; Merkouris, S.; Barde, Y.-A. Neurotrophin signalling in the human nervous system. Front. Mol. Neurosci. 2023, 16, 1225373. [Google Scholar] [CrossRef]
- Wang, C.S.; Kavalali, E.T.; Monteggia, L.M. BDNF signaling in context: From synaptic regulation to psychiatric disorders. Cell 2021, 185, 62–76. [Google Scholar] [CrossRef]
- Lu, B.; Nagappan, G.; Lu, Y. BDNF and Synaptic Plasticity, Cognitive Function, and Dysfunction. In Neurotrophic Factors; Lewin, G.R., Carter, B.D., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 223–250. [Google Scholar]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Palasz, E.; Wysocka, A.; Gasiorowska, A.; Chalimoniuk, M.; Niewiadomski, W.; Niewiadomska, G. BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1170. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of Huntingtin-Mediated BDNF Gene Transcription in Huntington’s Disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.-Y.A.; Byrne, L.M.; Rodrigues, F.B.; Tortelli, R.; Johnson, E.B.; Foiani, M.S.; Arridge, M.; De Vita, E.; Scahill, R.I.; Heslegrave, A.; et al. Brain-derived neurotrophic factor in cerebrospinal fluid and plasma is not a biomarker for Huntington’s disease. Sci. Rep. 2021, 11, 3481. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, J.; Noakes, P.G.; Bellingham, M.C. The Role of Altered BDNF/TrkB Signaling in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2019, 13, 368. [Google Scholar] [CrossRef] [PubMed]
- Riolo, G.; Ricci, C.; De Angelis, N.; Marzocchi, C.; Guerrera, G.; Borsellino, G.; Giannini, F.; Battistini, S. BDNF and Pro-BDNF in Amyotrophic Lateral Sclerosis: A New Perspective for Biomarkers of Neurodegeneration. Brain Sci. 2022, 12, 617. [Google Scholar] [CrossRef]
- Duman, R.S.; Monteggia, L.M. A Neurotrophic Model for Stress-Related Mood Disorders. Biol. Psychiatry 2006, 59, 1116–1127. [Google Scholar] [CrossRef]
- Martinowich, K.; Manji, H.; Lu, B. New insights into BDNF function in depression and anxiety. Nat. Neurosci. 2007, 10, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Arévalo, J.C.; Deogracias, R. Mechanisms Controlling the Expression and Secretion of BDNF. Biomolecules 2023, 13, 789. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Siao, C.-J.; Nagappan, G.; Marinic, T.; Jing, D.; McGrath, K.; Chen, Z.-Y.; Mark, W.; Tessarollo, L.; Lee, F.S.; et al. Neuronal release of proBDNF. Nat. Neurosci. 2009, 12, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Zagrebelsky, M.; Korte, M. Form follows function: BDNF and its involvement in sculpting the function and structure of synapses. Neuropharmacology 2014, 76, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Larsen, R.S.; Philpot, B.D.; Paulsen, O. Roles of Presynaptic NMDA Receptors in Neurotransmission and Plasticity. Trends Neurosci. 2015, 39, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Raja, S.N.; Carr, D.B.; Cohen, M.; Finnerup, N.B.; Flor, H.; Gibson, S.; Keefe, F.J.; Mogil, J.S.; Ringkamp, M.; Sluka, K.A.; et al. The revised International Association for the Study of Pain definition of pain: Concepts, challenges, and compromises. Pain 2020, 161, 1976–1982. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, S.A.; Herr, M.J. Physiology, Nociception. In StatPearls; StatPearls Publishing Copyright © 2023; StatPearls Publishing LLC: Treasure Island, FL, USA, 2023. [Google Scholar]
- Dubin, A.E.; Patapoutian, A. Nociceptors: The sensors of the pain pathway. J. Clin. Investig. 2010, 120, 3760–3772. [Google Scholar] [CrossRef] [PubMed]
- Frias, B.; Merighi, A. Capsaicin, Nociception and Pain. Molecules 2016, 21, 797. [Google Scholar] [CrossRef]
- Vogt, B.A. Pain and emotion interactions in subregions of the cingulate gyrus. Nat. Rev. Neurosci. 2005, 6, 533–544. [Google Scholar] [CrossRef]
- Merighi, A. Neuropeptides and Coexistence. In Neuroscience and Biobehavioral Psychology; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Grichnik, K.P.; Ferrante, F.M. The difference between acute and chronic pain. Mt. Sinai J. Med. N. Y. 1991, 58, 217–220. [Google Scholar]
- Merighi, A.; Salio, C.; Ghirri, A.; Lossi, L.; Ferrini, F.; Betelli, C.; Bardoni, R. BDNF as a pain modulator. Prog. Neurobiol. 2008, 85, 297–317. [Google Scholar] [CrossRef] [PubMed]
- Vardeh, D.; Naranjo, J.F. Peripheral and Central Sensitization. In Pain Medicine: An Essential Review; Yong, R.J., Nguyen, M., Nelson, E., Urman, R.D., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 15–17. [Google Scholar]
- Li, C.; Kim, H.J.; Back, S.K.; Na, H.S. Common and discrete mechanisms underlying chronic pain and itch: Peripheral and central sensitization. Pflüg. Arch. Eur. J. Physiol. 2021, 473, 1603–1615. [Google Scholar] [CrossRef] [PubMed]
- Seidel, M.F.; Hügle, T.; Morlion, B.; Koltzenburg, M.; Chapman, V.; MaassenVanDenBrink, A.; Lane, N.E.; Perrot, S.; Zieglgänsberger, W. Neurogenic inflammation as a novel treatment target for chronic pain syndromes. Exp. Neurol. 2022, 356, 114108. [Google Scholar] [CrossRef]
- Sikandar, S.; Minett, M.S.; Millet, Q.; Santana-Varela, S.; Lau, J.; Wood, J.N.; Zhao, J. Brain-derived neurotrophic factor derived from sensory neurons plays a critical role in chronic pain. Brain 2018, 141, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Altar, C.A.; Cai, N.; Bliven, T.; Juhasz, M.; Conner, J.M.; Acheson, A.L.; Lindsay, R.M.; Wiegand, S.J. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 1997, 389, 856–860. [Google Scholar] [CrossRef]
- von Bartheld, C.S.; Wang, X.; Butowt, R. Anterograde axonal transport, transcytosis, and recycling of neurotrophic factors: The concept of trophic currencies in neural networks. Mol. Neurobiol. 2001, 24, 1–28. [Google Scholar] [CrossRef]
- Zhou, X.; Song, X.; Zhong, J.; Barati, S.; Zhou, F.H.; Johnson, S.M. Distribution and localization of pro-brain-derived neurotrophic factor-like immunoreactivity in the peripheral and central nervous system of the adult rat. J. Neurochem. 2004, 91, 704–715. [Google Scholar] [CrossRef]
- Conner, J.M.; Lauterborn, J.C.; Yan, Q.; Gall, C.M.; Varon, S. Distribution of Brain-Derived Neurotrophic Factor (BDNF) Protein and mRNA in the Normal Adult Rat CNS: Evidence for Anterograde Axonal Transport. J. Neurosci. 1997, 17, 2295–2313. [Google Scholar] [CrossRef]
- Kohara, K.; Kitamura, A.; Morishima, M.; Tsumoto, T. Activity-Dependent Transfer of Brain-Derived Neurotrophic Factor to Postsynaptic Neurons. Science 2001, 291, 2419–2423. [Google Scholar] [CrossRef]
- Kerr, B.J.; Bradbury, E.J.; Bennett, D.L.H.; Trivedi, P.M.; Dassan, P.; French, J.; Shelton, D.B.; McMahon, S.B.; Thompson, S.W.N. Brain-Derived Neurotrophic Factor Modulates Nociceptive Sensory Inputs and NMDA-Evoked Responses in the Rat Spinal Cord. J. Neurosci. 1999, 19, 5138–5148. [Google Scholar] [CrossRef]
- Michael, G.J.; Averill, S.; Nitkunan, A.; Rattray, M.; Bennett, D.L.H.; Yan, Q.; Priestley, J.V. Nerve Growth Factor Treatment Increases Brain-Derived Neurotrophic Factor Selectively in TrkA-Expressing Dorsal Root Ganglion Cells and in Their Central Terminations within the Spinal Cord. J. Neurosci. 1997, 17, 8476–8490. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, M. Recent advances in understanding context-dependent mechanisms controlling neurotrophin signaling and function. F1000Research 2019, 8, 1658. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-F.; Rush, R. Endogenous brain-derived neurotrophic factor is anterogradely transported in primary sensory neurons. Neuroscience 1996, 74, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-F.; Chie, E.; Rush, R. Distribution of Brain-Derived Neurotrophic Factor in Cranial and Spinal Ganglia. Exp. Neurol. 1998, 149, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, H.; Terayama, R.; Yamaai, T.; Yan, Z.; Sugimoto, T. Brain-derived neurotrophic factor-immunoreactive neurons in the rat vagal and glossopharyngeal sensory ganglia; co-expression with other neurochemical substances. Brain Res. 2007, 1155, 93–99. [Google Scholar] [CrossRef]
- Price, J. An immunohistochemical and quantitative examination of dorsal root ganglion neuronal subpopulations. J. Neurosci. 1985, 5, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Lallemend, F.; Ernfors, P. Molecular interactions underlying the specification of sensory neurons. Trends Neurosci. 2012, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Cai, B.; Song, Y.; Chen, Y.; Zhang, X. Somatosensory neuron types and their neural networks as revealed via single-cell transcriptomics. Trends Neurosci. 2023, 46, 654–666. [Google Scholar] [CrossRef]
- Vermeiren, S.; Bellefroid, E.J.; Desiderio, S. Vertebrate Sensory Ganglia: Common and Divergent Features of the Transcriptional Programs Generating Their Functional Specialization. Front. Cell Dev. Biol. 2020, 8, 587699. [Google Scholar] [CrossRef]
- Capra, N.F.; Wax, T.D. Distribution and central projections of primary afferent neurons that innervate the masseter muscle and mandibular periodontium: A double-label study. J. Comp. Neurol. 1989, 279, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Cobo, J.; Solé-Magdalena, A.; Menéndez, I.; de Vicente, J.; Vega, J. Connections between the facial and trigeminal nerves: Anatomical basis for facial muscle proprioception. JPRAS Open 2017, 12, 9–18. [Google Scholar] [CrossRef]
- Delfini, M.-C.; Mantilleri, A.; Gaillard, S.; Hao, J.; Reynders, A.; Malapert, P.; Alonso, S.; François, A.; Barrere, C.; Seal, R.; et al. TAFA4, a Chemokine-like Protein, Modulates Injury-Induced Mechanical and Chemical Pain Hypersensitivity in Mice. Cell Rep. 2013, 5, 378–388. [Google Scholar] [CrossRef]
- Emery, E.C.; Ernfors, P. Dorsal Root Ganglion Neuron Types and Their Functional Specialization. In The Oxford Handbook of the Neurobiology of Pain; Wood, J.N., Ed.; Oxford University Press: London, UK, 2020; pp. 1–30. [Google Scholar]
- Kupari, J.; Häring, M.; Agirre, E.; Castelo-Branco, G.; Ernfors, P. An Atlas of Vagal Sensory Neurons and Their Molecular Specialization. Cell Rep. 2019, 27, 2508–2523.e4. [Google Scholar] [CrossRef] [PubMed]
- Averill, S.; McMahon, S.B.; Clary, D.O.; Reichardt, L.F.; Priestley, J.V. Immunocytochemical Localization of trkA Receptors in Chemically Identified Subgroups of Adult Rat Sensory Neurons. Eur. J. Neurosci. 1995, 7, 1484–1494. [Google Scholar] [CrossRef] [PubMed]
- Lawson, S.N. Phenotype and Function of Somatic Primary Afferent Nociceptive Neurones with C-, Aδ- or Aα/β-Fibres. Exp. Physiol. 2002, 87, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Merighi, A.; Polak, J.M.; Gibson, S.J.; Gulbenkian, S.; Valentino, K.L.; Peirone, S.M. Ultrastructural studies on calcitonin gene-related peptide-, tachykinins- and somatostatin-immunoreactive neurones in rat dorsal root ganglia: Evidence for the colocalisation of different peptides in single secretory granules. Cell Tissue Res. 1988, 254, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Merighi, A. The histology, physiology, neurochemistry, and circuitry of the substantia gelatinosa Rolandi (lamina II) in the mammalian spinal cord. Prog. Neurobiol. 2018, 169, 91–134. [Google Scholar] [CrossRef] [PubMed]
