
Geometry Optimization Algorithms in Conjunction with the Machine Learning Potential ANI-2x Facilitate the Structure-Based Virtual Screening and Binding Mode Prediction
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Methods
2.1. Workflow Overlook
2.2. Datasets Preparation
2.3. Structure Preparation and Docking
2.4. ANI-2x/CG-BS Calculation
2.5. Evaluation Metrics
2.5.1. Root-Mean-Square Deviation (RMSD)
2.5.2. Pearson’s Correlation Coefficient (R)
2.5.3. Spearman’s Rank Correlation Coefficient (ρ)
3. Results
3.1. Docking Power Evaluation
3.2. Ranking and Scoring Power Evaluations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McGibbon, M.; Money-Kyrle, S.; Blay, V.; Houston, D.R. SCORCH: Improving structure-based virtual screening with machine learning classifiers, data augmentation, and uncertainty estimation. J. Adv. Res. 2023, 46, 135–147. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y. Improving scoring-docking-screening powers of protein-ligand scoring functions using random forest. J. Comput. Chem. 2017, 38, 169–177. [Google Scholar] [CrossRef]
- Wang, D.; Cui, C.; Ding, X.; Xiong, Z.; Zheng, M.; Luo, X.; Jiang, H.; Chen, K. Improving the Virtual Screening Ability of Target-Specific Scoring Functions Using Deep Learning Methods. Front. Pharmacol. 2019, 10, 924. [Google Scholar] [CrossRef]
- Liu, J.; Wang, R. Classification of Current Scoring Functions. J. Chem. Inf. Model. 2015, 55, 475–482. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, Y. Delta Machine Learning to Improve Scoring-Ranking-Screening Performances of Protein–Ligand Scoring Functions. J. Chem. Inf. Model. 2022, 62, 2696–2712. [Google Scholar] [CrossRef]
- Su, M.; Yang, Q.; Du, Y.; Feng, G.; Liu, Z.; Li, Y.; Wang, R. Comparative Assessment of Scoring Functions: The CASF-2016 Update. J. Chem. Inf. Model. 2019, 59, 895–913. [Google Scholar] [CrossRef]
- Slater, O.; Kontoyianni, M. The compromise of virtual screening and its impact on drug discovery. Expert. Opin. Drug Discov. 2019, 14, 619–637. [Google Scholar] [CrossRef]
