
Molecular Mechanisms in Tumorigenesis of Hepatocellular Carcinoma and in Target Treatments—An Overview

Abstract
:1. Introduction—Definition, Epidemiology, and Etiology
2. Molecular Biology of HCC
2.1. Chromosomal Aberrations
2.2. Genetic Mutations, Altered Signaling Pathways, and Epigenetic Modifications Involved in Hepatocarcinogenesis
2.3. Altered Signaling Pathways in Hepatic Carcinogenesis
2.3.1. FGF
2.3.2. TGF
2.3.3. EGF
2.3.4. IGF
2.3.5. HGF/c-MET
2.3.6. VEGF
2.3.7. TP53
2.3.8. Cadherins
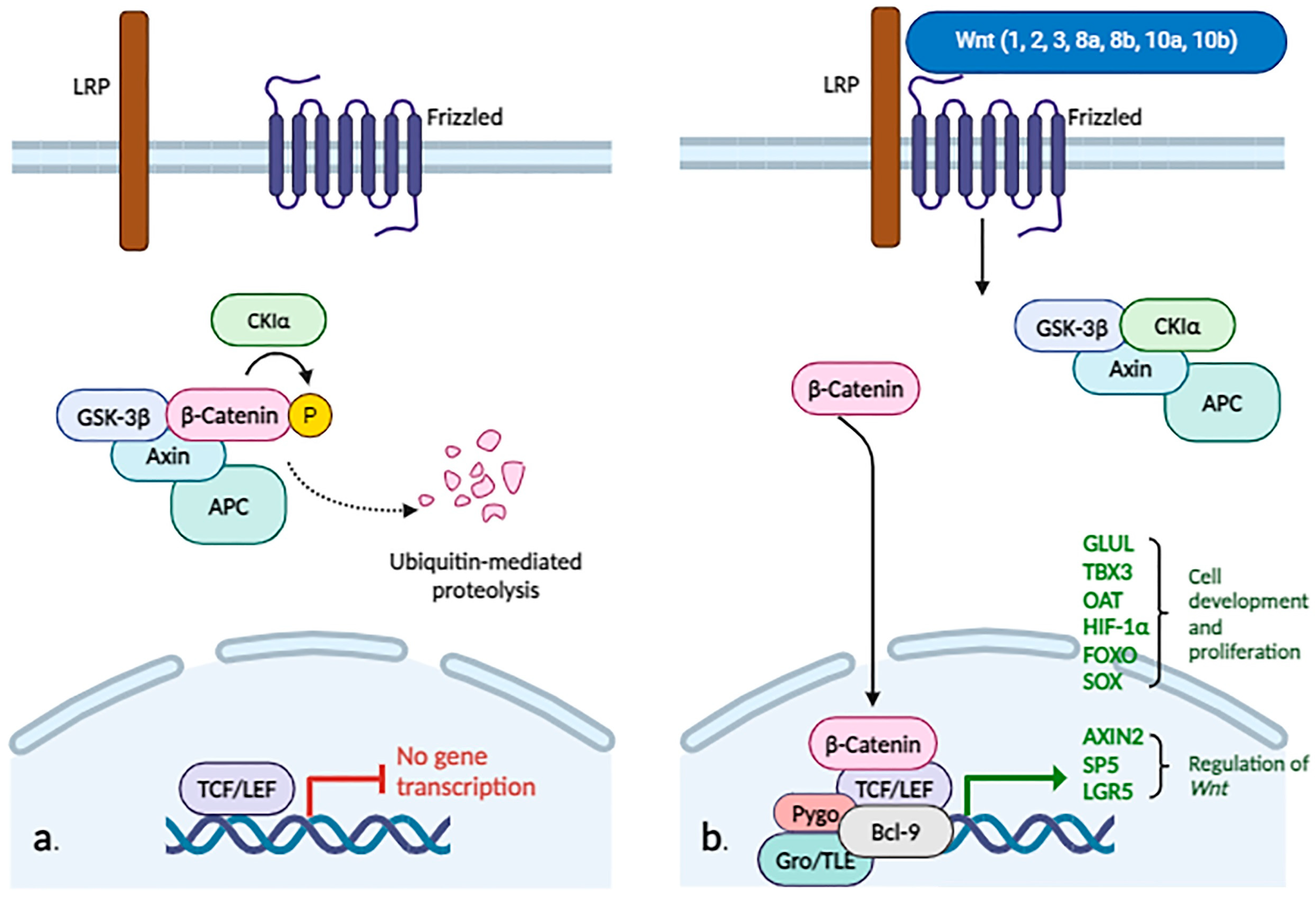
2.3.9. WNT/β-Catenin
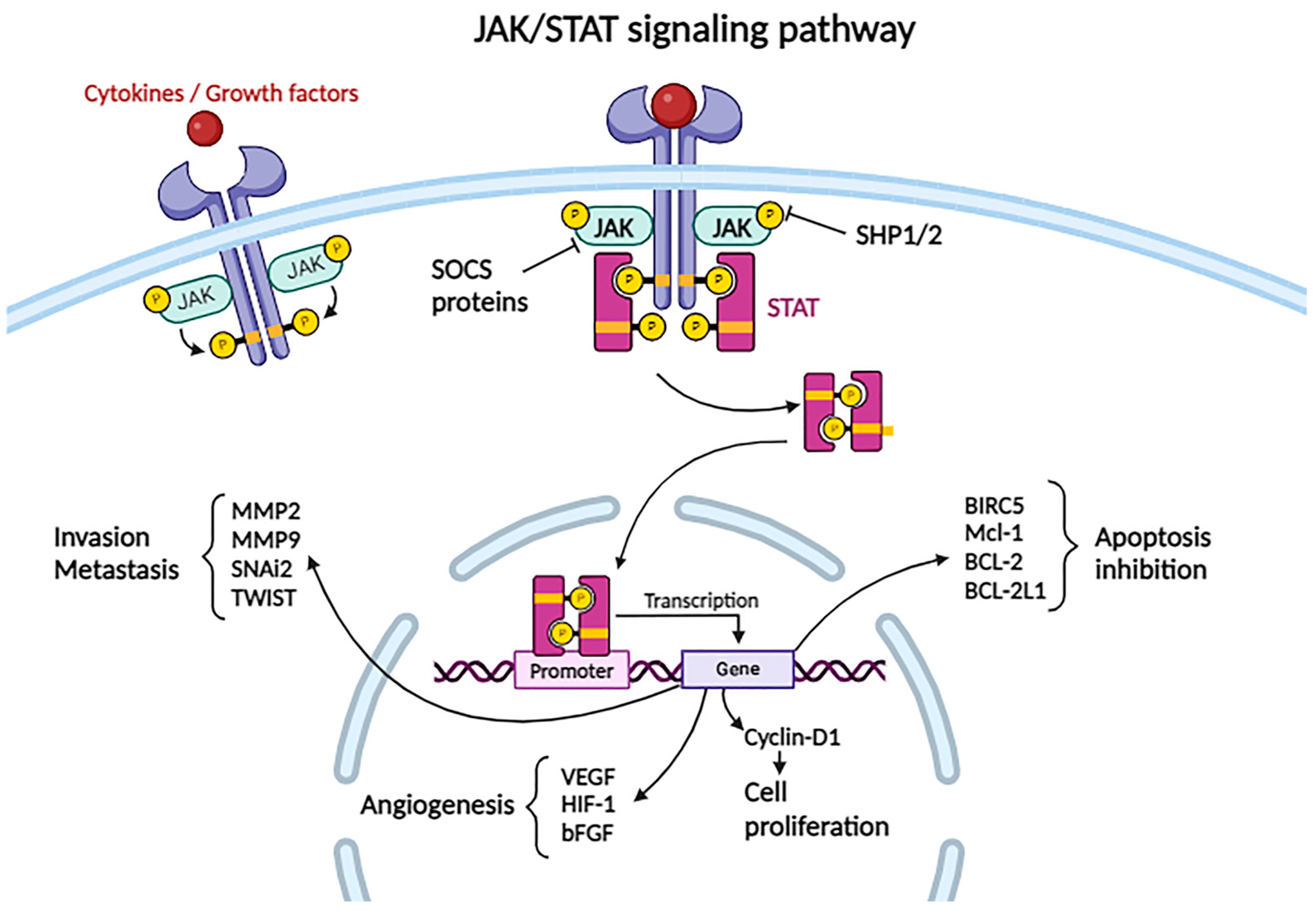
2.3.10. JAK/STAT Pathway
2.3.11. Hippo Signaling Pathway
2.3.12. Hedgehog (Hh) Signaling Pathway
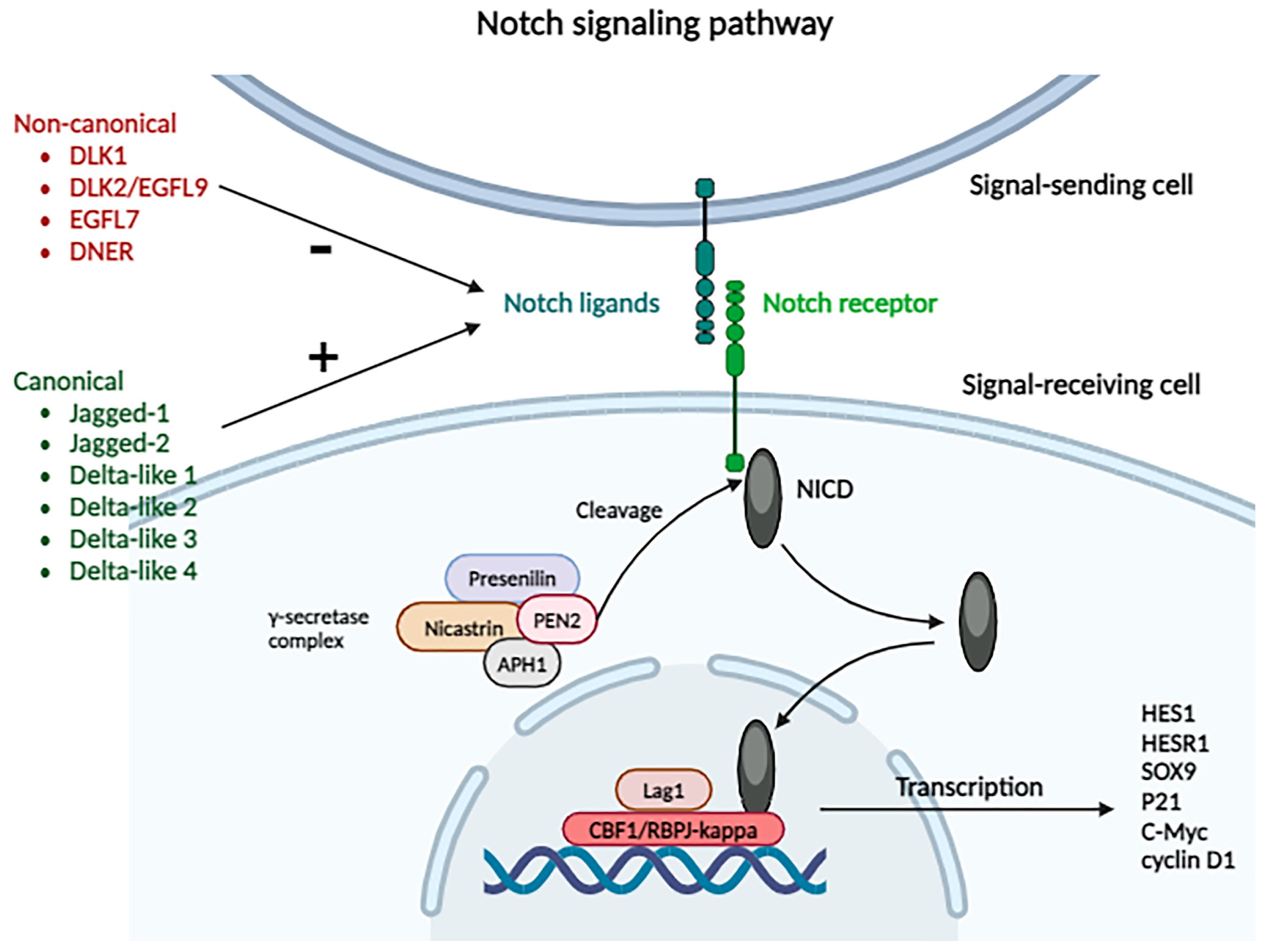
2.3.13. Notch Signaling Pathway
2.4. Nuclear Signaling Pathways Involved in Tumorigenesis
2.4.1. Telomere Shortening and Telomerase Reactivation
2.4.2. Epigenetic Mechanisms
DNA Methylation/Demethylation
Histone Modification
Non-Coding RNAs
miRNAs
piRNAs
snoRNAs
circRNAs
lncRNAs
3. Tumoral Microenvironment
3.1. Hypoxia
3.2. Inflammatory Cells
3.3. Cancer-Associated Fibroblasts
3.4. Cancer Stem Cells
3.5. Mesenchymal Stem Cells
3.6. Extracellular Vesicles
4. Current Treatment Guidelines for HCC, According to the NCCN Guidelines
- A Child–Pugh score classifying them as class A or B (for highly selected patients included in class B, limited hepatic resection is appropriate);
- No portal hypertension;
- Suitable tumor location;
- Adequate liver reserve;
- Suitable liver remnant;
- Other criteria such as an AFP level ≤ 1000 ng/mL and a tumor with a diameter of 2–5 cm or 2–3 tumors with a diameter of 1–3 cm and no macrovascular involvement.
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Torbenson, M.S.; Ng, I.O.L.; Park, Y.N.; Roncalli, M.; Sakarnato, M. Hepatocelullar carcinoma. In WHO Classification of Tumors Editorial Board. Digestive System Tumours, 5th ed.; Paradis, V., Fukayama, M., Park, Y.N., Schirmacher, P., Eds.; International Agency for Research on Cancer: Lyon, France, 2019; pp. 229–232. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Rizzo, G.E.M.; Cabibbo, G.; Craxì, A. Hepatitis B virus-associated hepatocellular carcinoma. Viruses 2022, 14, 986. [Google Scholar] [CrossRef]
- Wang, Q.; Luan, W.; Villanueva, G.A.; Rahbari, N.N.; Yee, H.T.; Manizate, F.; Hiotis, S.P. Clinical prognostic variables in young patients (under 40 years) with hepatitis B virus-associated hepatocellular carcinoma. J. Dig. Dis. 2012, 13, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.; Bruno, S.; Mondelli, M.U.; Maisonneuve, P. Hepatitis C virus genotype 1b as a risk factor for hepatocellular carcinoma development: A meta-analysis. J. Hepatol. 2009, 50, 1142–1154. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.R.; Ilyas, J.; Duan, Z.; El-Serag, H.B. HCV genotype 3 is associated with an increased risk of cirrhosis and hepatocellular cancer in a national sample of U.S. Veterans with HCV. Hepatology 2014, 60, 98–105. [Google Scholar] [CrossRef]
- Saud, L.R.C.; Chagas, A.L.; Maccali, C.; Pinto, P.V.A.; Horvat, N.; Alencar, R.S.S.M.; Tani, C.M.; Abdala, E.; Carrilho, F.J. Hepatocellular carcinoma in patients coinfected with hepatitis B or C and HIV: More aggressive tumor behavior? Eur. J. Gastroenterol. Hepatol. 2021, 33, 583–588. [Google Scholar] [CrossRef]
- Degasperi, E.; Colombo, M. Distinctive features of hepatocellular carcinoma in non-alcoholic fatty liver disease. Lancet Gastroenterol. Hepatol. 2016, 1, 156–164. [Google Scholar] [CrossRef]
- Kucukoglu, O.; Sowa, J.P.; Mazzolini, G.D.; Syn, W.K.; Canbay, A. Hepatokines and adipokines in NASH-related hepatocellular carcinoma. J. Hepatol. 2021, 74, 442–457. [Google Scholar] [CrossRef]
- Ferrell, L.D. Benign and malignant tumors of the Liver. In Surgical Pathology of the GI Tract, Liver, Biliary Tract and Pancreas, 2nd ed.; Odze, R.D., Goldblum, J.R., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2009; pp. 1291–1325. [Google Scholar] [CrossRef]
- Chu, Y.J.; Yang, H.I.; Wu, H.C.; Liu, J.; Wang, L.Y.; Lu, S.N.; Lee, M.H.; Jen, C.L.; You, S.L.; Santella, R.M.; et al. Aflatoxin B1 exposure increases the risk of cirrhosis and hepatocellular carcinoma in chronic hepatitis B virus carriers. Int. J. Cancer 2017, 141, 711–720. [Google Scholar] [CrossRef]
- Raney, V.M.; Harris, T.M.; Stone, M.P. DNA conformation mediates aflatoxin B1-DNA binding and the formation of guanine N7 adducts by aflatoxin B1 8,9-exo-epoxide. Chem. Res. Toxicol. 1993, 6, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.J.; Yang, H.I.; Wu, H.C.; Lee, M.H.; Liu, J.; Wang, L.Y.; Lu, S.N.; Jen, C.L.; You, S.L.; Santella, R.M.; et al. Aflatoxin B1 exposure increases the risk of hepatocellular carcinoma associated with hepatitis C virus infection or alcohol consumption. Eur. J. Cancer 2018, 94, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Paradis, V.; Zalinski, S.; Chelbi, E.; Guedj, N.; Degos, F.; Vilgrain, V.; Bedossa, P.; Belghiti, J. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: A pathological analysis. Hepatology 2009, 49, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Hao, J.; Zeng, J.; Sauter, E.R. SnapShot: FABP functions. Cell 2020, 182, 1066.e1. [Google Scholar] [CrossRef]