- Salio, C.; Lossi, L.; Ferrini, F.; Merighi, A. Ultrastructural evidence for a pre- and post-synaptic localization of full-length trkB receptors in substantia gelatinosa (lamina II) of rat and mouse spinal cord. Eur. J. Neurosci. 2005, 22, 1951–1966. [Google Scholar] [CrossRef]
- Salio, C.; Averill, S.; Priestley, J.; Merighi, A. Costorage of BDNF and neuropeptides within individual dense-core vesicles in central and peripheral neurons. Dev. Neurobiol. 2007, 67, 326–338. [Google Scholar] [CrossRef]
- Merighi, A.; Bardoni, R.; Salio, C.; Lossi, L.; Ferrini, F.; Prandini, M.; Zonta, M.; Gustincich, S.; Carmignoto, G. Presynaptic functional trkB receptors mediate the release of excitatory neurotransmitters from primary afferent terminals in lamina II (substantia gelatinosa) of postnatal rat spinal cord. Dev. Neurobiol. 2008, 68, 457–475. [Google Scholar] [CrossRef] [PubMed]
- Salio, C.; Ferrini, F. BDNF and GDNF expression in discrete populations of nociceptors. Ann. Anat.-Anat. Anz. 2015, 207, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.-M.; Gulick, M.A.; Yu, S.J.; Grider, J.R.; Murthy, K.S.; Kuemmerle, J.F.; Akbarali, H.I.; Qiao, L.-Y. Up-regulation of brain-derived neurotrophic factor in primary afferent pathway regulates colon-to-bladder cross-sensitization in rat. J. Neuroinflamm. 2012, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.O.; Kim, J.K.; Hong, H.S.; Kim, D.S.; Cho, H.J. Expression of brain-derived neurotrophic factor in rat dorsal root ganglia, spinal cord and gracile nuclei in experimental models of neuropathic pain. Neuroscience 2001, 107, 301–309. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Ro, L.-S.; Wang, H.-L.; Chen, J.-C. Up-regulation of dorsal root ganglia BDNF and trkB receptor in inflammatory pain: An in vivo and in vitrostudy. J. Neuroinflamm. 2011, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-J.; Kim, J.-K.; Zhou, X.-F.; Rush, R.A. Increased brain-derived neurotrophic factor immunoreactivity in rat dorsal root ganglia and spinal cord following peripheral inflammation. Brain Res. 1997, 764, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Tomotsuka, N.; Kaku, R.; Obata, N.; Matsuoka, Y.; Kanzaki, H.; Taniguchi, A.; Ishii, N.; Omiya, H.; Itano, Y.; Sato, T.; et al. Up-regulation of brain-derived neurotrophic factor in the dorsal root ganglion of the rat bone cancer pain model. J. Pain Res. 2014, 7, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Waite, P.M.E. CHAPTER 26—Trigeminal Sensory System. In The Rat Nervous System, 3rd ed.; Paxinos, G., Ed.; Academic Press: Burlington, ON, Canada, 2004; pp. 817–851. [Google Scholar]
- Ichikawa, H.; Yabuuchi, T.; Jin, H.; Terayama, R.; Yamaai, T.; Deguchi, T.; Kamioka, H.; Takano-Yamamoto, T.; Sugimoto, T. Brain-derived neurotrophic factor-immunoreactive primary sensory neurons in the rat trigeminal ganglion and trigeminal sensory nuclei. Brain Res. 2006, 1081, 113–118. [Google Scholar] [CrossRef]
- Buldyrev, I.; Tanner, N.M.; Hsieh, H.; Dodd, E.G.; Nguyen, L.T.; Balkowiec, A. Calcitonin gene-related peptide enhances release of native brain-derived neurotrophic factor from trigeminal ganglion neurons. J. Neurochem. 2006, 99, 1338–1350. [Google Scholar] [CrossRef]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lönnerberg, P.; Lou, D.; Hjerling-Leffler, J.; Haeggstrom, J.Z.; Kharchenko, O.; Kharchenko, P.V.; et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015, 18, 145–153. [Google Scholar] [CrossRef]
- Wang, J.; Kollarik, M.; Ru, F.; Sun, H.; McNeil, B.; Dong, X.; Stephens, G.; Korolevich, S.; Brohawn, P.; Kolbeck, R.; et al. Distinct and common expression of receptors for inflammatory mediators in vagal nodose versus jugular capsaicin-sensitive/TRPV1-positive neurons detected by low input RNA sequencing. PLoS ONE 2017, 12, e0185985. [Google Scholar] [CrossRef]
- Huerta, T.S.; Haider, B.; Adamovich-Zeitlin, R.; Chen, A.C.; Chaudhry, S.; Zanos, T.P.; Chavan, S.S.; Tracey, K.J.; Chang, E.H. Calcium imaging and analysis of the jugular-nodose ganglia enables identification of distinct vagal sensory neuron subsets. J. Neural Eng. 2023, 20, 026014. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.; Zaidi, S.I.A.; Mayer, C.; Katz, D.M. BDNF Is a Target-Derived Survival Factor for Arterial Baroreceptor and Chemoafferent Primary Sensory Neurons. J. Neurosci. 1999, 19, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.G.; Hasser, E.M.; Kunze, D.L.; Katz, D.M.; Kline, D.D. Endogenous Brain-Derived Neurotrophic Factor in the Nucleus Tractus Solitarius Tonically Regulates Synaptic and Autonomic Function. J. Neurosci. 2011, 31, 12318–12329. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liu, R.; Guo, F.; Wen, M.-Q.; Ma, X.-L.; Li, K.-Y.; Sun, H.; Xu, C.-L.; Li, Y.-Y.; Wu, M.-Y.; et al. Parabrachial nucleus circuit governs neuropathic pain-like behavior. Nat. Commun. 2020, 11, 5974. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, M.; Pawlowski, S.A.; Barthas, F.; Yalcin, I.; Kaufling, J.; Dardente, H.; Zachariou, V.; DiLeone, R.J.; Barrot, M.; Veinante, P. BDNF parabrachio-amygdaloid pathway in morphine-induced analgesia. Int. J. Neuropsychopharmacol. 2013, 16, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.K.; Bian, Z.-X.; Xu, H.-X.; Sung, J.J.-Y. Neonatal Maternal Separation Increases Brain-Derived Neurotrophic Factor and Tyrosine Kinase Receptor B Expression in the Descending Pain Modulatory System. Neurosignals 2009, 17, 213–221. [Google Scholar] [CrossRef]
- Thibault, K.; Lin, W.K.; Rancillac, A.; Fan, M.; Snollaerts, T.; Sordoillet, V.; Hamon, M.; Smith, G.M.; Lenkei, Z.; Pezet, S. BDNF-Dependent Plasticity Induced by Peripheral Inflammation in the Primary Sensory and the Cingulate Cortex Triggers Cold Allodynia and Reveals a Major Role for Endogenous BDNF As a Tuner of the Affective Aspect of Pain. J. Neurosci. 2014, 34, 14739–14751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, G.; Ma, J.; Liu, C.; Liu, X.; Zhan, Y.; Zhang, M. Brain-derived neurotrophic factor (BDNF) in the rostral anterior cingulate cortex (rACC) contributes to neuropathic spontaneous pain-related aversion via NR2B receptors. Brain Res. Bull. 2016, 127, 56–65. [Google Scholar] [CrossRef]
- Li, N.; Chen, B.; Jia, G.; Xu, R.; Xia, Y.; Lai, C.; Li, G.; Li, W.; Han, Y. Reduced BDNF expression in the auditory cortex contributed to neonatal pain-induced hearing impairment and dendritic pruning deficiency in mice. Reg. Anesth. Pain Med. 2022, 48, 85–92. [Google Scholar] [CrossRef]
- Ma, L.; Yue, L.; Zhang, Y.; Wang, Y.; Han, B.; Cui, S.; Liu, F.-Y.; Wan, Y.; Yi, M. Spontaneous Pain Disrupts Ventral Hippocampal CA1-Infralimbic Cortex Connectivity and Modulates Pain Progression in Rats with Peripheral Inflammation. Cell Rep. 2019, 29, 1579–1593.e6. [Google Scholar] [CrossRef] [PubMed]
- Obata, K.; Yamanaka, H.; Dai, Y.; Tachibana, T.; Fukuoka, T.; Tokunaga, A.; Yoshikawa, H.; Noguchi, K. Differential Activation of Extracellular Signal-Regulated Protein Kinase in Primary Afferent Neurons Regulates Brain-Derived Neurotrophic Factor Expression after Peripheral Inflammation and Nerve Injury. J. Neurosci. 2003, 23, 4117–4126. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Gulick, M.A.; Bowers, J.; Kuemmerle, J.F.; Grider, J.R. Differential changes in brain-derived neurotrophic factor and extracellular signal-regulated kinase in rat primary afferent pathways with colitis. Neurogastroenterol. Motil. 2008, 20, 928–938. [Google Scholar] [CrossRef]
- Garraway, S.M.; Petruska, J.C.; Mendell, L.M. BDNF sensitizes the response of lamina II neurons to high threshold primary afferent inputs. Eur. J. Neurosci. 2003, 18, 2467–2476. [Google Scholar] [CrossRef] [PubMed]
- Garraway, S.M.; Anderson, A.J.; Mendell, L.M. BDNF-Induced Facilitation of Afferent-Evoked Responses in Lamina II Neurons Is Reduced after Neonatal Spinal Cord Contusion Injury. J. Neurophysiol. 2005, 94, 1798–1804. [Google Scholar] [CrossRef]
- Yaksh, T.L. Calcium Channels As Therapeutic Targets in Neuropathic Pain. J. Pain 2006, 7, S13–S30. [Google Scholar] [CrossRef]
- Garraway, S.M.; Huie, J.R. Spinal Plasticity and Behavior: BDNF-Induced Neuromodulation in Uninjured and Injured Spinal Cord. Neural Plast. 2016, 2016, 9857201. [Google Scholar] [CrossRef] [PubMed]
- Pezet, S.; Malcangio, M.; McMahon, S.B. BDNF: A neuromodulator in nociceptive pathways? Brain Res. Brain Res. Rev. 2002, 40, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Merighi, A.; Carmignoto, G.; Gobbo, S.; Lossi, L.; Salio, C.; Vergnano, A.M.; Zonta, M. Neurotrophins in spinal cord nociceptive pathways. Prog. Brain Res. 2004, 146, 291–321. [Google Scholar]
- Pezet, S.; McMahon, S.B. NEUROTROPHINS: Mediators and Modulators of Pain. Annu. Rev. Neurosci. 2006, 29, 507–538. [Google Scholar] [CrossRef]
- Dembo, T.; Braz, J.M.; Hamel, K.A.; Kuhn, J.A.; Basbaum, A.I. Primary Afferent-Derived BDNF Contributes Minimally to the Processing of Pain and Itch. Eneuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L.; Merighi, A. The Use of ex Vivo Rodent Platforms in Neuroscience Translational Research with Attention to the 3Rs Philosophy. Front. Vet. Sci. 2018, 5, 164. [Google Scholar] [CrossRef] [PubMed]
- Lu, V.B.; Moran, T.D.; Balasubramanyan, S.; Alier, K.A.; Dryden, W.F.; Colmers, W.F.; Smith, P.A. Substantia Gelatinosa neurons in defined-medium organotypic slice culture are similar to those in acute slices from young adult rats. Pain 2006, 121, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Walwyn, W.; Ennes, H.S.; Kim, H.; McRoberts, J.A.; Marvizón, J.C.G. BDNF released during neuropathic pain potentiates NMDA receptors in primary afferent terminals. Eur. J. Neurosci. 2014, 39, 1439–1454. [Google Scholar] [CrossRef]
- Biggs, J.E.; Lu, V.B.; Kim, H.J.; Lai, A.; Todd, K.G.; Ballanyi, K.; Colmers, W.F.; Smith, P.A. Defined Medium Organotypic Cultures of Spinal Cord Put ‘Pain in a Dish’. In Isolated Central Nervous System Circuits; Ballanyi, K., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 405–436. [Google Scholar]
- Biggs, J.E.; Lu, V.B.; Stebbing, M.J.; Balasubramanyan, S.; Smith, P.A. Is BDNF sufficient for information transfer between microglia and dorsal horn neurons during the onset of central sensitization? Mol. Pain 2010, 6, 44. [Google Scholar] [CrossRef]
- Smith, P.A. BDNF: No gain without pain? Neuroscience 2014, 283, 107–123. [Google Scholar] [CrossRef]
- Li, C.; Lei, Y.; Tian, Y.; Xu, S.; Shen, X.; Wu, H.; Bao, S.; Wang, F. The etiological contribution of GABAergic plasticity to the pathogenesis of neuropathic pain. Mol. Pain 2019, 15, 1744806919847366. [Google Scholar] [CrossRef]