- Scardino, V.; Bollini, M.; Cavasotto, C.N. Combination of pose and rank consensus in docking-based virtual screening: The best of both worlds. RSC Adv. 2021, 11, 35383–35391. [Google Scholar] [CrossRef]
- Yang, C.; Chen, E.A.; Zhang, Y. Protein-Ligand Docking in the Machine-Learning Era. Molecules 2022, 27, 4568. [Google Scholar] [CrossRef]
- Lyu, J.; Wang, S.; Balius, T.E.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef]
- Smith, J.S.; Isayev, O.; Roitberg, A.E. ANI-1: An extensible neural network potential with DFT accuracy at force field computational cost. Chem. Sci. 2017, 8, 3192–3203. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Ramezanghorbani, F.; Isayev, O.; Smith, J.S.; Roitberg, A.E. TorchANI: A Free and Open Source PyTorch-Based Deep Learning Implementation of the ANI Neural Network Potentials. J. Chem. Inf. Model. 2020, 60, 3408–3415. [Google Scholar] [CrossRef] [PubMed]
- Devereux, C.; Smith, J.S.; Huddleston, K.K.; Barros, K.; Zubatyuk, R.; Isayev, O.; Roitberg, A.E. Extending the Applicability of the ANI Deep Learning Molecular Potential to Sulfur and Halogens. J. Chem. Theory Comput. 2020, 16, 4192–4202. [Google Scholar] [CrossRef] [PubMed]
- Hao, D.; He, X.; Roitberg, A.E.; Zhang, S.; Wang, J. Development and Evaluation of Geometry Optimization Algorithms in Conjunction with ANI Potentials. J. Chem. Theory Comput. 2022, 18, 978–991. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Hao, D.; He, X.; Wang, L.; Niu, T.; Wang, J. Distribution of Bound Conformations in Conformational Ensembles for X-ray Ligands Predicted by the ANI-2X Machine Learning Potential. J. Chem. Inf. Model. 2023, 63, 6608–6618. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2011, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Ji, B.; He, X.; Zhang, Y.; Zhai, J.; Man, V.H.; Liu, S.; Wang, J. Incorporating structural similarity into a scoring function to enhance the prediction of binding affinities. J. Cheminform. 2021, 13, 11. [Google Scholar] [CrossRef]