- Laouirem, S.; Sannier, A.; Norkowski, E.; Cauchy, F.; Doblas, S.; Rautou, P.E.; Albuquerque, M.; Garteiser, P.; Sognigbé, L.; Raffenne, J.; et al. Endothelial fatty liver binding protein 4: A new targetable mediator in hepatocellular carcinoma related to metabolic syndrome. Oncogene 2019, 38, 3033–3046. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Xiao, H.; Liao, R.; Chen, Q.; Peng, C.; Zhang, Y.; Mu, T.; Wu, Z. Fatty acid binding protein 5 promotes tumor angiogenesis and activates the IL6/STAT3/VEGFA pathway in hepatocellular carcinoma. Biomed. Pharmacother. 2018, 106, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Ohata, T.; Yokoo, H.; Kamiyama, T.; Fukai, M.; Aiyama, T.; Hatanaka, Y.; Hatanaka, K.; Wakayama, K.; Orimo, T.; Kakisaka, T.; et al. Fatty acid-binding protein 5 function in hepatocellular carcinoma through induction of epithelial-mesenchymal transition. Cancer Med. 2017, 6, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, S.; Yang, M. Hepatocellular carcinoma and obesity, type 2 diabetes mellitus, cardiovascular disease: Causing factors, molecular links, and treatment options. Front. Endocrinol. 2021, 12, 808526. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, S.; Aleksandrova, K.; Pischon, T.; Jenab, M.; Fedirko, V.; Trepo, E.; Overvad, K.; Roswall, N.; Tjønneland, A.; Boutron-Ruault, M.C.; et al. Diabetes mellitus, insulin treatment, diabetes duration, and risk of biliary tract cancer and hepatocellular carcinoma in a European cohort. Ann. Oncol. 2013, 24, 2449–2455. [Google Scholar] [CrossRef]
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 17 November 2023).
- GBD 2019 Risk Factors Collaborators. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1223–1249. [Google Scholar] [CrossRef]
- Kanel, G.C. Hepatic tumors, malignant: Hepatocelullar carcinoma. In Pathology of Liver Diseases; Wiley: Hoboken, NJ, USA, 2017; pp. 289–307. [Google Scholar]
- Tornesello, M.L.; Buonaguro, L.; Izzo, F.; Buonaguro, F.M. Molecular alterations in hepatocellular carcinoma associated with hepatitis B and hepatitis C infections. Oncotarget 2016, 7, 25087–25102. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Jiang, L.; Guan, X.Y. The genetic and epigenetic alterations in human hepatocellular carcinoma: A recent update. Protein Cell 2014, 5, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Losic, B.; Craig, A.J.; Villacorta-Martin, C.; Martins-Filho, S.N.; Akers, N.; Chen, X.; Ahsen, M.E.; von Felden, J.; Labgaa, I.; DʹAvola, D.; et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 2020, 11, 291. [Google Scholar] [CrossRef] [PubMed]
- Amirouchene-Angelozzi, N.; Swanton, C.; Bardelli, A. Tumor evolution as a therapeutic target. Cancer Discov. 2017, 7, 805–817. [Google Scholar] [CrossRef]
- Nia, A.; Dhanasekaran, R. Genomic landscape of HCC. Curr. Hepatol. Rep. 2020, 19, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lezana, T.; Lopez-Canovas, J.L.; Villanueva, A. Signaling pathways in hepatocellular carcinoma. Adv. Cancer Res. 2021, 149, 63–101. [Google Scholar] [CrossRef] [PubMed]
- Harimoto, N.; Taguchi, K.; Shirabe, K.; Adachi, E.; Sakaguchi, Y.; Toh, Y.; Okamura, T.; Kayashima, H.; Taketomi, A.; Maehara, Y. The significance of fibroblast growth factor receptor 2 expression in differentiation of hepatocellular carcinoma. Oncology 2010, 78, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Shigesawa, T.; Maehara, O.; Suda, G.; Natsuizaka, M.; Kimura, M.; Shimazaki, T.; Yamamoto, K.; Yamada, R.; Kitagataya, T.; Nakamura, A.; et al. Lenvatinib suppresses cancer stem-like cells in HCC by inhibiting FGFR1-3 signaling, but not FGFR4 signaling. Carcinogenesis 2021, 42, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Dituri, F.; Mancarella, S.; Cigliano, A.; Chieti, A.; Giannelli, G. TGF-β as multifaceted orchestrator in HCC progression: Signaling, EMT, immune microenvironment, and novel therapeutic perspectives. Semin. Liver Dis. 2019, 39, 53–69. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Dimri, M.; Satyanarayana, A. Molecular signaling pathways and therapeutic targets in hepatocellular carcinoma. Cancers 2020, 12, 491. [Google Scholar] [CrossRef] [PubMed]
- Delire, B.; Stärkel, P. The Ras/MAPK pathway and hepatocarcinoma: Pathogenesis and therapeutic implications. Eur. J. Clin. Investig. 2015, 45, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Komposch, K.; Sibilia, M. EGFR signaling in liver diseases. Int. J. Mol. Sci. 2016, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.H.; Guo, W.Z.; Jin, Y.; Zhang, H.P.; Pang, C.; Li, J.; Line, P.D.; Zhang, S.J. Recognition of HER2 expression in hepatocellular carcinoma and its significance in postoperative tumor recurrence. Cancer Med. 2019, 8, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Tovar, V.; Cornella, H.; Moeini, A.; Vidal, S.; Hoshida, Y.; Sia, D.; Peix, J.; Cabellos, L.; Alsinet, C.; Torrecilla, S.; et al. Tumour initiating cells and IGF/FGF signalling contribute to sorafenib resistance in hepatocellular carcinoma. Gut 2017, 66, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Adamek, A.; Kasprzak, A. Insulin-like growth factor (IGF) system in liver diseases. Int. J. Mol. Sci. 2018, 19, 1308. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Quetglas, I.; Pinyol, R.; Dauch, D.; Torrecilla, S.; Tovar, V.; Moeini, A.; Alsinet, C.; Portela, A.; Rodriguez-Carunchio, L.; Solé, M.; et al. IGF2 is up-regulated by epigenetic mechanisms in hepatocellular carcinomas and is an actionable oncogene product in experimental models. Gastroenterology 2016, 151, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Bouattour, M.; Raymond, E.; Qin, S.; Cheng, A.L.; Stammberger, U.; Locatelli, G.; Faivre, S. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology 2018, 67, 1132–1149. [Google Scholar] [CrossRef] [PubMed]
- Firtina Karagonlar, Z.; Koc, D.; Iscan, E.; Erdal, E.; Atabey, N. Elevated hepatocyte growth factor expression as an autocrine c-Met activation mechanism in acquired resistance to sorafenib in hepatocellular carcinoma cells. Cancer Sci. 2016, 107, 407–416. [Google Scholar] [CrossRef]
