
Unveiling the Druggable Landscape of Bacterial Peptidyl tRNA Hydrolase: Insights into Structure, Function, and Therapeutic Potential


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
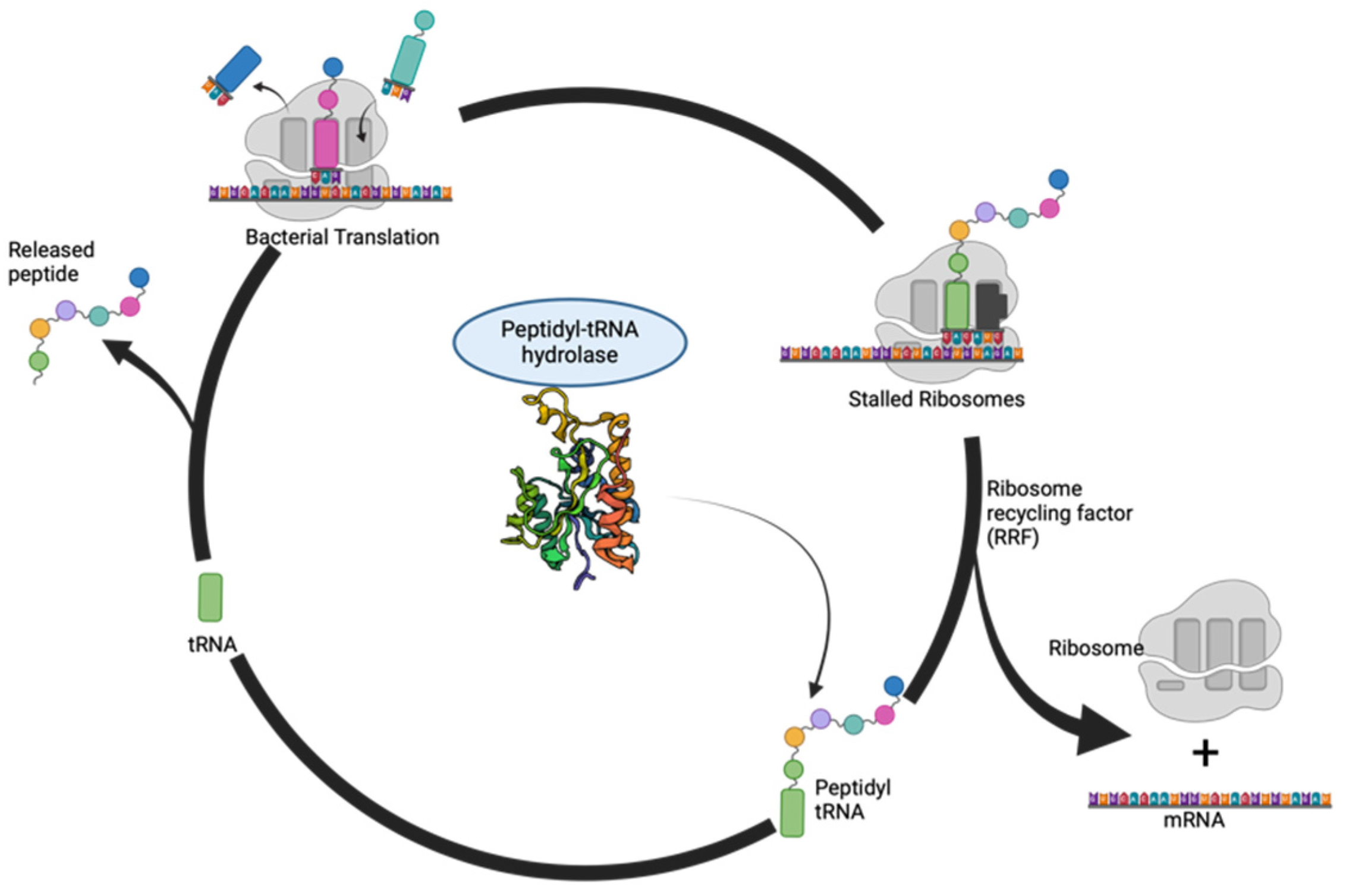
:1. Introduction
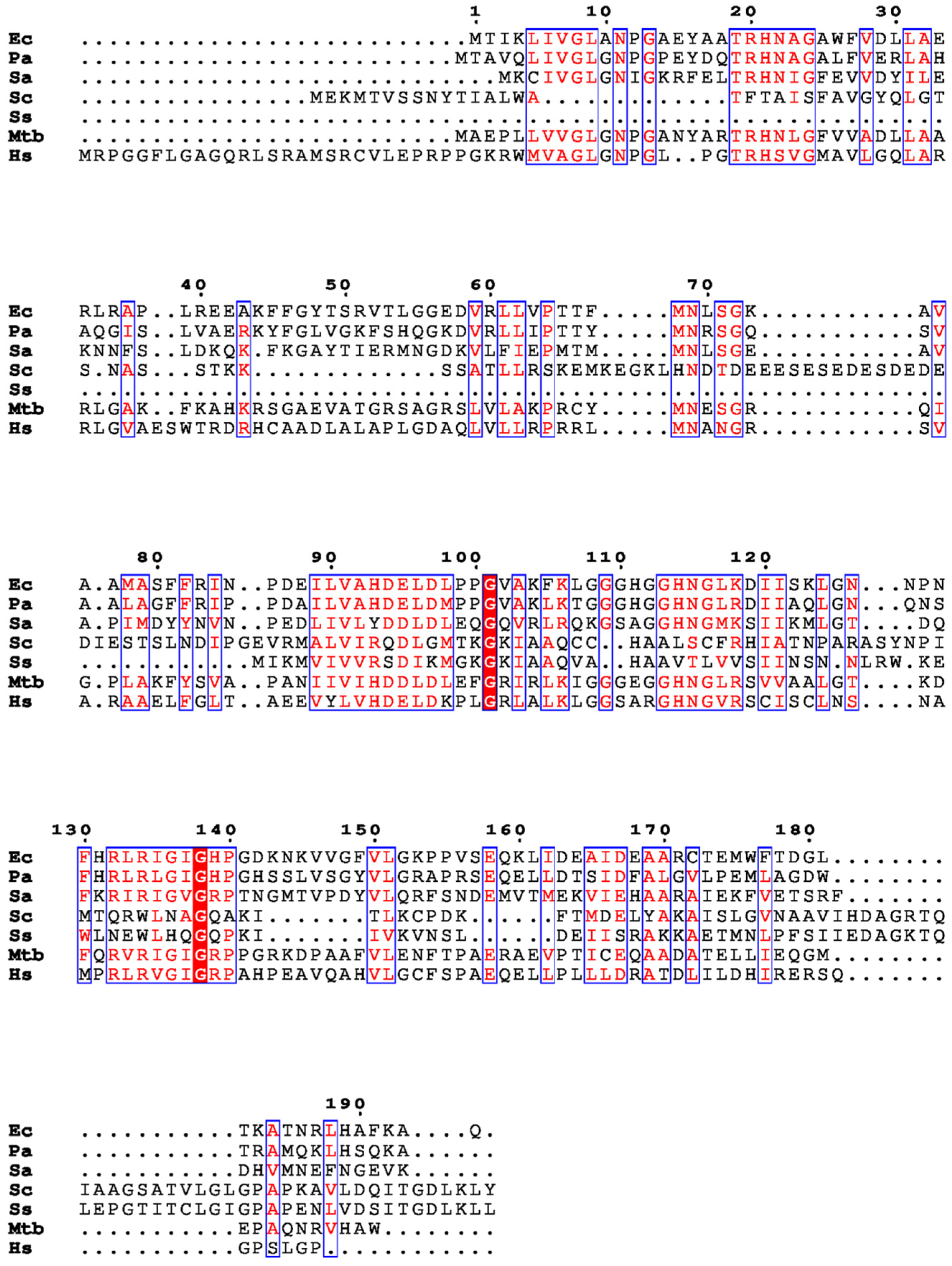