- Pezet, S.; Cunningham, J.; Patel, J.; Grist, J.; Gavazzi, I.; Lever, I.J.; Malcangio, M. BDNF Modulates Sensory Neuron Synaptic Activity by a Facilitation of GABA Transmission in the Dorsal Horn. Mol. Cell. Neurosci. 2002, 21, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Bardoni, R.; Ghirri, A.; Salio, C.; Prandini, M.; Merighi, A. BDNF-mediated modulation of GABA and glycine release in dorsal horn lamina II from postnatal rats. Dev. Neurobiol. 2007, 67, 960–975. [Google Scholar] [CrossRef]
- Lu, V.B.; Colmers, W.F.; Smith, P.A. Long-term actions of BDNF on inhibitory synaptic transmission in identified neurons of the rat substantia gelatinosa. J. Neurophysiol. 2012, 108, 441–452. [Google Scholar] [CrossRef]
- Dedek, A.; Xu, J.; Lorenzo, L.; Godin, A.G.; Kandegedara, C.M.; Glavina, G.; Landrigan, J.A.; Lombroso, P.J.; De Koninck, Y.; Tsai, E.C.; et al. Sexual dimorphism in a neuronal mechanism of spinal hyperexcitability across rodent and human models of pathological pain. Brain 2022, 145, 1124–1138. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Seereeram, A.; Nassar, M.A.; Levato, A.; Pezet, S.; Hathaway, G.; Morenilla-Palao, C.; Stirling, C.; Fitzgerald, M.; McMahon, S.B.; et al. Nociceptor-derived brain-derived neurotrophic factor regulates acute and inflammatory but not neuropathic pain. Mol. Cell. Neurosci. 2006, 31, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Groth, R.; Aanonsen, L. Spinal brain-derived neurotrophic factor (BDNF) produces hyperalgesia in normal mice while antisense directed against either BDNF or trkB, prevent inflammation-induced hyperalgesia. Pain 2002, 100, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Slack, S.E.; Thompson, S.W.N. Brain-derived neurotrophic factor induces NMDA receptor 1 phosphorylation in rat spinal cord. NeuroReport 2002, 13, 1967–1970. [Google Scholar] [CrossRef]
- Matayoshi, S.; Jiang, N.; Katafuchi, T.; Koga, K.; Furue, H.; Yasaka, T.; Nakatsuka, T.; Zhou, X.; Kawasaki, Y.; Tanaka, N.; et al. Actions of brain-derived neurotrophic factor on spinal nociceptive transmission during inflammation in the rat. J. Physiol. 2005, 569, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Lalisse, S.; Hua, J.; Lenoir, M.; Linck, N.; Rassendren, F.; Ulmann, L. Sensory neuronal P2RX4 receptors controls BDNF signaling in inflammatory pain. Sci. Rep. 2018, 8, 964. [Google Scholar] [CrossRef]
- Martin, K.K.; Noble, D.J.; Parvin, S.; Jang, K.; Garraway, S.M. Pharmacogenetic inhibition of TrkB signaling in adult mice attenuates mechanical hypersensitivity and improves locomotor function after spinal cord injury. Front. Cell. Neurosci. 2022, 16, 987236. [Google Scholar] [CrossRef] [PubMed]
- Bonomini, F.; Favero, G.; Castrezzati, S.; Borsani, E. Role of Neurotrophins in Orofacial Pain Modulation: A Review of the Latest Discoveries. Int. J. Mol. Sci. 2023, 24, 12438. [Google Scholar] [CrossRef] [PubMed]
- Bradnam, L.; Barry, C. The Role of the Trigeminal Sensory Nuclear Complex in the Pathophysiology of Craniocervical Dystonia. J. Neurosci. 2013, 33, 18358–18367. [Google Scholar] [CrossRef]
- Takeda, M.; Takahashi, M.; Kitagawa, J.; Kanazawa, T.; Nasu, M.; Matsumoto, S. Brain-Derived Neurotrophic Factor Enhances the Excitability of Small-Diameter Trigeminal Ganglion Neurons Projecting to the Trigeminal Nucleus Interpolaris/Caudalis Transition Zone following Masseter Muscle Inflammation. Mol. Pain 2013, 9, 49. [Google Scholar] [CrossRef]
- Grayson, M.; Arris, D.; Wu, P.; Merlo, J.; Ibrahim, T.; Fang-Mei, C.; Valenzuela, V.; Ganatra, S.; Ruparel, S. Oral squamous cell carcinoma–released brain-derived neurotrophic factor contributes to oral cancer pain by peripheral tropomyosin receptor kinase B activation. Pain 2021, 163, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wei, Y.; Pu, Y.; Jiang, D.; Jiang, X.; Zhang, Y.; Tao, J. Brain-derived neurotrophic factor stimulation of T-type Ca2+ channels in sensory neurons contributes to increased peripheral pain sensitivity. Sci. Signal. 2019, 12, eaaw2300. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chung, M.-K. Orthodontic force induces nerve injury-like transcriptomic changes driven by TRPV1-expressing afferents in mouse trigeminal ganglia. Mol. Pain 2020, 16, 1744806920973141. [Google Scholar] [CrossRef] [PubMed]
- Koshida, T.; Maruta, T.; Tanaka, N.; Hidaka, K.; Kurogi, M.; Nemoto, T.; Yanagita, T.; Takeya, R.; Tsuneyoshi, I. Changes in TRPV1 Receptor, CGRP, and BDNF Expression in Rat Dorsal Root Ganglion with Resiniferatoxin-Induced Neuropathic Pain: Modulation by Pulsed Radiofrequency Applied to the Sciatic Nerve. Acta Med. Okayama 2023, 77, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Kooshki, R.; Abbasnejad, M.; Mahani, S.E.; Raoof, M.; Aghtaei, M.M.M.; Dabiri, S. Orexin-A inhibits capsaicin-induced changes in cyclooxygenase-2 and brain-derived neurotrophic factor expression in trigeminal nucleus caudalis of rats. Korean J. Pain 2018, 31, 174–182. [Google Scholar] [CrossRef]
- Scarabelot, V.L.; de Oliveira, C.; Medeiros, L.F.; de Macedo, I.C.; Cioato, S.G.; Adachi, L.N.S.; Paz, A.H.; de Souza, A.; Caumo, W.; Torres, I.L.S. Transcranial direct-current stimulation reduces nociceptive behaviour in an orofacial pain model. J. Oral Rehabil. 2018, 46, 40–50. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, Y.; Yang, L.; Wang, Y.; Xiao, Z. PACAP6-38 improves nitroglycerin-induced central sensitization by modulating synaptic plasticity at the trigeminal nucleus caudalis in a male rat model of chronic migraine. J. Headache Pain 2023, 24, 66. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, Y.; Liu, Q.; Jiang, L.; Li, M.; Wang, S.; Long, T.; He, W.; Kong, X.; Qin, G.; et al. P2X4-receptor participates in EAAT3 regulation via BDNF-TrkB signaling in a model of trigeminal allodynia. Mol. Pain 2018, 14, 1744806918795930. [Google Scholar] [CrossRef]
- Luo, C.; Zhong, X.-L.; Zhou, F.H.; Li, J.-Y.; Zhou, P.; Xu, J.-M.; Song, B.; Li, C.-Q.; Zhou, X.-F.; Dai, R.-P. Peripheral Brain Derived Neurotrophic Factor Precursor Regulates Pain as an Inflammatory Mediator. Sci. Rep. 2016, 6, 27171. [Google Scholar] [CrossRef]
- Balkowiec, A.; Katz, D.M. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J. Neurosci. 2000, 20, 7417–7423. [Google Scholar] [CrossRef]
- Chen, Z.-Y.; Ieraci, A.; Teng, H.; Dall, H.; Meng, C.-X.; Herrera, D.G.; Nykjaer, A.; Hempstead, B.L.; Lee, F.S. Sortilin Controls Intracellular Sorting of Brain-Derived Neurotrophic Factor to the Regulated Secretory Pathway. J. Neurosci. 2005, 25, 6156–6166. [Google Scholar] [CrossRef] [PubMed]
- Ho, I.H.; Liu, X.; Zou, Y.; Liu, T.; Hu, W.; Chan, H.; Tian, Y.; Zhang, Y.; Li, Q.; Kou, S.; et al. A Novel Peptide Interfering with proBDNF-Sortilin Interaction Alleviates Chronic Inflammatory Pain. Theranostics 2019, 9, 1651–1665. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Liang, R.; Wang, S.; Meng, B.; Yang, C.-R.; Ding, H.-J.; Zhou, F.-H.; Han, C.-Y.; Zhang, X.-Y.; Zhang, X.-L.; et al. proBDNF/p75NTR promotes rheumatoid arthritis and inflammatory response by activating proinflammatory cytokines. FASEB J. 2022, 36, e22180. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, T.; Sun, J.; Zhao, S.; Wang, X.; Luo, W.; Luo, R.; Shen, W.; Luo, C.; Fu, D. Up-Regulation of ProBDNF/p75NTR Signaling in Spinal Cord Drives Inflammatory Pain in Male Rats. J. Inflamm. Res. 2023, 16, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Theodosiou, M.; Rush, A.R.; Zhou, F.X.; Hu, D.; Walker, S.J.; Tracey, J.D. Hyperalgesia due to nerve damage: Role of nerve growth factor. Pain 1999, 81, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Yajima, Y.; Narita, M.; Narita, M.; Matsumoto, N.; Suzuki, T. Involvement of a spinal brain-derived neurotrophic factor/full-length TrkB pathway in the development of nerve injury-induced thermal hyperalgesia in mice. Brain Res. 2002, 958, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Yajima, Y.; Narita, M.; Usui, A.; Kaneko, C.; Miyatake, M.; Narita, M.; Yamaguchi, T.; Tamaki, H.; Wachi, H.; Seyama, Y.; et al. Direct evidence for the involvement of brain-derived neurotrophic factor in the development of a neuropathic pain-like state in mice. J. Neurochem. 2005, 93, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Deng, Y..; Xian, C.J.; Zhong, J. Neurotrophins from dorsal root ganglia trigger allodynia after spinal nerve injury in rats. Eur. J. Neurosci. 2000, 12, 100–105. [Google Scholar] [CrossRef]
- Geng, S.-J.; Liao, F.-F.; Dang, W.-H.; Ding, X.; Liu, X.-D.; Cai, J.; Han, J.-S.; Wan, Y.; Xing, G.-G. Contribution of the spinal cord BDNF to the development of neuropathic pain by activation of the NR2B-containing NMDA receptors in rats with spinal nerve ligation. Exp. Neurol. 2010, 222, 256–266. [Google Scholar] [CrossRef]
- Miletic, G.; Miletic, V. Increases in the concentration of brain derived neurotrophic factor in the lumbar spinal dorsal horn are associated with pain behavior following chronic constriction injury in rats. Neurosci. Lett. 2002, 319, 137–140. [Google Scholar] [CrossRef]
- Obata, K.; Yamanaka, H.; Kobayashi, K.; Dai, Y.; Mizushima, T.; Katsura, H.; Fukuoka, T.; Tokunaga, A.; Noguchi, K. Role of Mitogen-Activated Protein Kinase Activation in Injured and Intact Primary Afferent Neurons for Mechanical and Heat Hypersensitivity after Spinal Nerve Ligation. J. Neurosci. 2004, 24, 10211–10222. [Google Scholar] [CrossRef] [PubMed]
- Obata, K.; Noguchi, K. BDNF in sensory neurons and chronic pain. Neurosci. Res. 2006, 55, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Obata, K.; Yamanaka, H.; Kobayashi, K.; Dai, Y.; Mizushima, T.; Katsura, H.; Fukuoka, T.; Tokunaga, A.; Noguchi, K. The effect of site and type of nerve injury on the expression of brain-derived neurotrophic factor in the dorsal root ganglion and on neuropathic pain behavior. Neuroscience 2005, 137, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Vanelderen, P.; Rouwette, T.; Kozicz, T.; Roubos, E.; Van Zundert, J.; Heylen, R.; Vissers, K. The role of brain-derived neurotrophic factor in different animal models of neuropathic pain. Eur. J. Pain 2010, 14, 473.e1–473.e9. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.M.; Mitchell, V.A.; White, D.M.; Rush, R.A.; Duggan, A.W. Release of immunoreactive brain-derived neurotrophic factor in the spinal cord of the rat following sciatic nerve transection. Brain Res. 2001, 899, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Cai, J.; Li, S.; Liu, X.-D.; Wan, Y.; Xing, G.-G. BDNF contributes to the development of neuropathic pain by induction of spinal long-term potentiation via SHP2 associated GluN2B-containing NMDA receptors activation in rats with spinal nerve ligation. Neurobiol. Dis. 2015, 73, 428–451. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.E.; Xu, J.; Dedek, A.; Li, Y.; Sengar, A.S.; Beggs, S.; Lombroso, P.J.; Salter, M.W. Potentiation of Synaptic GluN2B NMDAR Currents by Fyn Kinase Is Gated through BDNF-Mediated Disinhibition in Spinal Pain Processing. Cell Rep. 2016, 17, 2753–2765. [Google Scholar] [CrossRef] [PubMed]