- Boccaccio, C.; Comoglio, P.M. Invasive growth: A MET-driven genetic programme for cancer and stem cells. Nat. Rev. Cancer 2006, 6, 637–645. [Google Scholar] [CrossRef]
- Pinto, E.; Pelizzaro, F.; Farinati, F.; Russo, F.P. Angiogenesis and hepatocellular carcinoma: From molecular mechanisms to systemic therapies. Medicina 2023, 59, 1115. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007, 26, 1324–1337. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, M.M.; Day, M.L. Soluble E-cadherin: More than a symptom of disease. Front. Biosci. 2012, 17, 1948–1964. [Google Scholar] [CrossRef] [PubMed]
- Berx, G.; van Roy, F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a003129. [Google Scholar] [CrossRef] [PubMed]
- McCrea, P.D.; Maher, M.T.; Gottardi, C.J. Nuclear signaling from cadherin adhesion complexes. Curr. Top. Dev. Biol. 2015, 112, 129–196. [Google Scholar] [CrossRef]
- Hu, Q.P.; Kuang, J.Y.; Yang, Q.K.; Bian, X.W.; Yu, S.C. Beyond a tumor suppressor: Soluble E-cadherin promotes the progression of cancer. Int. J. Cancer 2016, 138, 2804–2812. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Jin, S.; Li, Y.; Khadaroo, P.A.; Dai, Y.; He, L.; Zhou, D.; Lin, H. Genetic and epigenetic regulation of E-cadherin signaling in human hepatocellular carcinoma. Cancer Manag. Res. 2019, 11, 8947–8963. [Google Scholar] [CrossRef] [PubMed]
- Anthony, C.C.; Robbins, D.J.; Ahmed, Y.; Lee, E. Nuclear regulation of Wnt/β-catenin signaling: It’s a complex situation. Genes 2020, 11, 886. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Alam, A.; Pant, R.; Chattopadhyay, S. Wnt signaling and its significance within the tumor microenvironment: Novel therapeutic insights. Front. Immunol. 2019, 10, 2872. [Google Scholar] [CrossRef]
- Liu, L.J.; Xie, S.X.; Chen, Y.T.; Xue, J.L.; Zhang, C.J.; Zhu, F. Aberrant regulation of Wnt signaling in hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 7486–7499. [Google Scholar] [CrossRef]
- Ackers, I.; Malgor, R. Interrelationship of canonical and non-canonical Wnt signalling pathways in chronic metabolic diseases. Diabetes Vasc. Dis. Res. 2018, 15, 3–13. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Tang, S. WNT/β-catenin signaling in the development of liver cancers. Biomed. Pharmacother. 2020, 132, 110851. [Google Scholar] [CrossRef] [PubMed]
- Gujral, T.S.; Chan, M.; Peshkin, L.; Sorger, P.K.; Kirschner, M.W.; MacBeath, G. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 2014, 159, 844–856. [Google Scholar] [CrossRef] [PubMed]
- White, B.D.; Chien, A.J.; Dawson, D.W. Dysregulation of Wnt/β-catenin signaling in gastrointestinal cancers. Gastroenterology 2012, 142, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, A.M.; Fuentes, D.; Morshid, A.I.; Burke, M.R.; Kaseb, A.O.; Hassan, M.; Hazle, J.D.; Elsayes, K.M. Role of Wnt/β-catenin signaling in hepatocellular carcinoma, pathogenesis, and clinical significance. J. Hepatocell. Carcinoma 2018, 5, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Lee, S.; Lee, J.; Moon, H.; Ro, S.W. Exploring the JAK/STAT signaling pathway in hepatocellular carcinoma: Unraveling signaling complexity and therapeutic implications. Int. J. Mol. Sci. 2023, 24, 13764. [Google Scholar] [CrossRef] [PubMed]
- Hin Tang, J.J.; Hao Thing, D.K.; Lim, J.J.; Toh, T.B. JAK/STAT signaling in hepatocellular carcinoma. Hepat. Oncol. 2020, 7, HEP18. [Google Scholar] [CrossRef] [PubMed]
- Durham, G.A.; Williams, J.J.L.; Nasim, M.T.; Palmer, T.M. Targeting SOCS proteins to control JAK-STAT signalling in disease. Trends Pharmacol. Sci. 2019, 40, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Hu, Y.; Lan, T.; Guan, K.L.; Luo, T.; Luo, M. The Hippo signalling pathway and its implications in human health and diseases. Signal Transduct. Target. Ther. 2022, 7, 376. [Google Scholar] [CrossRef]
- Kim, M.; Jho, E.H. Cross-talk between Wnt/β-catenin and Hippo signaling pathways: A brief review. BMB Rep. 2014, 47, 540–545. [Google Scholar] [CrossRef]
- Mranda, G.M.; Xiang, Z.P.; Liu, J.J.; Wei, T.; Ding, Y. Advances in prognostic and therapeutic targets for hepatocellular carcinoma and intrahepatic cholangiocarcinoma: The hippo signaling pathway. Front. Oncol. 2022, 12, 937957. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zeng, W.; Gai, X.; Xu, Q.; Li, C.; Liang, Z.; Tuo, H.; Liu, Q. Role of the Hedgehog pathway in hepatocellular carcinoma (review). Oncol. Rep. 2013, 30, 2020–2026. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, C.M.; Viscardi, G.; Papaccio, F.; Esposito, G.; Martini, G.; Ciardiello, D.; Martinelli, E.; Ciardiello, F.; Morgillo, F. Implication of the Hedgehog pathway in hepatocellular carcinoma. World J. Gastroenterol. 2017, 23, 4330–4340. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhang, Z.; Zhang, P.; Yu, M.; Yang, T. Role of canonical Hedgehog signaling pathway in liver. Int. J. Biol. Sci. 2018, 14, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Yoon, J.W.; Xiao, X.; Dean, N.M.; Monia, B.P.; Marcusson, E.G. Selective down-regulation of glioma-associated oncogene 2 inhibits the proliferation of hepatocellular carcinoma cells. Cancer Res. 2007, 67, 3583–3593. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, J.; Zheng, J.; Wei, A. The carcinogenic role of the Notch signaling pathway in the development of hepatocellular carcinoma. J. Cancer 2019, 10, 1570–1579. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.; Zhang, G.; Wu, Y.; Stawicki, S.; Liang, W.C.; Chanthery, Y.; Kowalski, J.; Watts, R.J.; Callahan, C.; Kasman, I.; et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 2006, 444, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Khan, S.K.; Gvozdenovic-Jeremic, J.; Kim, Y.; Dahlman, J.; Kim, H.; Park, O.; Ishitani, T.; Jho, E.H.; Gao, B.; et al. Hippo signaling interactions with Wnt/β-catenin and Notch signaling repress liver tumorigenesis. J. Clin. Investig. 2017, 127, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.; Yang, Y. Hepatic Hippo signaling inhibits development of hepatocellular carcinoma. Clin. Mol. Hepatol. 2020, 26, 742–750. [Google Scholar] [CrossRef]