- Santos, K.B.; Guedes, I.A.; Karl, A.L.M.; Dardenne, L.E. Highly Flexible Ligand Docking: Benchmarking of the DockThor Program on the LEADS-PEP Protein–Peptide Data Set. J. Chem. Inf. Model. 2020, 60, 667–683. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Windshügel, B. LEADS-PEP: A Benchmark Data Set for Assessment of Peptide Docking Performance. J. Chem. Inf. Model. 2016, 56, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cisneros, G.A.; Cruzeiro, V.W.D.; et al. Amber 2022; University of California: San Francisco, CA, USA, 2022. [Google Scholar]
- Schrödinger, LLC. LigPrep; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, K.; Grauffel, C.; Lim, C. How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Coutsias, E.A.; Wester, M.J. RMSD and Symmetry. J. Comput. Chem. 2019, 40, 1496–1508. [Google Scholar] [CrossRef] [PubMed]
- Al-Jabery, K.K.; Obafemi-Ajayi, T.; Olbricht, G.R.; Wunsch Ii, D.C. (Eds.) 7—Evaluation of cluster validation metrics. In Computational Learning Approaches to Data Analytics in Biomedical Applications; Academic Press: Cambridge, MA, USA, 2020; pp. 189–208. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Greene, D.; Xiao, L.; Qi, R.; Luo, R. Recent Developments and Applications of the MMPBSA Method. Front. Mol. Biosci. 2017, 4, 87. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; He, X.; Zhai, J.; Zhang, Y.; Man, V.H.; Wang, J. Machine learning on ligand-residue interaction profiles to significantly improve binding affinity prediction. Brief. Bioinform. 2021, 22, bbab054. [Google Scholar] [CrossRef] [PubMed]
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J.; et al. A practical guide to large-scale docking (vol 16, pg 4799, 2021). Nat. Protoc. 2022, 17, 177. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Receptor Name/Peptide Residue Number | PDB ID | Resolution | |
|---|---|---|---|
| 11 small molecule–macromolecule systems | 5-HT2AR | 6A93 | 3.00Å |
| A2AR | 3EML | 2.60 Å | |
| CB1 | 5XRA/6KQI | 2.80 Å/3.25 Å | |
| CFX | 1NFU | 2.05 Å | |
| D2R | 6CM4 | 2.87 Å | |
| ERK2 | 6SLG | 1.33 Å | |
| ER | 3OS8 | 2.03 Å | |
| M1R | 5CXV | 2.70 Å | |
| μOR | 5C1M | 2.10 Å | |