- Dedek, A.; Xu, J.; Kandegedara, C.M.; Lorenzo, L.; Godin, A.G.; De Koninck, Y.; Lombroso, P.J.; Tsai, E.C.; Hildebrand, M.E. Loss of STEP61 couples disinhibition to N-methyl-d-aspartate receptor potentiation in rodent and human spinal pain processing. Brain 2019, 142, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.T.-C.; Guo, D.; Campanelli, D.; Frattini, F.; Mayer, F.; Zhou, L.; Kuner, R.; Heppenstall, P.A.; Knipper, M.; Hu, J. Presynaptic GABAergic inhibition regulated by BDNF contributes to neuropathic pain induction. Nat. Commun. 2014, 5, 5331. [Google Scholar] [CrossRef]
- Lai, C.-Y.; Hsieh, M.-C.; Yeh, C.-M.; Yang, P.-S.; Cheng, J.-K.; Wang, H.-H.; Lin, K.-H.; Nie, S.-T.; Lin, T.-B.; Peng, H.-Y. MicroRNA-489-3p attenuates neuropathic allodynia by regulating oncoprotein DEK/TET1-dependent epigenetic modification in the dorsal horn. Neuropharmacology 2022, 210, 109028. [Google Scholar] [CrossRef]
- Phạm, T.L.; Noh, C.; Neupane, C.; Sharma, R.; Shin, H.J.; Park, K.D.; Lee, C.J.; Kim, H.-W.; Lee, S.Y.; Park, J.B. MAO-B Inhibitor, KDS2010, Alleviates Spinal Nerve Ligation-induced Neuropathic Pain in Rats Through Competitively Blocking the BDNF/TrkB/NR2B Signaling. J. Pain 2022, 23, 2092–2109. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, X.; Guo, H.; Liu, P.; Ma, M.; Wang, Y. Resolvin D1 Attenuates Mechanical Allodynia after Burn Injury: Involvement of Spinal Glia, p38 MAPK, and BDNF/TrkB signaling. Mol. Pain 2023, 19, 17448069231159970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Y.; Liu, F.; Fang, Z.-H.; Li, Y.-L.; Liao, H.-L.; Song, Q.-X.; Zhou, C.; Shen, J.-F. Differential roles of NMDAR subunits 2A and 2B in mediating peripheral and central sensitization contributing to orofacial neuropathic pain. Brain Behav. Immun. 2022, 106, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Finamor, F.; Scarabelot, V.L.; Medeiros, L.F.; Stein, D.J.; da Silva, M.D.; Callai, E.; Caumo, W.; de Souza, A.; Torres, I.L. Involvement of GABAergic, glutamatergic, opioidergic, and brain-derived neurotrophic factor systems in the trigeminal neuropathic pain process. Neurosci. Lett. 2023, 793, 136970. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, C.L.; Medeiros, L.F.; de Souza, V.S.; Lopes, B.C.; de Oliveira, F.F.; Marques, L.X.; Torres, I.L.d.S.; de Souza, A. Low-dose naltrexone reverses facial mechanical allodynia in a rat model of trigeminal neuralgia. Neurosci. Lett. 2020, 736, 135248. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Xu, Y.; Yu, Z.; Zhou, Y.; Wang, W.; Yang, J.; Wang, Y.; Bai, Q.; Li, Z. The cAMP Response Element- Binding Protein/Brain-Derived Neurotrophic Factor Pathway in Anterior Cingulate Cortex Regulates Neuropathic Pain and Anxiodepression Like Behaviors in Rats. Front. Mol. Neurosci. 2022, 15, 831151. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, B.; Acevedo, E.O. BDNF as a biomarker for neuropathic pain: Consideration of mechanisms of action and associated measurement challenges. Brain Behav. 2023, 13, e2903. [Google Scholar] [CrossRef] [PubMed]
- Malcangio, M. Microglia and chronic pain. Pain 2016, 157, 1002–1003. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-R.; Berta, T.; Nedergaard, M. Glia and pain: Is chronic pain a gliopathy? Pain 2013, 154, S10–S28. [Google Scholar] [CrossRef]
- Apfel, S.C.; Wright, D.E.; Wiideman, A.M.; Dormia, C.; Snider, W.D.; Kessler, J.A. Nerve Growth Factor Regulates the Expression of Brain-Derived Neurotrophic Factor mRNA in the Peripheral Nervous System. Mol. Cell. Neurosci. 1996, 7, 134–142. [Google Scholar] [CrossRef]
- Coull, J.A.M.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; De Koninck, Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 2005, 438, 1017–1021. [Google Scholar] [CrossRef]
- Ulmann, L.; Hatcher, J.P.; Hughes, J.P.; Chaumont, S.; Green, P.J.; Conquet, F.; Buell, G.N.; Reeve, A.J.; Chessell, I.P.; Rassendren, F. Up-Regulation of P2X4 Receptors in Spinal Microglia after Peripheral Nerve Injury Mediates BDNF Release and Neuropathic Pain. J. Neurosci. 2008, 28, 11263–11268. [Google Scholar] [CrossRef] [PubMed]
- Boakye, P.A.; Tang, S.-J.; Smith, P.A. Mediators of Neuropathic Pain; Focus on Spinal Microglia, CSF-1, BDNF, CCL21, TNF-α, Wnt Ligands, and Interleukin 1β. Front. Pain Res. 2021, 2, 698157. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Albini, M.; Krawczun-Rygmaczewska, A.; Cesca, F. Astrocytes and brain-derived neurotrophic factor (BDNF). Neurosci. Res. 2023, 197, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Gould, E.; Xu, J.; Kim, E.J.; Kim, J.H. Oligodendrocytes regulate presynaptic properties and neurotransmission through BDNF signaling in the mouse brainstem. eLife 2019, 8, e42156. [Google Scholar] [CrossRef] [PubMed]
- Atta, A.A.; Ibrahim, W.W.; Mohamed, A.F.; Abdelkader, N.F. Microglia polarization in nociplastic pain: Mechanisms and perspectives. Inflammopharmacology 2023, 31, 1053–1067. [Google Scholar] [CrossRef]
- Malcangio, M. Role of the immune system in neuropathic pain. Scand. J. Pain 2019, 20, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Trang, T.; Beggs, S.; Wan, X.; Salter, M.W. P2X4-Receptor-Mediated Synthesis and Release of Brain-Derived Neurotrophic Factor in Microglia Is Dependent on Calcium and p38-Mitogen-Activated Protein Kinase Activation. J. Neurosci. 2009, 29, 3518–3528. [Google Scholar] [CrossRef]
- Bonalume, V.; Caffino, L.; Castelnovo, L.F.; Faroni, A.; Giavarini, F.; Liu, S.; Caruso, D.; Schmelz, M.; Fumagalli, F.; Carr, R.W.; et al. Schwann Cell Autocrine and Paracrine Regulatory Mechanisms, Mediated by Allopregnanolone and BDNF, Modulate PKCε in Peripheral Sensory Neurons. Cells 2020, 9, 1874. [Google Scholar] [CrossRef]
- Ismail, C.A.N.; Suppian, R.; Ab Aziz, C.B.; Long, I. Ifenprodil Reduced Expression of Activated Microglia, BDNF and DREAM Proteins in the Spinal Cord Following Formalin Injection During the Early Stage of Painful Diabetic Neuropathy in Rats. J. Mol. Neurosci. 2020, 71, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.; Done, J.D.; Schaeffer, A.J.; Thumbikat, P. Experimental autoimmune prostatitis induces microglial activation in the spinal cord. Prostate 2014, 75, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.-T.; Wu, J.-R.; Chen, Z.-Y.; Liu, Z.-X.; Miao, B. Effects of dexmedetomidine on P2X4Rs, p38-MAPK and BDNF in spinal microglia in rats with spared nerve injury. Brain Res. 2014, 1568, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, K.D.; Dreyfus, C.F.; Black, I.B. Brain-Derived Neurotrophic Factor in Astrocytes, Oligodendrocytes, and Microglia/Macrophages after Spinal Cord Injury. Neurobiol. Dis. 2000, 7, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Jin, J.; Chen, K.; You, S.; Zhang, H.; Sideris, A.; Norcini, M.; Recio-Pinto, E.; Wang, J.; Gan, W.-B.; et al. BDNF produced by cerebral microglia promotes cortical plasticity and pain hypersensitivity after peripheral nerve injury. PLoS Biol. 2021, 19, e3001337. [Google Scholar] [CrossRef] [PubMed]
- Harty, B.L.; Monk, K.R. Unwrapping the unappreciated: Recent progress in Remak Schwann cell biology. Curr. Opin. Neurobiol. 2017, 47, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Wu, F.; Jin, Z.; Gu, Y.; Chen, Y.; Fei, Y.; Chen, H.; Wang, Y.; Xing, L.; Zhao, Y.; et al. Overexpression of P2X4 receptor in Schwann cells promotes motor and sensory functional recovery and remyelination via BDNF secretion after nerve injury. Glia 2018, 67, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Velázquez, K.T.; Mohammad, H.; Sweitzer, S.M. Protein kinase C in pain: Involvement of multiple isoforms. Pharmacol. Res. 2007, 55, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.-Y.; Zhou, T.-H.; Chen, B.-K.; Liu, Z.-X. Schwann cells and trigeminal neuralgia. Mol. Pain 2020, 16, 1744806920963809. [Google Scholar] [CrossRef]
- Crotti, A.; Ransohoff, R.M. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity 2016, 44, 505–515. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, Y.-Q.; Qadri, Y.J.; Serhan, C.N.; Ji, R.-R. Microglia in Pain: Detrimental and Protective Roles in Pathogenesis and Resolution of Pain. Neuron 2018, 100, 1292–1311. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.A. BDNF in Neuropathic Pain; the Culprit that Cannot be Apprehended. Neuroscience 2024, 543, 49–64. [Google Scholar] [CrossRef]
- Gruber-Schoffnegger, D.; Drdla-Schutting, R.; Hönigsperger, C.; Wunderbaldinger, G.; Gassner, M.; Sandkühler, J. Induction of Thermal Hyperalgesia and Synaptic Long-Term Potentiation in the Spinal Cord Lamina I by TNF-α and IL-1β is Mediated by Glial Cells. J. Neurosci. 2013, 33, 6540–6551. [Google Scholar] [CrossRef] [PubMed]
- Decosterd, I.; Ji, R.-R.; Suter, M.R.; Wen, Y.-R. Do glial cells control pain? Neuron Glia Biol. 2007, 3, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Ferrini, F.; De Koninck, Y. Microglia Control Neuronal Network Excitability via BDNF Signalling. Neural Plast. 2013, 2013, 429815. [Google Scholar] [CrossRef] [PubMed]
- Boakye, P.A.; Rancic, V.; Whitlock, K.H.; Simmons, D.; Longo, F.M.; Ballanyi, K.; Smith, P.A. Receptor dependence of BDNF actions in superficial dorsal horn: Relation to central sensitization and actions of macrophage colony stimulating factor 1. J. Neurophysiol. 2019, 121, 2308–2322. [Google Scholar] [CrossRef] [PubMed]
- Lu, V.B.; Ballanyi, K.; Colmers, W.F.; Smith, P.A. Neuron type-specific effects of brain-derived neurotrophic factor in rat superficial dorsal horn and their relevance to ‘central sensitization’. J. Physiol. 2007, 584, 543–563. [Google Scholar] [CrossRef] [PubMed]
- Lu, V.B.; Biggs, J.E.; Stebbing, M.J.; Balasubramanyan, S.; Todd, K.G.; Lai, A.Y.; Colmers, W.F.; Dawbarn, D.; Ballanyi, K.; Smith, P.A. Brain-derived neurotrophic factor drives the changes in excitatory synaptic transmission in the rat superficial dorsal horn that follow sciatic nerve injury. J. Physiol. 2009, 587, 1013–1032. [Google Scholar] [CrossRef]
- Baba, H.; Ji, R.-R.; Kohno, T.; Moore, K.A.; Ataka, T.; Wakai, A.; Okamoto, M.; Woolf, C.J. Removal of GABAergic inhibition facilitates polysynaptic A fiber-mediated excitatory transmission to the superficial spinal dorsal horn. Mol. Cell. Neurosci. 2003, 24, 818–830. [Google Scholar] [CrossRef]
- Hu, Z.; Yu, X.; Chen, P.; Jin, K.; Zhou, J.; Wang, G.; Yu, J.; Wu, T.; Wang, Y.; Lin, F.; et al. BDNF-TrkB signaling pathway-mediated microglial activation induces neuronal KCC2 downregulation contributing to dynamic allodynia following spared nerve injury. Mol. Pain 2023, 19, 17448069231185439. [Google Scholar] [CrossRef]