- Nault, J.C.; Ningarhari, M.; Rebouissou, S.; Zucman-Rossi, J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 544–558. [Google Scholar] [CrossRef]
- Lorbeer, F.K.; Hockemeyer, D. TERT promoter mutations and telomeres during tumorigenesis. Curr. Opin. Genet. Dev. 2020, 60, 56–62. [Google Scholar] [CrossRef]
- In der Stroth, L.; Tharehalli, U.; Günes, C.; Lechel, A. Telomeres and telomerase in the development of liver Cancer. Cancers 2020, 12, 2048. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Barrena, M.G.; Arechederra, M.; Colyn, L.; Berasain, C.; Avila, M.A. Epigenetics in hepatocellular carcinoma development and therapy: The tip of the iceberg. JHEP Rep. 2020, 2, 100167. [Google Scholar] [CrossRef] [PubMed]
- Gentilini, D.; Scala, S.; Gaudenzi, G.; Garagnani, P.; Capri, M.; Cescon, M.; Grazi, G.L.; Bacalini, M.G.; Pisoni, S.; Dicitore, A.; et al. Epigenome-wide association study in hepatocellular carcinoma: Identification of stochastic epigenetic mutations through an innovative statistical approach. Oncotarget 2017, 8, 41890–41902. [Google Scholar] [CrossRef] [PubMed]
- Braghini, M.R.; Lo Re, O.; Romito, I.; Fernandez-Barrena, M.G.; Barbaro, B.; Pomella, S.; Rotta, R.; Vinciguerra, M.; Avila, M.A.; Alisi, A. Epigenetic remodelling in human hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 107. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Cai, Y.; Liu, D.; Li, M.; Sha, Y.; Zhang, W.; Wang, K.; Gong, J.; Tang, N.; Huang, A.; et al. Pharmacological or transcriptional inhibition of both HDAC1 and 2 leads to cell cycle blockage and apoptosis via p21Waf1/Cip1 and p19INK4d upregulation in hepatocellular carcinoma. Cell Prolif. 2018, 51, e12447. [Google Scholar] [CrossRef] [PubMed]
- Al-Yhya, N.; Khan, M.F.; Almeer, R.S.; Alshehri, M.M.; Aldughaim, M.S.; Wadaan, M.A. Pharmacological inhibition of HDAC1/3-interacting proteins induced morphological changes, and hindered the cell proliferation and migration of hepatocellular carcinoma cells. Environ. Sci. Pollut. Res. Int. 2021, 28, 49000–49013. [Google Scholar] [CrossRef] [PubMed]
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Georgakopoulou, V.E.; Sarantis, P.; Antoniou, E.A.; Karamouzis, M.V.; Nonni, A.; Schizas, D.; Diamantis, E.; et al. Histone deacetylase inhibitors in the treatment of hepatocellular carcinoma: Current evidence and future opportunities. J. Pers. Med. 2021, 11, 223. [Google Scholar] [CrossRef] [PubMed]
- Rikimaru, T.; Taketomi, A.; Yamashita, Y.; Shirabe, K.; Hamatsu, T.; Shimada, M.; Maehara, Y. Clinical significance of histone deacetylase 1 expression in patients with hepatocellular carcinoma. Oncology 2007, 72, 69–74. [Google Scholar] [CrossRef]
- Goehringer, N.; Peng, Y.; Nitzsche, B.; Biermann, H.; Pradhan, R.; Schobert, R.; Herling, M.; Höpfner, M.; Biersack, B. Improved anticancer activities of a new pentafluorothio-substituted Vorinostat-type histone deacetylase inhibitor. Pharmaceuticals 2021, 14, 1319. [Google Scholar] [CrossRef]
- Jiang, C.; He, Z.L.; Hu, X.H.; Ma, P.Y. MiRNA-15a-3p inhibits the metastasis of hepatocellular carcinoma by interacting with HMOX1. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 12694–12700. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.B.; Tan, X.L.; Dong, K.S.; Zhang, H.W.; Chen, X.P.; Chu, L.; Zhang, B.X. miRNA-448 inhibits cell growth by targeting BCL-2 in hepatocellular carcinoma. Dig. Liver Dis. 2019, 51, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.; Cho, K.J.; Ju, H.L.; Moon, H.; Choi, S.H.; Chung, S.I.; Park, J.Y.; Choi, K.H.; Kim, D.Y.; Ahn, S.H.; et al. Analysis of miRNA expression patterns in human and mouse hepatocellular carcinoma cells. Hepatol. Res. 2015, 45, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Du, J.; Liu, Z.; Zhang, D.; Yao, X.; Yang, Y. MiR-6875-3p promotes the proliferation, invasion and metastasis of hepatocellular carcinoma via BTG2/FAK/Akt pathway. J. Exp. Clin. Cancer Res. 2019, 38, 7. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Zhang, H.; Liu, Y.; Liu, X.; Wang, X.; Sun, X.; Cheng, Y. miR-99b-3p promotes hepatocellular carcinoma metastasis and proliferation by targeting protocadherin 19. Gene 2019, 698, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Sui, C.; Xu, F.; Shen, W.; Geng, L.; Xie, F.; Dai, B.; Lu, J.; Zhang, M.; Yang, J. Overexpression of miR-218 inhibits hepatocellular carcinoma cell growth through RET. Tumour Biol. 2015, 36, 1511–1518. [Google Scholar] [CrossRef]
- Xie, F.; Yuan, Y.; Xie, L.; Ran, P.; Xiang, X.; Huang, Q.; Qi, G.; Guo, X.; Xiao, C.; Zheng, S. miRNA-320a inhibits tumor proliferation and invasion by targeting c-Myc in human hepatocellular carcinoma. OncoTargets Ther. 2017, 10, 885–894. [Google Scholar] [CrossRef]
- Liu, D.; Dong, L.; Liu, Y.; Wen, D.; Gao, D.; Sun, H.; Fan, J.; Wu, W.A. c-Myc/miR-17-5p feedback loop regulates metastasis and invasion of hepatocellular carcinoma. Tumour Biol. 2016, 37, 5039–5047. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Xu, Q.; Xiao, S.; Wu, Z.; Gong, J.; Liu, C.; Ren, G.; Wu, H. MicroRNA-424-5p acts as a potential biomarker and inhibits proliferation and invasion in hepatocellular carcinoma by targeting TRIM29. Life Sci. 2019, 224, 1–11. [Google Scholar] [CrossRef]