| rRNA | 2F4S | 2.80 Å | |
| VEGFR2 | 2OH4 | 2.05 Å | |
| 12 peptide–protein systems | 4 | 3VQG | 1.35 Å |
| 3 | 3BS4 | 1.60 Å | |
| 8 | 1N7F | 1.80 Å | |
| 6 | 3IDG | 1.86 Å | |
| 5 | 2HPL | 1.80 Å | |
| 6 | 4Q6H | 1.90 Å | |
| 5 | 4V3I | 1.50 Å | |
| 8 | 3CH8 | 1.90 Å | |
| 11 | 2XFX | 1.90 Å | |
| 9 | 4N7H | 1.70 Å | |
| 12 | 4J8S | 1.55 Å | |
| 11 | 4EIK | 1.60 Å |
| Receptor Name /PDB ID | RMSD (Å) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ANI-2x/CG-BS | Glide Docking | ||||||||
| Top 1 | Top 3 | Top 5 | Top 10 | Top 1 | Top 3 | Top 5 | Top 10 | ||
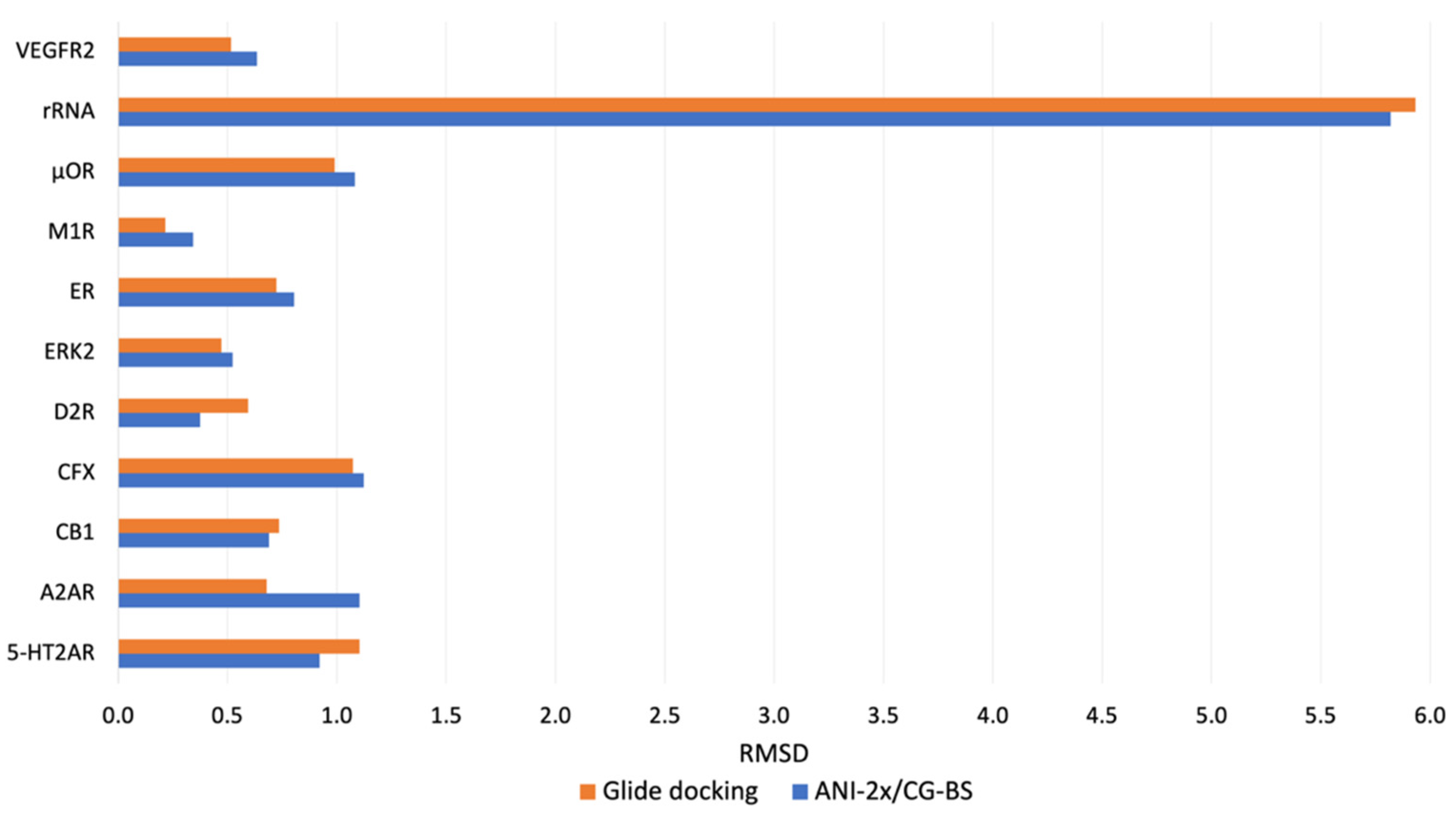
| 11 small molecule–macromolecule systems | 5-HT2AR | 2.96 | 0.92 | 0.92 | 0.92 | 2.98 | 2.25 | 1.56 | 1.10 |
| A2AR | 1.43 | 1.10 | 1.10 | 1.10 | 1.85 | 1.85 | 0.68 | 0.68 | |
| CB1 | 0.88 | 0.69 | 0.69 | 0.69 | 0.82 | 0.74 | 0.74 | 0.74 | |
| CFX | 8.69 | 8.48 | 1.62 | 1.12 | 8.55 | 8.27 | 1.47 | 1.07 | |
| D2R | 5.34 | 0.38 | 0.38 | 0.38 | 1.98 | 0.59 | 0.59 | 0.59 | |
| ERK2 | 0.88 | 0.52 | 0.52 | 0.52 | 0.47 | 0.47 | 0.47 | 0.47 | |
| ER | 1.46 | 1.15 | 1.13 | 0.81 | 1.12 | 1.12 | 1.12 | 0.72 | |
| M1R | 0.89 | 0.34 | 0.34 | 0.34 | 1.15 | 0.22 | 0.22 | 0.22 | |
| μOR | 1.20 | 1.08 | 1.08 | 1.08 | 1.08 | 1.07 | 0.99 | 0.99 | |
| rRNA | 5.92 | 5.83 | 5.82 | 5.82 | 6.11 | 5.96 | 5.96 | 5.93 | |
| VEGFR2 | 0.96 | 0.63 | 0.63 | 0.63 | 0.52 | 0.52 | 0.52 | 0.52 | |
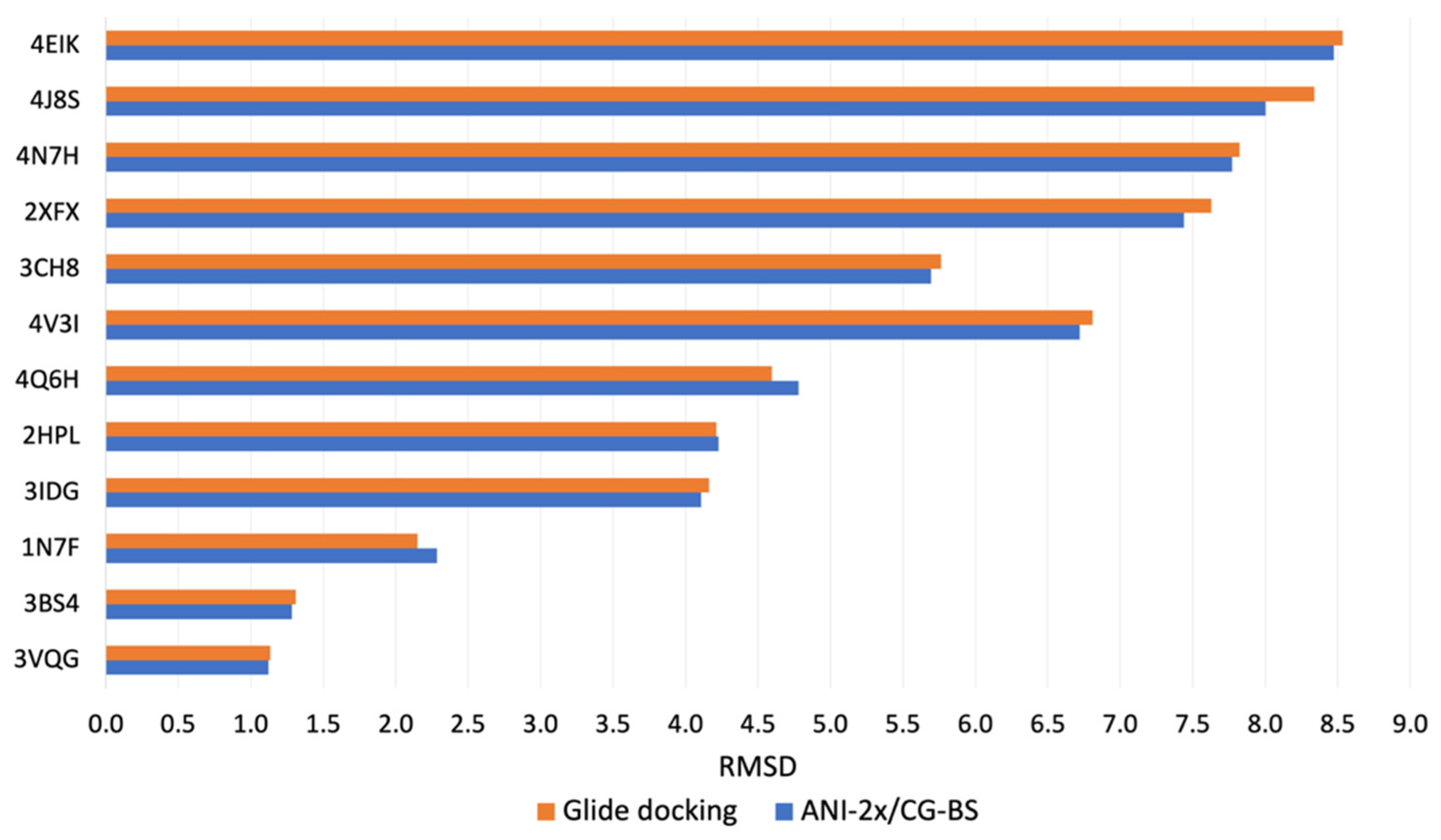
| 12 peptide–protein systems | 3VQG | 1.21 | 1.12 | 1.12 | 1.12 | 1.14 | 1.13 | 1.13 | 1.13 |
| 3BS4 | 1.33 | 1.28 | 1.28 | 1.28 | 1.31 | 1.31 | 1.31 | 1.31 | |
| 1N7F | 2.29 | 2.29 | 2.29 | 2.29 | 2.15 | 2.15 | 2.15 | 2.15 | |
| 3IDG | 6.82 | 6.82 | 4.46 | 4.11 | 6.97 | 4.16 | 4.16 | 4.16 | |
| 2HPL | 4.30 | 4.23 | 4.23 | 4.23 | 4.27 | 4.21 | 4.21 | 4.21 | |
| 4Q6H | 5.93 | 4.78 | 4.78 | 4.78 | 6.05 | 4.63 | 4.60 | 4.60 | |
| 4V3I | 6.72 | 6.72 | 6.72 | 6.72 | 8.08 | 8.08 | 8.08 | 6.81 | |
| 3CH8 | 12.99 | 11.97 | 7.17 | 5.69 | 7.07 | 7.07 | 7.07 | 5.76 | |
| 2XFX | 14.52 | 7.44 | 7.44 | 7.44 | 10.38 | 10.38 | 7.63 | 7.63 | |