- Zarei, M.; Sabetkasaei, M.; Zanjani, T.M.; Vaighan, N.S. The effect of microglial inhibition on the expression of BDNF, KCC2, and GABAA receptor before and after the establishment of CCI-induced neuropathic pain model. Fundam. Clin. Pharmacol. 2021, 36, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.-J.; Peng, J.; Xu, Y.-N.; Zeng, W.-J.; Zhang, J.; Wei, X.; Mai, C.-L.; Lin, Z.-J.; Liu, Y.; Murugan, M.; et al. Microglia Are Indispensable for Synaptic Plasticity in the Spinal Dorsal Horn and Chronic Pain. Cell Rep. 2019, 27, 3844–3859.e6. [Google Scholar] [CrossRef]
- Mapplebeck, J.C.; Beggs, S.; Salter, M.W. Sex differences in pain. Pain 2016, 157, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Paige, C.; Plasencia-Fernandez, I.; Kume, M.; Papalampropoulou-Tsiridou, M.; Lorenzo, L.-E.; David, E.T.; He, L.; Mejia, G.L.; Driskill, C.; Ferrini, F.; et al. A Female-Specific Role for Calcitonin Gene-Related Peptide (CGRP) in Rodent Pain Models. J. Neurosci. 2022, 42, 1930–1944. [Google Scholar] [CrossRef]
- Gardell, L.R.; Vanderah, T.W.; Gardell, S.E.; Wang, R.; Ossipov, M.H.; Lai, J.; Porreca, F. Enhanced Evoked Excitatory Transmitter Release in Experimental Neuropathy Requires Descending Facilitation. J. Neurosci. 2003, 23, 8370–8379. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Chen, J.; Su, M.; Lin, Z.; Zhan, H.; Yang, F.; Li, W.; Xie, J.; Huang, Y.; Liu, X.; et al. BDNF promotes activation of astrocytes and microglia contributing to neuroinflammation and mechanical allodynia in cyclophosphamide-induced cystitis. J. Neuroinflamm. 2020, 17, 19. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, L.; Xu, D.; Zhang, W.; Zhang, Y.; Zhou, J.; Huang, W. Interaction between astrocytic colony stimulating factor and its receptor on microglia mediates central sensitization and behavioral hypersensitivity in chronic post ischemic pain model. Brain Behav. Immun. 2018, 68, 248–260. [Google Scholar] [CrossRef]
- Guan, Z.; Kuhn, J.A.; Wang, X.; Colquitt, B.; Solorzano, C.; Vaman, S.; Guan, A.K.; Evans-Reinsch, Z.; Braz, J.; Devor, M.; et al. Injured sensory neuron–derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat. Neurosci. 2015, 19, 94–101. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, Y.; Peng, Y.; Zhong, L.; Zhu, S.; Zhang, W.; Tang, S.-J. Neuron activity–induced Wnt signaling up-regulates expression of brain-derived neurotrophic factor in the pain neural circuit. J. Biol. Chem. 2018, 293, 15641–15651. [Google Scholar] [CrossRef]
- Okubo, M.; Yamanaka, H.; Kobayashi, K.; Dai, Y.; Kanda, H.; Yagi, H.; Noguchi, K. Macrophage-Colony Stimulating Factor Derived from Injured Primary Afferent Induces Proliferation of Spinal Microglia and Neuropathic Pain in Rats. PLoS ONE 2016, 11, e0153375. [Google Scholar] [CrossRef]
- Cho, Y.-H.; Seo, T.-B. The timing point of exercise intervention regulates neuropathic pain-related molecules in the ipsilateral dorsal root ganglion neurons after sciatic nerve injury. J. Exerc. Rehabil. 2022, 18, 286–293. [Google Scholar] [CrossRef]
- Charles, A.C.; Merrill, J.E.; Dirksen, E.R.; Sandersont, M.J. Intercellular signaling in glial cells: Calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron 1991, 6, 983–992. [Google Scholar] [CrossRef]
- Nedergaard, M.; Ransom, B.; Goldman, S.A. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003, 26, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Zafra, F.; Lindholm, D.; Castren, E.; Hartikka, J.; Thoenen, H. Regulation of brain-derived neurotrophic factor and nerve growth factor mRNA in primary cultures of hippocampal neurons and astrocytes. J. Neurosci. 1992, 12, 4793–4799. [Google Scholar] [CrossRef]
- Schwartz, J.P.; Sheng, J.G.; Mitsuo, K.; Shirabe, S.; Nishiyama, N. Trophic Factor Production by Reactive Astrocytes in Injured Brain. Ann. N. Y. Acad. Sci. 1993, 679, 226–234. [Google Scholar] [CrossRef]
- Schwartz, J.P.; Nishiyama, N. Neurotrophic factor gene expression in astrocytes during development and following injury. Brain Res. Bull. 1994, 35, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Alderson, R.F.; Curtis, R.; Alterman, A.L.; Lindsay, R.M.; DiStefano, P.S. Truncated TrkB mediates the endocytosis and release of BDNF and neurotrophin-4/5 by rat astrocytes and Schwann cells in vitro. Brain Res. 2000, 871, 210–222. [Google Scholar] [CrossRef]
- Cao, T.; Matyas, J.J.; Renn, C.L.; Faden, A.I.; Dorsey, S.G.; Wu, J. Function and Mechanisms of Truncated BDNF Receptor TrkB.T1 in Neuropathic Pain. Cells 2020, 9, 1194. [Google Scholar] [CrossRef] [PubMed]
- Kinboshi, M.; Mukai, T.; Nagao, Y.; Matsuba, Y.; Tsuji, Y.; Tanaka, S.; Tokudome, K.; Shimizu, S.; Ito, H.; Ikeda, A.; et al. Inhibition of Inwardly Rectifying Potassium (Kir) 4.1 Channels Facilitates Brain-Derived Neurotrophic Factor (BDNF) Expression in Astrocytes. Front. Mol. Neurosci. 2017, 10, 408. [Google Scholar] [CrossRef]
- Milligan, E.D.; Watkins, L.R. Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci. 2009, 10, 23–36. [Google Scholar] [CrossRef]
- Renn, C.L.; Leitch, C.C.; Dorsey, S.G. In Vivo Evidence that Truncated Trkb.T1 Participates in Nociception. Mol. Pain 2009, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Renn, C.L.; Faden, A.I.; Dorsey, S.G. TrkB.T1 Contributes to Neuropathic Pain after Spinal Cord Injury through Regulation of Cell Cycle Pathways. J. Neurosci. 2013, 33, 12447–12463. [Google Scholar] [CrossRef]
- Matyas, J.J.; O’Driscoll, C.M.; Yu, L.; Coll-Miro, M.; Daugherty, S.; Renn, C.L.; Faden, A.I.; Dorsey, S.G.; Wu, J. Truncated TrkB.T1-Mediated Astrocyte Dysfunction Contributes to Impaired Motor Function and Neuropathic Pain after Spinal Cord Injury. J. Neurosci. 2017, 37, 3956–3971. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-L.; Wang, X.; Shao, L.; Jiang, G.-T.; Min, J.-W.; Mei, X.-Y.; He, X.-H.; Liu, W.-H.; Huang, W.-X.; Peng, B.-W. TRPV1 mediates astrocyte activation and interleukin-1β release induced by hypoxic ischemia (HI). J. Neuroinflamm. 2019, 16, 114. [Google Scholar] [CrossRef] [PubMed]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Malta, I.; Moraes, T.; Rodrigues, G.; Franco, P.; Galdino, G. The role of oligodendrocytes in chronic pain: Cellular and molecular mechanisms. J. Physiol. Pharmacol. 2019, 70, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Dubner, R. Interactions between the immune and nervous systems in pain. Nat. Med. 2010, 16, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Nakahashi, T.; Fujimura, H.; Altar, C.; Li, J.; Kambayashi, J.-I.; Tandon, N.N.; Sun, B. Vascular endothelial cells synthesize and secrete brain-derived neurotrophic factor. FEBS Lett. 2000, 470, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.A. Neuropathic pain; what we know and what we should do about it. Front. Pain Res. 2023, 4, 1220034. [Google Scholar] [CrossRef]
- Dimmek, D.J.; Korallus, C.; Buyny, S.; Christoph, G.; Lichtinghagen, R.; Jacobs, R.; Nugraha, B. Brain-Derived Neurotrophic Factor and Immune Cells in Osteoarthritis, Chronic Low Back Pain, and Chronic Widespread Pain Patients: Association with Anxiety and Depression. Medicina 2021, 57, 327. [Google Scholar] [CrossRef]
- Staszkiewicz, R.; Gładysz, D.; Gralewski, M.; Garczarek, M.; Gadzieliński, M.; Grabarek, B.O. Pathomechanism of the IVDs Degeneration and the Role of Neurotrophic Factors and Concentration of Selected Elements in Genesis of Low Back Pain. Curr. Pharm. Biotechnol. 2023, 24, 1164–1177. [Google Scholar] [CrossRef] [PubMed]
- Lisiewski, L.E.; Jacobsen, H.E.; Viola, D.C.M.; Kenawy, H.M.; Kiridly, D.N.; Chahine, N.O. Intradiscal inflammatory stimulation induces spinal pain behavior and intervertebral disc degeneration in vivo. FASEB J. 2023, 38, e23364. [Google Scholar] [CrossRef]
- Gheorghe, R.-O.; Grosu, A.V.; Magercu, M.; Ghenghea, M.-S.; Zbarcea, C.E.; Tanase, A.; Negres, S.; Filippi, A.; Chiritoiu, G.; Gherghiceanu, M.; et al. Switching Rat Resident Macrophages from M1 to M2 Phenotype by Iba1 Silencing Has Analgesic Effects in SNL-Induced Neuropathic Pain. Int. J. Mol. Sci. 2023, 24, 15831. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Muley, M.M.; Beggs, S.; Salter, M.W. Microglia-independent peripheral neuropathic pain in male and female mice. Pain 2022, 163, e1129–e1144. [Google Scholar] [CrossRef] [PubMed]
- Buskila, D. Genetics of chronic pain states. Best Pract. Res. Clin. Rheumatol. 2007, 21, 535–547. [Google Scholar] [CrossRef]
- Fillingim, R.; Wallace, M.R.; Herbstman, D.; Ribeiro-Dasilva, M.; Staud, R. Genetic contributions to pain: A review of findings in humans. Oral Dis. 2008, 14, 673–682. [Google Scholar] [CrossRef]
- Cappoli, N.; Tabolacci, E.; Aceto, P.; Russo, C.D. The emerging role of the BDNF-TrkB signaling pathway in the modulation of pain perception. J. Neuroimmunol. 2020, 349, 577406. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met Polymorphism Affects Activity-Dependent Secretion of BDNF and Human Memory and Hippocampal Function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Hempstead, B.L. Brain-Derived Neurotrophic Factor: Three Ligands, Many Actions. Trans. Am. Clin. Climatol. Assoc. 2015, 126, 9–19. [Google Scholar]
- Tian, Y.; Liu, X.; Jia, M.; Yu, H.; Lichtner, P.; Shi, Y.; Meng, Z.; Kou, S.; Ho, I.H.T.; Jia, B.; et al. Targeted Genotyping Identifies Susceptibility Locus in Brain-derived Neurotrophic Factor Gene for Chronic Postsurgical Pain. Anesthesiology 2018, 128, 587–597. [Google Scholar] [CrossRef]
- Vossen, H.; Kenis, G.; Rutten, B.; van Os, J.; Hermens, H.; Lousberg, R. The Genetic Influence on the Cortical Processing of Experimental Pain and the Moderating Effect of Pain Status. PLoS ONE 2010, 5, e13641. [Google Scholar] [CrossRef] [PubMed]
- Polli, A.; Ghosh, M.; Bakusic, J.; Ickmans, K.; Monteyne, D.; Velkeniers, B.; Bekaert, B.; Godderis, L.; Nijs, J. DNA Methylation and Brain-Derived Neurotrophic Factor Expression Account for Symptoms and Widespread Hyperalgesia in Patients with Chronic Fatigue Syndrome and Comorbid Fibromyalgia. Arthritis Rheumatol. 2020, 72, 1936–1944. [Google Scholar] [CrossRef] [PubMed]
- Chau, C.; Cepeda, I.; Devlin, A.; Weinberg, J.; Grunau, R. The Val66Met brain-derived neurotrophic factor gene variant interacts with early pain exposure to predict cortisol dysregulation in 7-year-old children born very preterm: Implications for cognition. Neuroscience 2015, 342, 188–199. [Google Scholar] [CrossRef]
- Yu, H.; Wang, D.-D.; Wang, Y.; Liu, T.; Lee, F.S.; Chen, Z.-Y. Variant Brain-Derived Neurotrophic Factor Val66Met Polymorphism Alters Vulnerability to Stress and Response to Antidepressants. J. Neurosci. 2012, 32, 4092–4101. [Google Scholar] [CrossRef]
- Gonçalves, F.d.T.; Pacheco-Barrios, K.; Rebello-Sanchez, I.; Castelo-Branco, L.; de Melo, P.S.; Parente, J.; Cardenas-Rojas, A.; Firigato, I.; Pessotto, A.V.; Imamura, M.; et al. Association of Mu opioid receptor (A118G) and BDNF (G196A) polymorphisms with rehabilitation-induced cortical inhibition and analgesic response in chronic osteoarthritis pain. Int. J. Clin. Health Psychol. 2022, 23, 100330. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, F.d.T.; Marques, L.M.; Pessotto, A.V.; Barbosa, S.P.; Imamura, M.; Simis, M.; Fregni, F.; Battistella, L. OPRM1 and BDNF polymorphisms associated with a compensatory neurophysiologic signature in knee osteoarthritis patients. Neurophysiol. Clin. 2023, 53, 102917. [Google Scholar] [CrossRef] [PubMed]