- Han, Y.N.; Li, Y.; Xia, S.Q.; Zhang, Y.Y.; Zheng, J.H.; Li, W. PIWI Proteins and PIWI-Interacting RNA: Emerging roles in cancer. Cell Physiol. Biochem. 2017, 44, 1–20. [Google Scholar] [CrossRef]
- Law, P.T.; Qin, H.; Ching, A.K.; Lai, K.P.; Co, N.N.; He, M.; Lung, R.W.; Chan, A.W.; Chan, T.F.; Wong, N. Deep sequencing of small RNA transcriptome reveals novel non-coding RNAs in hepatocellular carcinoma. J. Hepatol. 2013, 58, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Wang, J.; Zhang, P.; Yuan, L.X.; Ju, L.L.; Wang, H.X.; Chen, L.; Cao, Y.L.; Cai, W.H.; Ni, Y.; et al. PIWIL1 interacting RNA piR-017724 inhibits proliferation, invasion, and migration, and inhibits the development of HCC by silencing PLIN3. Front. Oncol. 2023, 13, 1203821. [Google Scholar] [CrossRef] [PubMed]
- Koduru, S.V.; Leberfinger, A.N.; Kawasawa, Y.I.; Mahajan, M.; Gusani, N.J.; Sanyal, A.J.; Ravnic, D.J. Non-coding RNAs in various stages of liver disease leading to hepatocellular carcinoma: Differential expression of miRNAs, piRNAs, lncRNAs, circRNAs, and sno/mt-RNAs. Sci. Rep. 2018, 8, 7967. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Fan, T.; Zhao, Y.; Hu, R.; Yan, D.; Sun, D.; Gao, L.; Qin, L.; Xue, X. Circular RNA hsa_circ_0001306 functions as a competing endogenous RNA to regulate FBXW7 expression by sponging miR-527 in hepatocellular carcinoma. J. Cancer 2021, 12, 6531–6542. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Li, E.; Liu, W.; Yang, Q.; Xie, C.; Ai, J.; Zhou, F.; Liao, W.; Wu, L. Circular RNA MYLK promotes hepatocellular carcinoma progression through the miR29a/KMT5C signaling pathway. Onco Targets Ther. 2020, 13, 8615–8627. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.G.; Wang, T.; Ding, M.; Xiang, S.H.; Shi, M.; Zhai, B. hsa_circ_0091570 acts as a ceRNA to suppress hepatocellular cancer progression by sponging hsa-miR-1307. Cancer Lett. 2019, 460, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.; Mirzaei, S.; Zandieh, M.A.; Rezaei, S.; Kakavand, A.; Dehghanpour, A.; Esmaeili, N.; Ghahremanzade, A.; Saebfar, H.; Heidari, H.; et al. Long non-coding RNAs (lncRNAs) in hepatocellular carcinoma progression: Biological functions and new therapeutic targets. Prog. Biophys. Mol. Biol. 2023, 177, 207–228. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, K.; Zou, X.; Hua, Z.; Wang, H.; Bian, W.; Wang, H.; Chen, F.; Dai, T. LncRNA DHRS4-AS1 ameliorates hepatocellular carcinoma by suppressing proliferation and promoting apoptosis via miR-522-3p/SOCS5 axis. Bioengineered 2021, 12, 10862–10877. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Qiu, W.; Kong, Z.; Wu, S.; Liu, Y.; Zhu, C.; Qin, X. LncRNA GSTM3TV2 Promotes cell proliferation and invasion via miR-597/FOSL2 axis in hepatocellular carcinoma. BioMed Res. Int. 2021, 2021, 3445970. [Google Scholar] [CrossRef]
- Zhang, N.; Shen, H.; Huang, S.; Wang, F.; Liu, H.; Xie, F.; Jiang, L.; Chen, X. LncRNA FGD5-AS1 functions as an oncogene to upregulate GTPBP4 expression by sponging miR-873-5p in hepatocellular carcinoma. Eur. J. Histochem. 2021, 65, 3300. [Google Scholar] [CrossRef]
- Wu, Y.H.; Yu, B.; Chen, W.X.; Ai, X.; Zhang, W.; Dong, W.; Shao, Y.J. Downregulation of lncRNA SBF2-AS1 inhibits hepatocellular carcinoma proliferation and migration by regulating the miR-361-5p/TGF-β1 signaling pathway. Aging 2021, 13, 19260–19271. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Shi, H.; Yang, J.; Jiang, N. Long non-coding RNA NEAT1 promoted hepatocellular carcinoma cell proliferation and reduced apoptosis through the regulation of Let-7b-IGF-1R Axis. Onco Targets Ther. 2019, 12, 10401–10413. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, Y.; Liu, Y.; Fang, L.; Li, L.; Sun, J.; Pan, Z.; Xin, W.; Huang, P. HIF-2α activated lncRNA NEAT1 promotes hepatocellular carcinoma cell invasion and metastasis by affecting the epithelial-mesenchymal transition. J. Cell Biochem. 2018, 119, 3247–3256. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Sahu, B.D.; Mugale, M.N. Role of lncRNAs in hepatocellular carcinoma. Life Sci. 2023, 325, 121751. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; You, J.; Zheng, Q.; Zhu, Y. Downregulation of lncRNA OGFRP1 inhibits hepatocellular carcinoma progression by AKT/mTOR and Wnt/β-catenin signaling pathways. Cancer Manag. Res. 2018, 10, 1817–1826. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yu, X.; Sun, Z.; He, Y.; Guo, W. Roles of lncRNAs mediating Wnt/β-catenin signaling in HCC. Front. Oncol. 2022, 12, 831366. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Zheng, X.; Zhang, J. Long non-coding RNA CRNDE promotes heptaocellular carcinoma cell proliferation by regulating PI3K/Akt /β-catenin signaling. Biomed. Pharmacother. 2018, 103, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, Y.; Qin, X.; Geng, H.; Zuo, D.; Zhao, Q. PI3K/AKT/mTOR pathway-related long non-coding RNAs: Roles and mechanisms in hepatocellular carcinoma. Pharmacol. Res. 2020, 160, 105195. [Google Scholar] [CrossRef]
- Hjazi, A.; Obaid, R.F.; Ali, S.S.; Abdullaev, B.; Alsaab, H.O.; Huldani, H.; Romero-Parra, R.M.; Mustafa, Y.F.; Hussien, B.M.; Saadoon, S.J. The cross-talk between LncRNAs and JAK-STAT signaling pathway in cancer. Pathol. Res. Pract. 2023, 248, 154657. [Google Scholar] [CrossRef]
- Bu, W.J.; Fang, Z.; Li, W.L.; Wang, X.; Dong, M.J.; Tao, Q.Y.; Zhang, L.; Xu, Y.Q. LINC00240 sponges miR-4465 to promote proliferation, migration, and invasion of hepatocellular carcinoma cells via HGF/c-MET signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 10452–10461. [Google Scholar] [CrossRef]
- Pu, J.; Li, W.; Wang, A.; Zhang, Y.; Qin, Z.; Xu, Z.; Wang, J.; Lu, Y.; Tang, Q.; Wei, H. Long non-coding RNA HOMER3-AS1 drives hepatocellular carcinoma progression via modulating the behaviors of both tumor cells and macrophages. Cell Death Dis. 2021, 12, 1103. [Google Scholar] [CrossRef]