| 4N7H | 11.02 | 7.77 | 7.77 | 7.77 | 9.99 | 9.64 | 7.82 | 7.82 | |
| 4J8S | 8.83 | 8.83 | 8.29 | 8.00 | 8.71 | 8.71 | 8.71 | 8.34 | |
| 4EIK | 12.50 | 8.47 | 8.47 | 8.47 | 8.53 | 8.53 | 8.53 | 8.53 | |
| The number of systems with minimal RMSD occurrences in each category | 2 | 15 | 1 | 5 | 5 | 6 | 5 | 7 | |
| Top rank identification success rate (%) | 74% | 48% | |||||||
| 5-HT2AR | A2AR | CB1 | CFX | D2R | ERK2 | ER | M1R | μOR | rRNA | VEGFR2 | Average | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Docking | 0.23 | 0.29 | 0.19 | 0.10 | 0.11 | 0.60 | 0.60 | 0.21 | 0.16 | 0.24 | 0.48 | 0.29 |
| ANI-2x/CG-BS | 0.20 | 0.02 | 0.23 | 0.04 | 0.12 | 0.54 | 0.60 | 0.31 | 0.16 | 0.85 | 0.45 | 0.32 |
| 5-HT2AR | A2AR | CB1 | CFX | D2R | ERK2 | ER | M1R | μOR | rRNA | VEGFR2 | Average | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Docking | 0.21 | 0.30 | 0.21 | 0.29 | 0.10 | 0.57 | 0.65 | 0.30 | 0.14 | 0.14 | 0.47 | 0.30 |
| ANI-2x/CG-BS | 0.19 | 0.07 | 0.25 | 0.06 | 0.14 | 0.36 | 0.61 | 0.47 | 0.16 | 0.69 | 0.46 | 0.31 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; He, X.; Ji, B.; Han, F.; Niu, T.; Cai, L.; Zhai, J.; Hao, D.; Wang, J. Geometry Optimization Algorithms in Conjunction with the Machine Learning Potential ANI-2x Facilitate the Structure-Based Virtual Screening and Binding Mode Prediction. Biomolecules 2024, 14, 648. https://doi.org/10.3390/biom14060648
Wang L, He X, Ji B, Han F, Niu T, Cai L, Zhai J, Hao D, Wang J. Geometry Optimization Algorithms in Conjunction with the Machine Learning Potential ANI-2x Facilitate the Structure-Based Virtual Screening and Binding Mode Prediction. Biomolecules. 2024; 14(6):648. https://doi.org/10.3390/biom14060648
Chicago/Turabian StyleWang, Luxuan, Xibing He, Beihong Ji, Fengyang Han, Taoyu Niu, Lianjin Cai, Jingchen Zhai, Dongxiao Hao, and Junmei Wang. 2024. "Geometry Optimization Algorithms in Conjunction with the Machine Learning Potential ANI-2x Facilitate the Structure-Based Virtual Screening and Binding Mode Prediction" Biomolecules 14, no. 6: 648. https://doi.org/10.3390/biom14060648