- Janssen, L.P.; Medeiros, L.F.; DE Souza, A.; DA Silva, J. Fibromyalgia: A Review of Related Polymorphisms and Clinical Relevance. An. Acad. Bras. Cienc. 2021, 93, e20210618. [Google Scholar] [CrossRef] [PubMed]
- Behnoush, A.H.; Khalaji, A.; Khanmohammadi, S.; Alehossein, P.; Saeedian, B.; Shobeiri, P.; Teixeira, A.L.; Rezaei, N. Brain-derived neurotrophic factor in fibromyalgia: A systematic review and meta-analysis of its role as a potential biomarker. PLoS ONE 2023, 18, e0296103. [Google Scholar] [CrossRef] [PubMed]
- Baumbauer, K.M.; Ramesh, D.; Perry, M.; Carney, K.B.; Julian, T.; Glidden, N.B.; Dorsey, S.G.R.; Starkweather, A.R.R.; Young, E.E. Contribution of COMT and BDNF Genotype and Expression to the Risk of Transition from Acute to Chronic Low Back Pain. Clin. J. Pain 2020, 36, 430–439. [Google Scholar] [CrossRef]
- Yamada, A.S.; Antunes, F.T.T.; Ferraz, C.; de Souza, A.H.; Simon, D. The genetic influence of the brain-derived neurotrophic factor Val66Met polymorphism in chronic low back pain. Adv. Rheumatol. 2021, 61, 24. [Google Scholar] [CrossRef]
- Goto, T.; Von Ah, D.; Li, X.; Xiang, L.; Kwiat, C.; Nguyen, C.; Hsiao, C.-P.; Saligan, L.N. Brain-Derived Neurotrophic Factor rs6265 polymorphism is associated with severe cancer-related fatigue and neuropathic pain in female cancer survivors. J. Cancer Surviv. 2023, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Azoulay, D.; Abed, S.; Sfadi, A.; Sheleg, O.; Shaoul, E.; Shehadeh, M.; Kaykov, E.; Nodelman, M.; Bashkin, A. Low brain-derived neurotrophic factor protein levels and single-nucleotide polymorphism Val66Met are associated with peripheral neuropathy in type II diabetic patients. Acta Diabetol. 2020, 57, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, R.; Price, T.J. The pharmacology of nociceptor priming. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 227, pp. 15–37. [Google Scholar] [CrossRef]
- Melemedjian, O.K.; Tillu, D.V.; Asiedu, M.N.; Mandell, E.K.; Moy, J.K.; Blute, V.M.; Taylor, C.J.; Ghosh, S.; Price, T.J. BDNF Regulates Atypical PKC at Spinal Synapses to Initiate and Maintain a Centralized Chronic Pain State. Mol. Pain 2013, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Gouin, O.; L’herondelle, K.; Lebonvallet, N.; Le Gall-Ianotto, C.; Sakka, M.; Buhé, V.; Plée-Gautier, E.; Carré, J.-L.; Lefeuvre, L.; Misery, L.; et al. TRPV1 and TRPA1 in cutaneous neurogenic and chronic inflammation: Pro-inflammatory response induced by their activation and their sensitization. Protein Cell 2017, 8, 644–661. [Google Scholar] [CrossRef] [PubMed]
- Schmelz, M. Itch and pain. Neurosci. Biobehav. Rev. 2010, 34, 171–176. [Google Scholar] [CrossRef]
- Ohtori, S.; Takahashi, K.; Moriya, H. Inflammatory pain mediated by a phenotypic switch in brain-derived neurotrophic factor-immunoreactive dorsal root ganglion neurons innervating the lumbar facet joints in rats. Neurosci. Lett. 2002, 323, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-F.; Chie, E.; Deng, Y.-S.; Zhong, J.-H.; Xue, Q.; Rush, R.; Xian, C. Injured primary sensory neurons switch phenotype for brain-derived neurotrophic factor in the rat. Neuroscience 1999, 92, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Mannion, R.J.; Costigan, M.; Decosterd, I.; Amaya, F.; Ma, Q.-P.; Holstege, J.C.; Ji, R.-R.; Acheson, A.; Lindsay, R.M.; Wilkinson, G.A.; et al. Neurotrophins: Peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc. Natl. Acad. Sci. USA 1999, 96, 9385–9390. [Google Scholar] [CrossRef] [PubMed]
- DiStefano, P.S.; Friedman, B.; Radziejewski, C.; Alexander, C.; Boland, P.; Schick, C.M.; Lindsay, R.M.; Wiegand, S.J. The neurotrophins BDNF, NT-3, and NGF display distinct patterns of retrograde axonal transport in peripheral and central neurons. Neuron 1992, 8, 983–993. [Google Scholar] [CrossRef]
- Heppenstall, P.A.; Lewin, G.R. Neurotrophins, nociceptors and pain. Curr. Opin. Anaesthesiol. 2000, 13, 573–576. [Google Scholar] [CrossRef]
- Park, S.Y.; Lee, J.Y.; Choi, J.Y.; Park, M.J.; Kim, D.S. Nerve growth factor activates brain-derived neurotrophic factor promoter IV via extracellular signal-regulated protein kinase 1/2 in PC12 cells. Mol. Cells 2006, 21, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Paterson, S.; Schmelz, M.; McGlone, F.; Turner, G.; Rukwied, R. Facilitated neurotrophin release in sensitized human skin. Eur. J. Pain 2009, 13, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Dubner, R. Pain Facilitation and Activity-Dependent Plasticity in Pain Modulatory Circuitry: Role of BDNF-TrkB Signaling and NMDA Receptors. Mol. Neurobiol. 2007, 35, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-G.; Zhou, L.-J. Long-term Potentiation at Spinal C-fiber Synapses: A Target for Pathological Pain. Curr. Pharm. Des. 2014, 21, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cai, J.; Feng, Z.-B.; Jin, Z.-R.; Liu, B.-H.; Zhao, H.-Y.; Jing, H.-B.; Wei, T.-J.; Yang, G.-N.; Liu, L.-Y.; et al. BDNF Contributes to Spinal Long-Term Potentiation and Mechanical Hypersensitivity Via Fyn-Mediated Phosphorylation of NMDA Receptor GluN2B Subunit at Tyrosine 1472 in Rats Following Spinal Nerve Ligation. Neurochem. Res. 2017, 42, 2712–2729. [Google Scholar] [CrossRef] [PubMed]
- Sandkühler, J.; Gruber-Schoffnegger, D. Hyperalgesia by synaptic long-term potentiation (LTP): An update. Curr. Opin. Pharmacol. 2012, 12, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.-J.; Zhong, Y.; Yang, T.; Wei, X.-H.; Xin, W.-J.; Liu, C.-C.; Zhou, L.-H.; Li, Y.-Y.; Liu, X.-G. Limited BDNF contributes to the failure of injury to skin afferents to produce a neuropathic pain condition. Pain 2010, 148, 148–157. [Google Scholar] [CrossRef]
- Slack, S.E.; Pezet, S.; McMahon, S.B.; Thompson, S.W.N.; Malcangio, M. Brain-derived neurotrophic factor induces NMDA receptor subunit one phosphorylation via ERK and PKC in the rat spinal cord. Eur. J. Neurosci. 2004, 20, 1769–1778. [Google Scholar] [CrossRef]
- Zhou, L.-J.; Zhong, Y.; Ren, W.-J.; Li, Y.-Y.; Zhang, T.; Liu, X.-G. BDNF induces late-phase LTP of C-fiber evoked field potentials in rat spinal dorsal horn. Exp. Neurol. 2008, 212, 507–514. [Google Scholar] [CrossRef]
- Bao, Y.; Hou, W.; Liu, R.; Gao, Y.; Kong, X.; Yang, L.; Shi, Z.; Li, W.; Zheng, H.; Jiang, S.; et al. PAR2-Mediated Upregulation of BDNF Contributes to Central Sensitization in Bone Cancer Pain. Mol. Pain 2014, 10, 28. [Google Scholar] [CrossRef]
- Zhou, L.-J.; Yang, T.; Wei, X.; Liu, Y.; Xin, W.-J.; Chen, Y.; Pang, R.-P.; Zang, Y.; Li, Y.-Y.; Liu, X.-G. Brain-derived neurotrophic factor contributes to spinal long-term potentiation and mechanical hypersensitivity by activation of spinal microglia in rat. Brain Behav. Immun. 2011, 25, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Retamal, J.; Reyes, A.; Ramirez, P.; Bravo, D.; Hernandez, A.; Pelissier, T.; Villanueva, L.; Constandil, L. Burst-Like Subcutaneous Electrical Stimulation Induces BDNF-Mediated, Cyclotraxin B-Sensitive Central Sensitization in Rat Spinal Cord. Front. Pharmacol. 2018, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Arconada, I.; Benedet, T.; Roza, C.; Torres, B.; Barrio, J.; Krzyzanowska, A.; Avendaño, C.; Mellström, B.; Lopez-Garcia, J.A.; Naranjo, J.R. DREAM Regulates BDNF-Dependent Spinal Sensitization. Mol. Pain 2010, 6, 95. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Lee, K.H.; Grau, J.W. Complete spinal cord injury (SCI) transforms how brain derived neurotrophic factor (BDNF) affects nociceptive sensitization. Exp. Neurol. 2017, 288, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Price, T.J.; Ghosh, S. ZIPping to Pain Relief: The Role (or Not) of PKMζ in Chronicc Pain. Mol. Pain 2013, 9, 6. [Google Scholar] [CrossRef]
- Sacktor, T.C. How does PKMζ maintain long-term memory? Nat. Rev. Neurosci. 2010, 12, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Croll, S.D.; Chesnutt, C.R.; Rudge, J.S.; Acheson, A.; Ryan, T.E.; Siuciak, J.A.; DiStefano, P.S.; Wiegand, S.J.; Lindsay, R.M. Co-infusion with a TrkB-Fc Receptor Body Carrier Enhances BDNF Distribution in the Adult Rat Brain. Exp. Neurol. 1998, 152, 20–33. [Google Scholar] [CrossRef]
- Morgado, C.; Pereira-Terra, P.; Cruz, C.D.; Tavares, I. Minocycline completely reverses mechanical hyperalgesia in diabetic rats through microglia-induced changes in the expression of the potassium chloride co-transporter 2 (KCC2) at the spinal cord. Diabetes Obes. Metab. 2010, 13, 150–159. [Google Scholar] [CrossRef]
- Moy, J.K.; Szabo-Pardi, T.; Tillu, D.V.; Megat, S.; Pradhan, G.; Kume, M.; Asiedu, M.N.; Burton, M.D.; Dussor, G.; Price, T.J. Temporal and sex differences in the role of BDNF/TrkB signaling in hyperalgesic priming in mice and rats. Neurobiol. Pain 2018, 5, 100024. [Google Scholar] [CrossRef]
- Rosenthal, A.; Lin, J.C. Modulation of neurotrophin signaling by monoclonal antibodies. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2014; Volume 220, pp. 497–512. [Google Scholar] [CrossRef]
- Cazorla, M.; Prémont, J.; Mann, A.; Girard, N.; Kellendonk, C.; Rognan, D. Identification of a low–molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J. Clin. Investig. 2011, 121, 1846–1857. [Google Scholar] [CrossRef]
- Cazorla, M.; Arrang, J.; Prémont, J. Pharmacological characterization of six trkB antibodies reveals a novel class of functional agents for the study of the BDNF receptor. Br. J. Pharmacol. 2011, 162, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Ferrini, F.; Trang, T.; Mattioli, T.-A.M.; Laffray, S.; Del’Guidice, T.; Lorenzo, L.-E.; Castonguay, A.; Doyon, N.; Zhang, W.; Godin, A.G.; et al. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl− homeostasis. Nat. Neurosci. 2013, 16, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Cazorla, M.; Jouvenceau, A.; Rose, C.; Guilloux, J.-P.; Pilon, C.; Dranovsky, A.; Prémont, J. Cyclotraxin-B, the First Highly Potent and Selective TrkB Inhibitor, Has Anxiolytic Properties in Mice. PLoS ONE 2010, 5, e9777. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.-C.; Kuan, Y.-H.; Shyu, B.-C. Targeting brain-derived neurotrophic factor in the medial thalamus for the treatment of central poststroke pain in a rodent model. Pain 2017, 158, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Constandil, L.; Goich, M.; Hernández, A.; Bourgeais, L.; Cazorla, M.; Hamon, M.; Villanueva, L.; Pelissier, T. Cyclotraxin-B, a New TrkB Antagonist, and Glial Blockade by Propentofylline, Equally Prevent and Reverse Cold Allodynia Induced by BDNF or Partial Infraorbital Nerve Constriction in Mice. J. Pain 2012, 13, 579–589. [Google Scholar] [CrossRef] [PubMed]