- Lu, L.; Huang, J.; Mo, J.; Da, X.; Li, Q.; Fan, M.; Lu, H. Exosomal lncRNA TUG1 from cancer-associated fibroblasts promotes liver cancer cell migration, invasion, and glycolysis by regulating the miR-524-5p/SIX1 axis. Cell Mol. Biol. Lett. 2022, 27, 17. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Du, Y.; Guan, X.Y.; Yan, Q. The current status of tumor microenvironment and cancer stem cells in sorafenib resistance of hepatocellular carcinoma. Front. Oncol. 2023, 13, 1204513. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Sun, G.; Zhang, Y.; Kong, X.; Rong, D.; Song, J.; Tang, W.; Wang, X. Targeting immune cells in the tumor microenvironment of HCC: New opportunities and challenges. Front. Cell Dev. Biol. 2021, 9, 775462. [Google Scholar] [CrossRef]
- Sun, L.Y.; Zhang, K.J.; Xie, Y.M.; Liu, J.W.; Xiao, Z.Q. Immunotherapies for advanced hepatocellular carcinoma. Front. Pharmacol. 2023, 14, 1138493. [Google Scholar] [CrossRef]
- Hou, K.; Xu, X.; Ge, X.; Jiang, J.; Ouyang, F. Blockade of PD-1 and CTLA-4: A potent immunotherapeutic approach for hepatocellular carcinoma. BioFactors 2024, 50, 250–265. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Koletsa, T.; Mitroulis, I.; Germanidis, G. Tumor-associated macrophages in hepatocellular carcinoma pathogenesis, prognosis and therapy. Cancers 2022, 14, 226. [Google Scholar] [CrossRef]
- Xiong, S.; Wang, R.; Chen, Q.; Luo, J.; Wang, J.; Zhao, Z.; Li, Y.; Wang, Y.; Wang, X.; Cheng, B. Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Am. J. Cancer Res. 2018, 8, 302–316. [Google Scholar] [PubMed]
- Ji, J.; Eggert, T.; Budhu, A.; Forgues, M.; Takai, A.; Dang, H.; Ye, Q.; Lee, J.S.; Kim, J.H.; Greten, T.F.; et al. Hepatic stellate cell and monocyte interaction contributes to poor prognosis in hepatocellular carcinoma. Hepatology 2015, 62, 481–495. [Google Scholar] [CrossRef]
- Zheng, H.; Pomyen, Y.; Hernandez, M.O.; Li, C.; Livak, F.; Tang, W.; Dang, H.; Greten, T.F.; Davis, J.L.; Zhao, Y.; et al. Single-cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma. Hepatology 2018, 68, 127–140. [Google Scholar] [CrossRef]
- Yamashita, T.; Wang, X.W. Cancer stem cells in the development of liver cancer. J. Clin. Investig. 2013, 123, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, N.; Zhu, Y.; Wen, W. The role of mesenchymal stem cells in the occurrence, development, and therapy of hepatocellular carcinoma. Cancer Med. 2022, 11, 931–943. [Google Scholar] [CrossRef]
- Zong, C.; Zhang, H.; Yang, X.; Gao, L.; Hou, J.; Ye, F.; Jiang, J.; Yang, Y.; Li, R.; Han, Z.; et al. The distinct roles of mesenchymal stem cells in the initial and progressive stage of hepatocarcinoma. Cell Death Dis. 2018, 9, 345. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.W.; Li, J.Y.; Zhang, X.C.; Liu, Y.; Liu, Q.Y.; Xiao, L.; Zhang, W.J.; Wu, H.Y.; Deng, K.Y.; Xin, H.B. Human amniotic mesenchymal stem cells inhibit hepatocellular carcinoma in tumour-bearing mice. J. Cell Mol. Med. 2020, 24, 10525–10541. [Google Scholar] [CrossRef]
- Liu, C.; Liu, Y.; Xu, X.X.; Guo, X.; Sun, G.W.; Ma, X.J. Mesenchymal stem cells enhance the metastasis of 3D-cultured hepatocellular carcinoma cells. BMC Cancer 2016, 16, 566. [Google Scholar] [CrossRef] [PubMed]
- Seay, T.W.; Suo, Z. Roles of extracellular vesicles on the progression and metastasis of hepatocellular carcinoma. Cells 2023, 12, 1879. [Google Scholar] [CrossRef]
- Wang, H.C.; Yin, W.X.; Jiang, M.; Han, J.Y.; Kuai, X.W.; Sun, R.; Sun, Y.F.; Ji, J.L. Function and biomedical implications of exosomal microRNAs delivered by parenchymal and nonparenchymal cells in hepatocellular carcinoma. World J. Gastroenterol. 2023, 29, 5435–5451. [Google Scholar] [CrossRef]
- Liu, C.; Wu, H.; Mao, Y.; Chen, W.; Chen, S. Exosomal microRNAs in hepatocellular carcinoma. Cancer Cell Int. 2021, 21, 254. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. NCCN Guidelines: Hepatocellular Carcinoma; Version 1.2024; National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2024. [Google Scholar]
- Ho, W.J.; Zhu, Q.; Durham, J.; Popovic, A.; Xavier, S.; Leatherman, J.; Mohan, A.; Mo, G.; Zhang, S.; Gross, N.; et al. Neoadjuvant Cabozantinib and Nivolumab converts locally advanced HCC into resectable disease with enhanced antitumor immunity. Nat. Cancer 2021, 2, 891–903. [Google Scholar] [CrossRef]
- Peng, Z.; Fan, W.; Zhu, B.; Wang, G.; Sun, J.; Xiao, C.; Huang, F.; Tang, R.; Cheng, Y.; Huang, Z.; et al. Lenvatinib combined with transarterial chemoembolization as first-line treatment for advanced hepatocellular carcinoma: A phase III, randomized clinical trial (LAUNCH). J. Clin. Oncol. 2023, 41, 117–127. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Tang, W.; Qian, X.; Li, X.; Cheng, F.; Wang, K.; Zhang, F.; Zhang, C.; Li, D.; Song, J.; et al. Efficacy and safety of Camrelizumab plus Apatinib during the perioperative period in resectable hepatocellular carcinoma: A single-arm, open label, phase II clinical trial. J. Immunother. Cancer 2022, 10, e004656. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and safety of Nivolumab plus Ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: The Checkmate 040 randomized clinical trial. JAMA Oncol. 2020, 6, e204564, Erratum in JAMA Oncol. 2021, 7, 140. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Sangro, B.; Harris, W.; Ikeda, M.; Okusaka, T.; Kang, Y.K.; Qin, S.; Tai, D.W.; Lim, H.Y.; Yau, T.; et al. Safety, efficacy, and pharmacodynamics of Tremelimumab plus Durvalumab for patients with unresectable hepatocellular carcinoma: Randomized expansion of a phase I/II study. J. Clin. Oncol. 2021, 39, 2991–3001. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Mei, J.; Cheng, Y.; Li, Q.; Wang, Q.X.; Fang, C.K.; Lei, Q.C.; Huang, H.K.; Cao, M.R.; Luo, R.; et al. Postoperative adjuvant hepatic arterial infusion chemotherapy with FOLFOX in hepatocellular carcinoma with microvascular invasion: A multicenter, phase III, randomized study. J. Clin. Oncol. 2023, 41, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Kudo, M.; Meyer, T.; Bai, Y.; Guo, Y.; Meng, Z.; Satoh, T.; Marino, D.; Assenat, E.; Li, S.; et al. Tislelizumab vs Sorafenib as first-line treatment for unresectable hepatocellular carcinoma: A phase 3 randomized clinical trial. JAMA Oncol. 2023, 9, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Verset, G.; Borbath, I.; Karwal, M.; Verslype, C.; Van Vlierberghe, H.; Kardosh, A.; Zagonel, V.; Stal, P.; Sarker, D.; Palmer, D.H.; et al. Pembrolizumab monotherapy for previously untreated advanced hepatocellular carcinoma: Data from the open-label, phase II KEYNOTE-224 trial. Clin. Cancer Res. 2022, 28, 2547–2554. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; He, M.; Bu, X.; Xu, Y.; Huang, Y.; Wen, D.; Li, Q.; Xu, L.; Zhang, Y.; Wei, W.; et al. Lenvatinib, Toripalimab plus hepatic arterial infusion chemotherapy in patients with high-risk advanced hepatocellular carcinoma: A biomolecular exploratory, phase II trial. Eur. J. Cancer 2022, 174, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shen, J.; Gu, S.; Zhang, Y.; Wu, L.; Wu, J.; Shao, G.; Zhang, Y.; Xu, L.; Yin, T.; et al. Camrelizumab in combination with Apatinib in patients with advanced hepatocellular carcinoma (RESCUE): A nonrandomized, open-label, phase II trial. Clin. Cancer Res. 2021, 27, 1003–1011. [Google Scholar] [CrossRef]
- Finn, R.S.; Ikeda, M.; Zhu, A.X.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. Phase Ib study of Lenvatinib plus Pembrolizumab in patients with unresectable hepatocellular carcinoma. J. Clin. Oncol. 2020, 38, 2960–2970. [Google Scholar] [CrossRef]
- Qiao, Q.; Han, C.; Ye, S.; Li, J.; Shao, G.; Bai, Y.; Xu, A.; Sun, M.; Wang, W.; Wu, J.; et al. The efficacy and safety of Cadonilimab combined with Lenvatinib for first-line treatment of advanced hepatocellular carcinoma (COMPASSION-08): A phase Ib/II single-arm clinical trial. Front. Immunol. 2023, 14, 1238667. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chuang, J.; Cho, M.; Toomey, K.; Hendifar, A.; Li, D. Molecular targets, pathways, and therapeutic implications for hepatocellular carcinoma. Int. J. Mol. Sci. 2020, 21, 5232. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Berton, D.; Curigliano, G.; Sabatier, R.; Tinker, A.V.; Oaknin, A.; Ellard, S.; de Braud, F.; Arkenau, H.T.; Trigo, J.; et al. Antitumor activity and safety of Dostarlimab monotherapy in patients with mismatch repair deficient solid tumors: A nonrandomized controlled trial. JAMA Netw. Open 2023, 6, e2341165. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.K.H.; Hui, R.W.H.; Mak, L.Y.; Fung, J.; Seto, W.K. Hepatocellular carcinoma: Advances in systemic therapies. F1000Research 2024, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Zeng, Z.M.; Zhuo, M.Y.; Luo, J.R.; Zhuang, X.H.; Xu, J.N.; Zeng, J.; Ma, J.; Lin, H.F. Anlotinib combined with Toripalimab as first-line therapy for unresectable hepatocellular carcinoma: A prospective, multicenter, phase II study. Oncologist 2023, 28, e1239–e1247. [Google Scholar] [CrossRef] [PubMed]
- France, N.L.; Blair, H.A. Tremelimumab: A review in advanced or unresectable hepatocellular carcinoma. Target. Oncol. 2024, 19, 115–123. [Google Scholar] [CrossRef]
- Subbiah, V.; Wolf, J.; Konda, B.; Kang, H.; Spira, A.; Weiss, J.; Takeda, M.; Ohe, Y.; Khan, S.; Ohashi, K.; et al. Tumour-agnostic efficacy and safety of Selpercatinib in patients with RET fusion-positive solid tumours other than lung or thyroid tumours (LIBRETTO-001): A phase 1/2, open-label, basket trial. Lancet Oncol. 2022, 23, 1261–1273. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Yan, Y.; Li, J.; Han, J.; Hou, N.; Song, Y.; Dong, L. Chlorogenic acid enhances the effects of 5-fluorouracil in human hepatocellular carcinoma cells through the inhibition of extracellular signal-regulated kinases. Anti-Cancer Drugs 2015, 26, 540–546. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Growth factors and specific signaling pathway | Epidermal growth factor (EGF) and its receptor (EGFR) Fibroblast growth factor (FGF) and its receptors (FGFR) Transforming growth factor (TGF) and its receptors (TGFR) Insulin-like growth factor (IGF) and its receptors (IGFR) Hepatocyte growth factor (HGF)/c-MET RAF/ERK/MAPK PI3K-AKT-mTOR (and PTEN) VEGF and its receptor (VEGFR) |
| Cell differentiation and development | WNT-β-catenin JAK/STAT signaling pathway Hippo signaling pathway Hedgehog signaling pathway Notch signaling pathway |
| Nuclear signaling pathways | Cell cycle (TP53) Telomere shortening (TERT/TERC) Epigenetics (DNA methylation/demethylation, histone modification, non-coding RNAs) |
| Expression | Function | Gene |
|---|---|---|
| Hypermethylation repressed/induced | Cell cycle and cell growth regulation | APC, CDH1, CDKN1A, CDKN2A, CDKN2B, PTGS2 |
| Cell signaling regulation—proliferation, survival, and metastasis of HCC cells | DAB2IP, DKK,3 GNA14, HHIP, RASSF1A, SFRP2, SOCS1 | |
| Gene transcription | ESR1, HOXA9, RUNX3, SALL3, TP73, WT1 | |
| Metabolic regulation | CPS1, FBP1, GSTP1, IGFBP5, MAT1A | |
| Matrix remodeling | MMP9, MMP12 | |
| Hypomethylation induced | Gene transcription | C/EBPb |
| Metabolic and signaling regulation | IGF2, NOX4, SPINK1 | |
| Chemotaxis and angiogenesis | CCL20, ESM1 |
| Readers | Writers | Erasers |