- M’dahoma, S.; Barthélemy, S.; Tromilin, C.; Jeanson, T.; Viguier, F.; Michot, B.; Pezet, S.; Hamon, M.; Bourgoin, S. Respective pharmacological features of neuropathic-like pain evoked by intrathecal BDNF versus sciatic nerve ligation in rats. Eur. Neuropsychopharmacol. 2015, 25, 2118–2130. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhang, Y.; Gu, Q.; Yuan, T.; Fan, B.; Xia, J.; Wu, J.; Xia, Y.; Li, W.; Han, Y. Alteration of neural activity and neuroinflammatory factors in the insular cortex of mice with corneal neuropathic pain. Genes Brain Behav. 2023, 22, e12842. [Google Scholar] [CrossRef]
- Mohamed, T.; Colciago, A.; Marelli, M.M.; Moretti, R.M.; Magnaghi, V. Protein kinase C epsilon activation regulates proliferation, migration, and epithelial to mesenchymal-like transition in rat Schwann cells. Front. Cell. Neurosci. 2023, 17, 1237479. [Google Scholar] [CrossRef]
- Fan, F.; Tang, Y.; Dai, H.; Cao, Y.; Sun, P.; Chen, Y.; Chen, A.; Lin, C. Blockade of BDNF signalling attenuates chronic visceral hypersensitivity in an IBS-like rat model. Eur. J. Pain 2020, 24, 839–850. [Google Scholar] [CrossRef]
- Ma, Y.; Zhao, W.; Chen, D.; Zhou, D.; Gao, Y.; Bian, Y.; Xu, Y.; Xia, S.-H.; Fang, T.; Yang, J.-X.; et al. Disinhibition of Mesolimbic Dopamine Circuit by the Lateral Hypothalamus Regulates Pain Sensation. J. Neurosci. 2023, 43, 4525–4540. [Google Scholar] [CrossRef]
- Ye, Y.; Yan, X.; Wang, L.; Xu, J.; Li, T. Transcranial direct current stimulation attenuates chronic pain in knee osteoarthritis by modulating BDNF/TrkB signaling in the descending pain modulation system. Neurosci. Lett. 2023, 810, 137320. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yang, Y.; Wang, C.; Huang, J.; Wang, X.; Liu, Y.; Wang, H. Exploring the role and mechanisms of diallyl trisulfide and diallyl disulfide in chronic constriction-induced neuropathic pain in rats. Korean J. Pain 2020, 33, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Lesnak, J.B.; Plumb, A.N.; Janowski, A.J.; Smith, A.F.; Hill, J.K.; Sluka, K.A. Brain-derived neurotrophic factor contributes to activity-induced muscle pain in male but not female mice. bioRxiv 2023. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, L.; Liu, B.; Sun, P.; Chen, Z.; Huang, Y.; Ai-Qin, C.; Chen, Y.; Lin, C. Spinal P2X4 Receptors Involved in Visceral Hypersensitivity of Neonatal Maternal Separation Rats. Purinergic Signal. 2022, 19, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, H.W.; Girard, B.M.; Ratkovits, L.; Campbell, S.E.; Vizzard, M.A. Effects of pharmacological neurotrophin receptor inhibition on bladder function in female mice with cyclophosphamide-induced cystitis. Front. Urol. 2022, 2, 1037511. [Google Scholar] [CrossRef] [PubMed]
- Long, T.; He, W.; Pan, Q.; Zhang, S.; Zhang, D.; Qin, G.; Chen, L.; Zhou, J. Microglia P2X4R-BDNF signalling contributes to central sensitization in a recurrent nitroglycerin-induced chronic migraine model. J. Headache Pain 2020, 21, 4. [Google Scholar] [CrossRef] [PubMed]
- Chodroff, L.; Bendele, M.; Valenzuela, V.; Henry, M.; Ruparel, S. BDNF signaling contributes to oral cancer pain in a preclinical orthotopic rodent model. Mol. Pain 2016, 12, 1744806916666841. [Google Scholar] [CrossRef]
- Wang, X.-M.; Pan, W.; Xu, N.; Zhou, Z.-Q.; Zhang, G.-F.; Shen, J.-C. Environmental enrichment improves long-term memory impairment and aberrant synaptic plasticity by BDNF/TrkB signaling in nerve-injured mice. Neurosci. Lett. 2018, 694, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.-Y.; Sun, Y.; Shi, X.-Y.; Sahbaie, P.; Clark, J.D. Epigenetic Regulation of Spinal Cord Gene Expression Controls Opioid-Induced Hyperalgesia. Mol. Pain 2014, 10, 59. [Google Scholar] [CrossRef]
- Fang, X.; Yang, C.; Li, S.; Zhan, G.; Zhang, J.; Huang, N.; Du, X.; Xu, H.; Hashimoto, K.; Luo, A. Brain-derived neurotrophic factor-TrkB signaling in the medial prefrontal cortex plays a role in the anhedonia-like phenotype after spared nerve injury. Eur. Arch. Psychiatry Clin. Neurosci. 2018, 270, 195–205. [Google Scholar] [CrossRef]
- Sahbaie, P.; Liang, D.-Y.; Shi, X.-Y.; Sun, Y.; Clark, J.D. Epigenetic regulation of spinal cord gene expression contributes to enhanced postoperative pain and analgesic tolerance subsequent to continuous opioid exposure. Mol. Pain 2016, 12, 1744806916641950. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Lin, Y.M.; Winston, J.H.; Radhakrishnan, R.; Huang, L.M.; Shi, X.Z. Role of brain-derived neurotrophic factor in the pathogenesis of distention-associated abdominal pain in bowel obstruction. Neurogastroenterol. Motil. 2018, 30, e13373. [Google Scholar] [CrossRef] [PubMed]
- Tillu, D.V.; Hassler, S.N.; Burgos-Vega, C.C.; Quinn, T.L.; Sorge, R.E.; Dussor, G.; Boitano, S.; Vagner, J.; Price, T.J. Protease-activated receptor 2 activation is sufficient to induce the transition to a chronic pain state. Pain 2015, 156, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, F.; Liu, X.; Ma, C.; Zhao, J. Surgical incision induces learning impairment in mice partially through inhibition of the brain-derived neurotrophic factor signaling pathway in the hippocampus and amygdala. Mol. Pain 2018, 14, 1744806918805902. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Lakkaniga, N.R.; Carlomagno, F.; Santoro, M.; McDonald, N.Q.; Lv, F.; Gunaganti, N.; Frett, B.; Li, H.-Y. Insights into Current Tropomyosin Receptor Kinase (TRK) Inhibitors: Development and Clinical Application. J. Med. Chem. 2018, 62, 1731–1760. [Google Scholar] [CrossRef] [PubMed]
- Stachel, S.J.; Sanders, J.M.; Henze, D.A.; Rudd, M.T.; Su, H.-P.; Li, Y.; Nanda, K.K.; Egbertson, M.S.; Manley, P.J.; Jones, K.L.G.; et al. Maximizing Diversity from a Kinase Screen: Identification of Novel and Selective pan-Trk Inhibitors for Chronic Pain. J. Med. Chem. 2014, 57, 5800–5816. [Google Scholar] [CrossRef] [PubMed]
- Angeles, T.S.; Yang, S.X.; Steffler, C.; Dionne, C.A. Kinetics of trkA Tyrosine Kinase Activity and Inhibition by K-252a. Arch. Biochem. Biophys. 1998, 349, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.H.; Toma, H.; Shenoy, M.; He, Z.-J.; Zou, L.; Xiao, S.-Y.; Micci, M.-A.; Pasricha, P.J. Acute pancreatitis results in referred mechanical hypersensitivity and neuropeptide up-regulation that can be suppressed by the protein kinase inhibitor k252a. J. Pain 2003, 4, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Frias, B.; Allen, S.; Dawbarn, D.; Charrua, A.; Cruz, F.; Cruz, C. Brain-derived neurotrophic factor, acting at the spinal cord level, participates in bladder hyperactivity and referred pain during chronic bladder inflammation. Neuroscience 2013, 234, 88–102. [Google Scholar] [CrossRef]
- Miletic, G.; Miletic, V. Loose ligation of the sciatic nerve is associated with TrkB receptor-dependent decreases in KCC2 protein levels in the ipsilateral spinal dorsal horn. Pain 2008, 137, 532–539. [Google Scholar] [CrossRef]
- Zhang, H.-H.; Zhang, X.-Q.; Xue, Q.-S.; Luo, Y.; Huang, J.-L.; Zhang, S.; Shao, H.-J.; Lu, H.; Wang, W.-Y.; Yu, B.-W. The BDNF/TrkB Signaling Pathway Is Involved in Heat Hyperalgesia Mediated by Cdk5 in Rats. PLoS ONE 2014, 9, e85536. [Google Scholar] [CrossRef] [PubMed]
- Yasui, M.; Shiraishi, Y.; Ozaki, N.; Hayashi, K.; Hori, K.; Ichiyanagi, M.; Sugiura, Y. Nerve growth factor and associated nerve sprouting contribute to local mechanical hyperalgesia in a rat model of bone injury. Eur. J. Pain 2011, 16, 953–965. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wang, F.; Wang, L.; Xu, Y.; Lv, L.; Duan, W.; Bai, R.; Meng, Z.; Shao, X. Involvement of the BDNF–TrkB–KCC2 pathway in neuropathic pain after brachial plexus avulsion. Brain Behav. 2022, 12, e2464. [Google Scholar] [CrossRef] [PubMed]
- Hiroki, T.; Suto, T.; Ohta, J.; Saito, S.; Obata, H. Spinal γ-Aminobutyric Acid Interneuron Plasticity Is Involved in the Reduced Analgesic Effects of Morphine on Neuropathic Pain. J. Pain 2021, 23, 547–557. [Google Scholar] [CrossRef]
- dos Reis, R.C.; Kopruszinski, C.M.; Nones, C.F.M.; Aguiar, D.A.; Chichorro, J.G. The opposing contribution of neurotrophin-3 and nerve growth factor to orofacial heat hyperalgesia in rats. Behav. Pharmacol. 2020, 31, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Miao, B.; Yin, Y.; Mao, G.; Zhao, B.; Wu, J.; Shi, H.; Fei, S. The implication of transient receptor potential canonical 6 in BDNF-induced mechanical allodynia in rat model of diabetic neuropathic pain. Life Sci. 2021, 273, 119308. [Google Scholar] [CrossRef]
- Bailey, J.J.; Jaworski, C.; Tung, D.; Wängler, C.; Wängler, B.; Schirrmacher, R. Tropomyosin receptor kinase inhibitors: An updated patent review for 2016–2019. Expert Opin. Ther. Pat. 2020, 30, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.J.; Schirrmacher, R.; Farrell, K.; Bernard-Gauthier, V. Tropomyosin receptor kinase inhibitors: An updated patent review for 2010–2016—Part I. Expert Opin. Ther. Pat. 2017, 27, 733–751. [Google Scholar] [CrossRef] [PubMed]
- Hameed, A.; Al-Rashida, M.; Uroos, M.; Ali, S.A.; Khan, K.M. Schiff bases in medicinal chemistry: A patent review (2010–2015). Expert Opin. Ther. Pat. 2017, 27, 63–79. [Google Scholar] [CrossRef]
- Wang, T.; Yu, D.; Lamb, M.L. Trk kinase inhibitors as new treatments for cancer and pain. Expert Opin. Ther. Pat. 2009, 19, 305–319. [Google Scholar] [CrossRef]
- Ghilardi, J.R.; Freeman, K.T.; Jimenez-Andrade, J.M.; Mantyh, W.G.; Bloom, A.P.; Bouhana, K.S.; Trollinger, D.; Winkler, J.; Lee, P.; Andrews, S.W.; et al. Sustained blockade of neurotrophin receptors TrkA, TrkB and TrkC reduces non-malignant skeletal pain but not the maintenance of sensory and sympathetic nerve fibers. Bone 2011, 48, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Skerratt, S.E.; Andrews, M.; Bagal, S.K.; Bilsland, J.; Brown, D.; Bungay, P.J.; Cole, S.; Gibson, K.R.; Jones, R.; Morao, I.; et al. The discovery of a potent, selective, and peripherally restricted pan-Trk inhibitor (PF-06273340) for the treatment of pain. J. Med. Chem. 2016, 59, 10084–10099. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, N.; Oyama, S.; Higashi, R.; Yanagida, K. Efficacy, Safety, and Tolerability of ONO-4474, an Orally Available Pan-Tropomyosin Receptor Kinase Inhibitor, in Japanese Patients with Moderate to Severe Osteoarthritis of the Knee: A Randomized, Placebo-Controlled, Double-Blind, Parallel-Group Comparative Study. J. Clin. Pharmacol. 2019, 60, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Boulle, F.; Kenis, G.; Cazorla, M.; Hamon, M.; Steinbusch, H.W.; Lanfumey, L.; Hove, D.L.v.D. TrkB inhibition as a therapeutic target for CNS-related disorders. Prog. Neurobiol. 2012, 98, 197–206. [Google Scholar] [CrossRef]