|---|---|---|
| MBD (methyl-CpG binding domain)-containing proteins | DNA methyltransferases | DNA demethylation system |
| Chromo domain-containing proteins | Histone methyltransferases: lysine | Histone demethylases |
| Tudor domain-containing proteins | Histone methyltransferases: protein arginine | Histone deacetylases (HDACs) |
| Malignant brain tumor- containing proteins | Histone acetyltransferases (HATs) | |
| Plant homeodomain- containing proteins | Serine-threonine and tyrosine kinases | |
| Bromodomain-containing proteins | ||
| Yeats domain-containing proteins |
| First-Line Systemic Therapy | ||
|---|---|---|
| Options | Other recommended regimens | Useful in certain circumstances |
| Atezolizumab + bevacizumab or | Durvalumab | None |
| Tremelimumab-actl + durvalumab | Lenvatinib | |
| Sorafenib | ||
| Pembrolizumab | ||
| Subsequent-line systemic therapy for disease in progression | ||
| Options | Other recommended regimens | Useful in certain circumstances |
| Cabozantinib | Nivolumab + Ipilimumab | Ramucirumab (AFP ≥ 400 ng/mL) |
| Lenvatinib | Pembrolizumab | Nivolumab |
| Sorafenib | Dostarlimab-gxly (for MSI-H/dMMR tumors) | |
| Selpercatinib (for RET gene fusion-positive tumors) | ||
| Treatment | Trial Phase | NCT Number | Research Team |
|---|---|---|---|
| Cabozantinib + Nivolumab | 1b | NCT03299946 | Ho et al., 2021 [134] |
| Levatinib + TACE | 3 | NCT03905967 | Peng et al., 2023 [135] |
| Nivolumab | 3 | NCT02576509 | Yau et al., 2022 [136] |
| Camrelizumab + Apatinib | 2 | NCT04297202 | Xia et al., 2022 [137] |
| Nivolumab + Ipilimumab | 1–2 | NCT01658878 | Yau et al., 2021 [138] |
| Tremelimumab + Durvalumab | 1–2 | NCT02519348 | Kelley et al., 2021 [139] |
| 5-Fluorouracil + Oxaliplatin hepatic arterial infusion chemotherapy | 3 | NCT03192618 | Li et al., 2023 [140] |
| Tislelizumab | 3 | NCT03412773 | Qin et. al., 2023 [141] |
| Pembrolizumab | 2 | NCT02702414 | Verset et al., 2022 [142] |
| Lenvatinib + Toripalimab + hepatic arterial infusion chemotherapy (HAIC) with Oxaliplatin, Leucovorin, and 5-Fluorouracil (FOLFOX) | 2 | NCT04044313 | Lai et al., 2022 [143] |
| Camrelizumab + Apatinib | 2 | NCT03463876 | Xu et al., 2021 [144] |
| Lenvatinib + Pembrolizumab | 1b | NCT03006926 | Finn et al., 2020 [145] |
| Cadonilimab + Lenvatinib | 1b-2 | NCT04444167 | Qiao et al., 2023 [146] |
| Antineoplastic Agents | Drug | Mechanism of Action |
|---|---|---|
| Monoclonal antibodies | Atezolizumab | IgG1 monoclonal antibody that targets PD-L1 and inhibits the interaction between PD-L1 and its receptors, PD-1 and B7-1 (also known as CD80) [147] |
| Bevacizumab | Recombinant humanized monoclonal IgG1 antibody that binds to and inhibits the biologic activity of human VEGF [147] | |
| Cadonilimab | Humanized, bispecific antibody against PD-1 and CTLA-4 [146] | |
| Camrelizumab | Humanized monoclonal IgG4-κ antibody against the PD-1 receptor [137] | |
| Dostarlimab-gxly | Human IgG4 monoclonal antibody against the PD-1 receptor [148] | |
| Durvalumab | Human monoclonal antibody (IgG1κ) that binds to the PD-L1 protein and blocks the interaction of PD-L1 with the PD-1 and CD80 proteins [149] | |
| Ipilimumab | Human IgG monoclonal antibody against CTLA-4 [147] | |
| Nivolumab | Human IgG4 monoclonal antibody against PD-1 [147] | |
| Pembrolizumab | PD-1 inhibitor humanized PD-1 monoclonal antibody inhibitor [132,147] | |
| Ramucirumab | Human IgG1 monoclonal antibody against VEGFR2 [147] | |
| Tislelizumab | Human monoclonal antibody directed against PD-1 [141] | |
| Toripalimab | Recombinant human monoclonal antibody against-PD-1 [143,150] | |
| Tremelimumab-actl | Human monoclonal antibody that targets the activity of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) [149,151] | |
| Protein kinase inhibitors | Apatinib | Tyrosine kinase inhibitor that selectively inhibits VEGFR2 [137] |
| Cabozantinib | A receptor tyrosine kinase inhibitor that acts on MET, VEGFR, RET, AXL (GAS6 receptor), KIT, and FLT3 [147] | |
| Lenvatinib | Multi-kinase inhibitor that inhibits VEGFR1–3 as well as FGF receptors 1–4 along with platelet-derived growth factor receptor (PDGFR), c-KIT [147] | |
| Regorafenib | Multi-kinase inhibitor with anti-angiogenic activity against VEGFR2 [149] | |
| Selpercatinib | Inhibitor of tyrosine kinase receptor encoded by the RET gene (rearranged during transfection) [152] | |
| Sorafenib | Inhibitor of the serine-threonine kinases Raf-1 and B-Raf and the receptor tyrosine kinase activity of VEGF receptors 1, 2, and 3 and PDGFR-β [153] | |
| Others | 5-Fluorouracil (5-FU) | Heterocyclic aromatic organic compound which interferes with nucleoside metabolism and can be incorporated into RNA and DNA, leading to cytotoxicity and cell death [140,154] |
| Oxaliplatin | Oxalato(trans-l-1,2-diaminocyclohexane) platinum that induces apoptosis by DNA damage, blocking DNA synthesis, the suppression of RNA synthesis, and the initiation of immune responses [140] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szilveszter, R.-M.; Muntean, M.; Florea, A. Molecular Mechanisms in Tumorigenesis of Hepatocellular Carcinoma and in Target Treatments—An Overview. Biomolecules 2024, 14, 656. https://doi.org/10.3390/biom14060656
Szilveszter R-M, Muntean M, Florea A. Molecular Mechanisms in Tumorigenesis of Hepatocellular Carcinoma and in Target Treatments—An Overview. Biomolecules. 2024; 14(6):656. https://doi.org/10.3390/biom14060656
Chicago/Turabian StyleSzilveszter, Raluca-Margit, Mara Muntean, and Adrian Florea. 2024. "Molecular Mechanisms in Tumorigenesis of Hepatocellular Carcinoma and in Target Treatments—An Overview" Biomolecules 14, no. 6: 656. https://doi.org/10.3390/biom14060656