- Beggs, S.; Trang, T.; Salter, M.W. P2X4R+ microglia drive neuropathic pain. Nat. Neurosci. 2012, 15, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Doyon, N.; Ferrini, F.; Gagnon, M.; De Koninck, Y. Treating pathological pain: Is KCC2 the key to the gate? Expert Rev. Neurother. 2013, 13, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.; Bergeron, M.J.; Lavertu, G.; Castonguay, A.; Tripathy, S.; Bonin, R.P.; Perez-Sanchez, J.; Boudreau, D.; Wang, B.; Dumas, L.; et al. Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat. Med. 2013, 19, 1524–1528. [Google Scholar] [CrossRef] [PubMed]
- Laske, C.; Stransky, E.; Eschweiler, G.W.; Klein, R.; Wittorf, A.; Leyhe, T.; Richartz, E.; Köhler, N.; Bartels, M.; Buchkremer, G.; et al. Increased BDNF serum concentration in fibromyalgia with or without depression or antidepressants. J. Psychiatr. Res. 2007, 41, 600–605. [Google Scholar] [CrossRef] [PubMed]
- de Zanette, S.A.; Vercelino, R.; Laste, G.; Rozisky, J.R.; Schwertner, A.; Machado, C.B.; Xavier, F.; de Souza, I.C.C.; Deitos, A.; Torres, I.L.S.; et al. Melatonin analgesia is associated with improvement of the descending endogenous pain-modulating system in fibromyalgia: A phase II, randomized, double-dummy, controlled trial. BMC Pharmacol. Toxicol. 2014, 15, 40. [Google Scholar] [CrossRef]
- Jablochkova, A.; Bäckryd, E.; Kosek, E.; Mannerkorpi, K.; Ernberg, M.; Gerdle, B.; Ghafouri, B. Unaltered low nerve growth factor and high brain-derived neurotrophic factor levels in plasma from patients with fibromyalgia after a 15-week progressive resistance exercise. J. Rehabil. Med. 2019, 51, 779–787. [Google Scholar] [CrossRef]
- Brietzke, A.P.; Zortea, M.; Carvalho, F.; Sanches, P.R.; Silva, D.P.J.; Torres, I.L.d.S.; Fregni, F.; Caumo, W. Large Treatment Effect with Extended Home-Based Transcranial Direct Current Stimulation Over Dorsolateral Prefrontal Cortex in Fibromyalgia: A Proof of Concept Sham-Randomized Clinical Study. J. Pain 2020, 21, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Caumo, W.; Alves, R.L.; Vicuña, P.; Alves, C.F.d.S.; Ramalho, L.; Sanches, P.R.S.; Silva, D.P.; Torres, I.L.d.S.; Fregni, F. Impact of Bifrontal Home-Based Transcranial Direct Current Stimulation in Pain Catastrophizing and Disability due to Pain in Fibromyalgia: A Randomized, Double-Blind Sham-Controlled Study. J. Pain 2021, 23, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Stefani, L.C.; Leite, F.M.; Tarragó, M.d.G.L.; Zanette, S.A.; de Souza, A.; Castro, S.M.; Caumo, W. BDNF and serum S100B levels according the spectrum of structural pathology in chronic pain patients. Neurosci. Lett. 2019, 706, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Wolmeister, A.S.; Schiavo, C.L.; Nazário, K.C.K.; Castro, S.M.d.J.; de Souza, A.; Caetani, R.P.; Caumo, W.; Stefani, L.C. The Brief Measure of Emotional Preoperative Stress (B-MEPS) as a new predictive tool for postoperative pain: A prospective observational cohort study. PLoS ONE 2020, 15, e0227441. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.C.S.; Zortea, M.; Souza, A.; Santos, V.; Biazús, J.V.; Torres, I.L.S.; Fregni, F.; Caumo, W. Clinical impact of melatonin on breast cancer patients undergoing chemotherapy; effects on cognition, sleep and depressive symptoms: A randomized, double-blind, placebo-controlled trial. PLoS ONE 2020, 15, e0231379. [Google Scholar] [CrossRef] [PubMed]
- Dall’agnol, L.; Medeiros, L.F.; Torres, I.L.; Deitos, A.; Brietzke, A.; Laste, G.; de Souza, A.; Vieira, J.L.; Fregni, F.; Caumo, W. Repetitive Transcranial Magnetic Stimulation Increases the Corticospinal Inhibition and the Brain-Derived Neurotrophic Factor in Chronic Myofascial Pain Syndrome: An Explanatory Double-Blinded, Randomized, Sham-Controlled Trial. J. Pain 2014, 15, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Chassot, M.; Dussan-Sarria, J.A.; Sehn, F.C.; Deitos, A.; de Souza, A.; Vercelino, R.; Torres, I.L.; Fregni, F.; Caumo, W. Electroacupuncture analgesia is associated with increased serum brain-derived neurotrophic factor in chronic tension-type headache: A randomized, sham controlled, crossover trial. BMC Complement. Altern. Med. 2015, 15, 144. [Google Scholar] [CrossRef]
- Jiang, Y.-H.; Liu, H.-T.; Kuo, H.-C. Decrease of Urinary Nerve Growth Factor but Not Brain-Derived Neurotrophic Factor in Patients with Interstitial Cystitis/Bladder Pain Syndrome Treated with Hyaluronic Acid. PLoS ONE 2014, 9, e91609. [Google Scholar] [CrossRef]
- Medeiros, L.F.; Caumo, W.; Dussán-Sarria, J.; Deitos, A.; Brietzke, A.; Laste, G.; Campos-Carraro, C.; de Souza, A.; Scarabelot, V.L.; Cioato, S.G.; et al. Effect of Deep Intramuscular Stimulation and Transcranial Magnetic Stimulation on Neurophysiological Biomarkers in Chronic Myofascial Pain Syndrome. Pain Med. 2015, 17, 122–135. [Google Scholar] [CrossRef]
- Saxena, A.K.; Lakshman, K.; Sharma, T.; Gupta, N.; Banerjee, B.D.; Singal, A.; Maurer, A.J.; Lissounov, A.; Knezevic, I.; Candido, K.D.; et al. Modulation of serum BDNF levels in postherpetic neuralgia following pulsed radiofrequency of intercostal nerve and pregabalin. Pain Manag. 2016, 6, 217–227. [Google Scholar] [CrossRef]
- Jiang, M.; Wang, M.-H.; Wang, X.-B.; Liu, L.; Wu, J.-L.; Yang, X.-L.; Liu, X.-R.; Zhang, C.-X. Effect of intraoperative application of ketamine on postoperative depressed mood in patients undergoing elective orthopedic surgery. J. Anesth. 2015, 30, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, H.; Sesterhenn, R.B.; de Souza, A.; de Souza, A.C.; Alves, M.; Machado, J.C.; Burger, N.B.; Torres, I.L.d.S.; Stefani, L.C.; Fregni, F.; et al. Preoperative transcranial direct current stimulation: Exploration of a novel strategy to enhance neuroplasticity before surgery to control postoperative pain. A randomized sham-controlled study. PLoS ONE 2017, 12, e0187013. [Google Scholar] [CrossRef]
- Zhao, C.-G.; Sun, W.; Ju, F.; Wang, H.; Sun, X.-L.; Mou, X.; Yuan, H. Analgesic Effects of Directed Repetitive Transcranial Magnetic Stimulation in Acute Neuropathic Pain After Spinal Cord Injury. Pain Med. 2019, 21, 1216–1223. [Google Scholar] [CrossRef]
- Govil, M.; Mukhopadhyay, N.; Holwerda, T.; Sluka, K.; Rakel, B.; Schutte, D.L. Effects of genotype on TENS effectiveness in controlling knee pain in persons with mild to moderate osteoarthritis. Eur. J. Pain 2019, 24, 398–412. [Google Scholar] [CrossRef]
- Serrano, G.B.; Rodrigues, L.P.; Schein, B.; Zortea, M.; Torres, I.L.d.S.; Fregni, F.; Caumo, W. The Hypnotic Analgesia Suggestion Mitigated the Effect of the Transcranial Direct Current Stimulation on the Descending Pain Modulatory System: A Proof of Concept Study. J. Pain Res. 2020, 13, 2297–2311. [Google Scholar] [CrossRef]
- de Paula, T.M.H.; Castro, M.S.; Medeiros, L.F.; Paludo, R.H.; Couto, F.F.; da Costa, T.R.; Fortes, J.P.; Salbego, M.d.O.; Behnck, G.S.; de Moura, T.A.M.; et al. Association of low-dose naltrexone and transcranial direct current stimulation in fibromyalgia: A randomized, double-blinded, parallel clinical trial. Braz. J. Anesthesiol. (Engl. Ed.) 2023, 73, 409–417. [Google Scholar] [CrossRef]
- Montero-Marin, J.; Andrés-Rodríguez, L.; Tops, M.; Luciano, J.V.; Navarro-Gil, M.; Feliu-Soler, A.; López-Del-Hoyo, Y.; Garcia-Campayo, J. Effects of attachment-based compassion therapy (ABCT) on brain-derived neurotrophic factor and low-grade inflammation among fibromyalgia patients: A randomized controlled trial. Sci. Rep. 2019, 9, 15639. [Google Scholar] [CrossRef] [PubMed]
- Di Bonaventura, S.; Carnero, J.F.; Ferrer-Peña, R. Can a specific biobehavioral-based therapeutic education program lead to changes in pain perception and brain plasticity biomarkers in chronic pain patients? A study protocol for a randomized clinical trial. PLoS ONE 2024, 19, e0289430. [Google Scholar] [CrossRef] [PubMed]
- Polacchini, A.; Metelli, G.; Francavilla, R.; Baj, G.; Florean, M.; Mascaretti, L.G.; Tongiorgi, E. A method for reproducible measurements of serum BDNF: Comparison of the performance of six commercial assays. Sci. Rep. 2015, 5, 17989. [Google Scholar] [CrossRef]
- Salio, C.; Ferrini, F.; Muthuraju, S.; Merighi, A. Presynaptic Modulation of Spinal Nociceptive Transmission by Glial Cell Line-Derived Neurotrophic Factor (GDNF). J. Neurosci. 2014, 34, 13819–13833. [Google Scholar] [CrossRef]
- Ferrini, F.; Salio, C.; Boggio, E.M.; Merighi, A. Interplay of BDNF and GDNF in the Mature Spinal Somatosensory System and Its Potential Therapeutic Relevance. Curr Neuropharmacol 2021, 19, 1225–1245. [Google Scholar] [CrossRef] [PubMed]
- Merighi, A. Cross Talk of BDNF and GDNF in Spinal Substantia Gelatinosa (Lamina II): Focus on Circuitry. Adv. Exp. Med. Biol. 2021, 1331, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.M. Somatic Pain. In Encyclopedia of Pain; Schmidt, R.F., Willis, W.D., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 2190–2192. [Google Scholar]
- Price, D.D. Psychological and Neural Mechanisms of the Affective Dimension of Pain. Science 2000, 288, 1769–1772. [Google Scholar] [CrossRef] [PubMed]
- Michael, G.J.; Averill, S.; Shortland, P.J.; Yan, Q.; Priestley, J.V. Axotomy results in major changes in BDNF expression by dorsal root ganglion cells: BDNF expression in large trkB and trkC cells, in pericellular baskets, and in projections to deep dorsal horn and dorsal column nuclei. Eur. J. Neurosci. 1999, 11, 3539–3551. [Google Scholar] [CrossRef]
- Hudspith, M.J.; Siddall, P.J.; Munglani, R. Chapter 23—Physiology of pain. In Foundations of Anesthesia, 2nd ed.; Hemmings, H.C., Hopkins, P.M., Eds.; Mosby: Edinburgh, Scotland, 2006; pp. 267–285. [Google Scholar]
- Kim, H.Y.; Wang, J.; Gwak, Y.S. Gracile Neurons Contribute to the Maintenance of Neuropathic Pain in Peripheral and Central Neuropathic Models. J. Neurotrauma 2012, 29, 2587–2592. [Google Scholar] [CrossRef]
- Gebhart, G.F. Visceral Pain. In Encyclopedia of Neuroscience; Squire, L.R., Ed.; Academic Press: Oxford, UK, 2009; pp. 189–194. [Google Scholar]
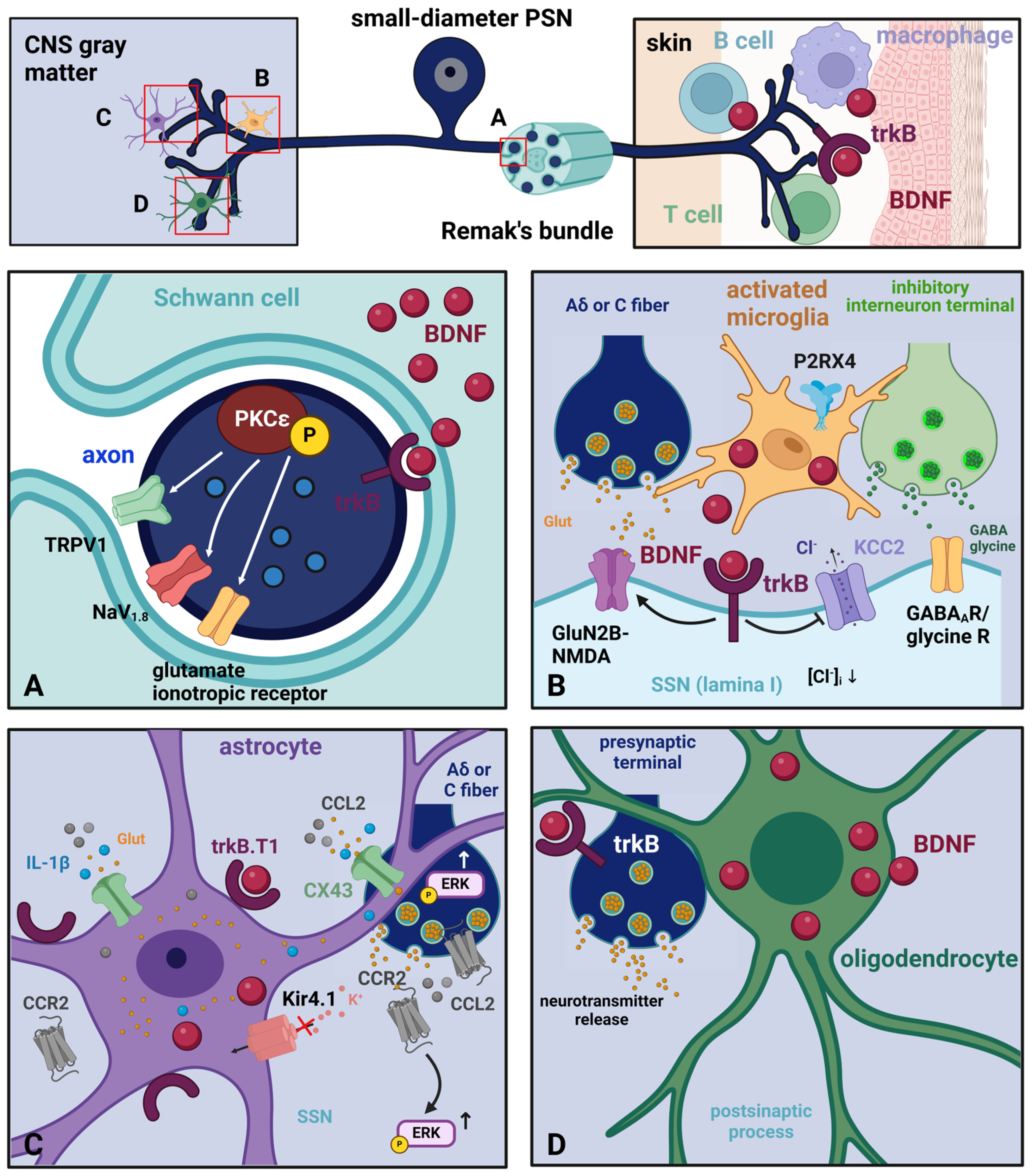
| Location | Type of Cells | Function | Ref |
|---|---|---|---|
| PNS | Schwann cells | Modulation of nociception | [157] |
| DH | Activated microglia (OX-42+/Iba1+) | Inflammatory/neuropathic pain | [158,159,160] |
| Oligodendrocytes (APC+) | Response to injury | [161] | |
| SNTN | Microglia (Iba1+) | Nociception (allodynia) | [115] |
| Au1 | Astrocytes (GFAP+) | Chronic pain | [76] |
| ACC | Astrocytes (GFAP+) | ||
| S1 | Astrocytes (GFAP+) Microglia (Iba1+) | Inflammatory/neuropathic pain | [74,162] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merighi, A. Brain-Derived Neurotrophic Factor, Nociception, and Pain. Biomolecules 2024, 14, 539. https://doi.org/10.3390/biom14050539
Merighi A. Brain-Derived Neurotrophic Factor, Nociception, and Pain. Biomolecules. 2024; 14(5):539. https://doi.org/10.3390/biom14050539
Chicago/Turabian StyleMerighi, Adalberto. 2024. "Brain-Derived Neurotrophic Factor, Nociception, and Pain" Biomolecules 14, no. 5: 539. https://doi.org/10.3390/biom14050539
APA StyleMerighi, A. (2024). Brain-Derived Neurotrophic Factor, Nociception, and Pain. Biomolecules, 14(5), 539. https://doi.org/10.3390/biom14050539