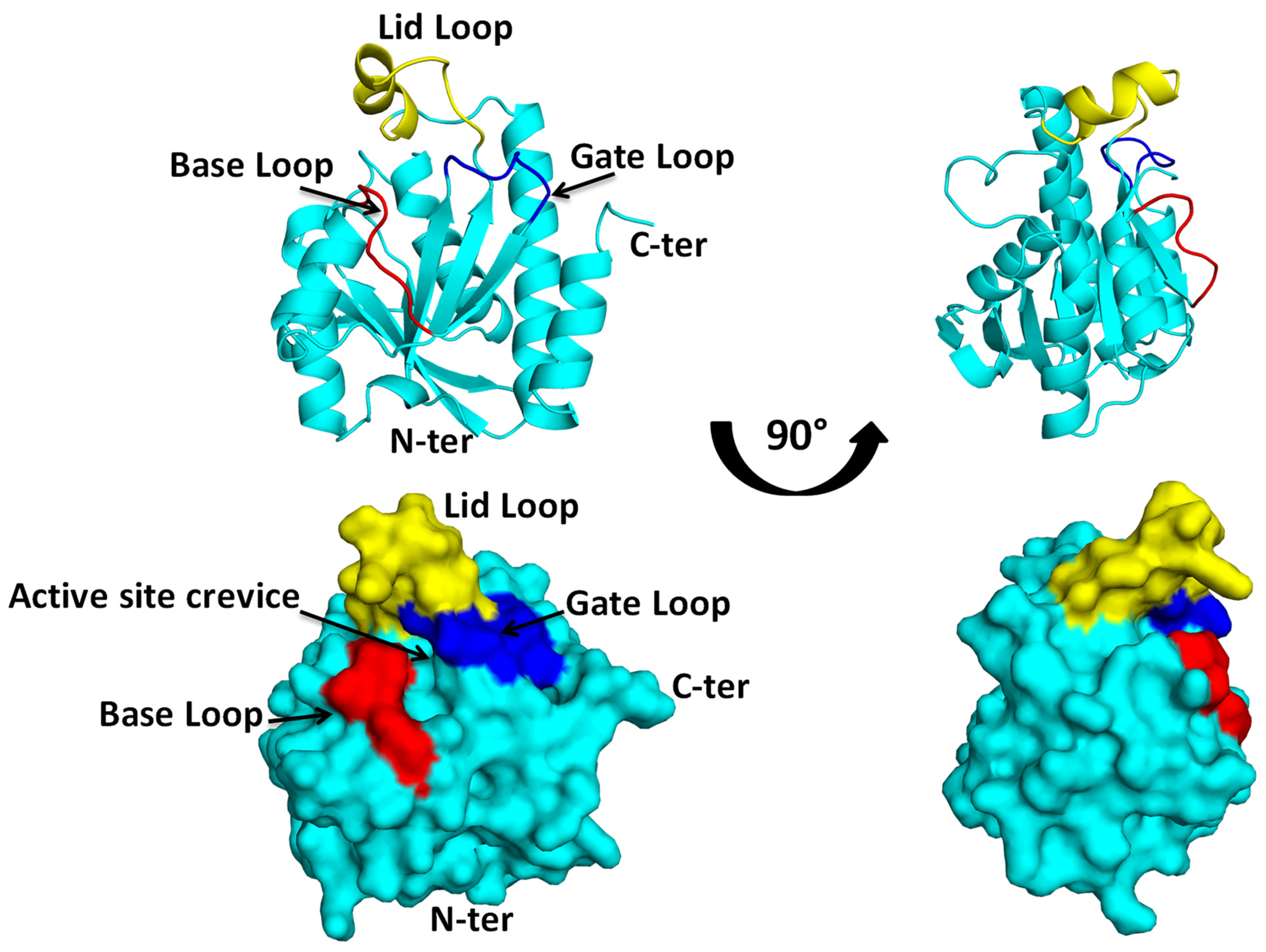
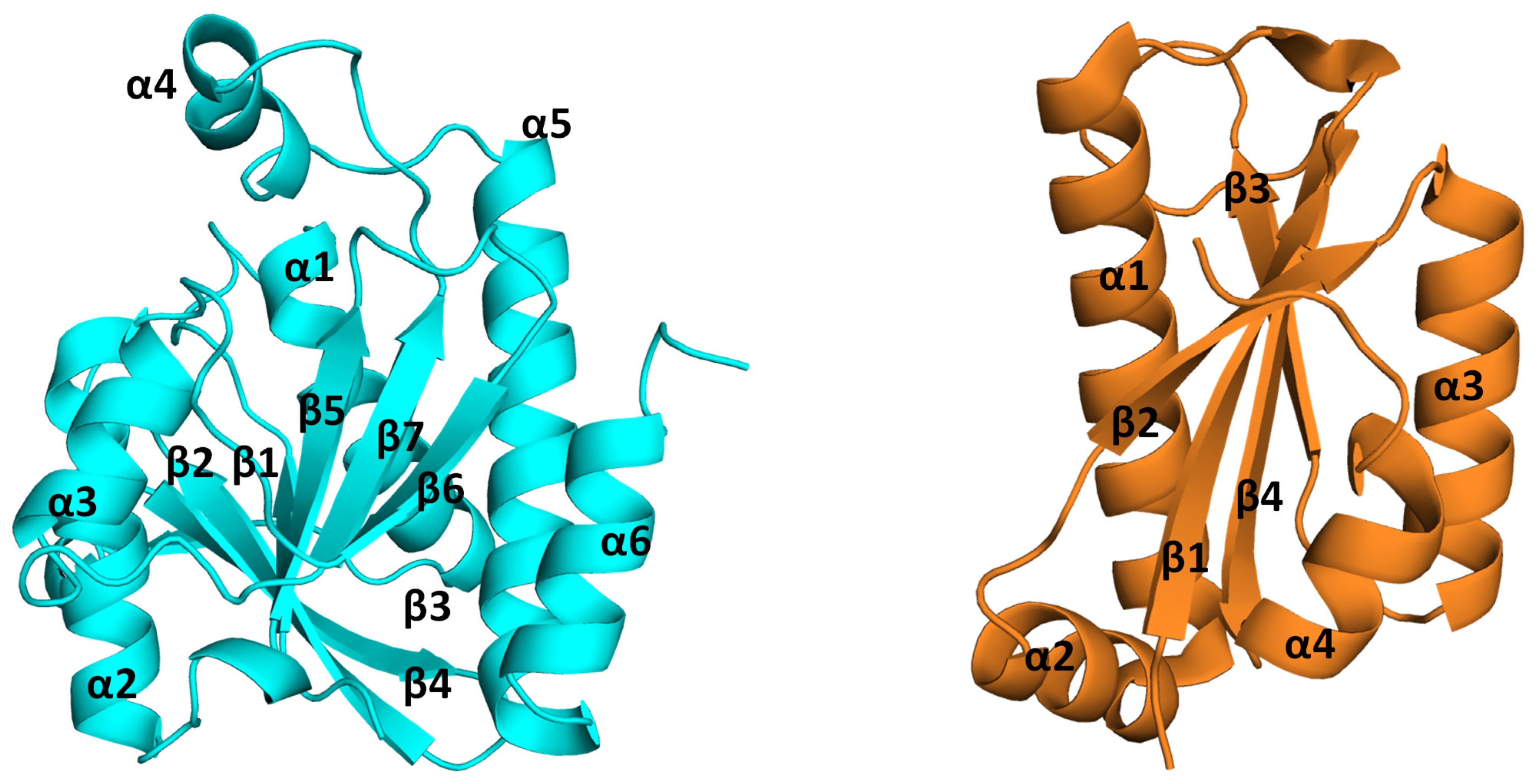
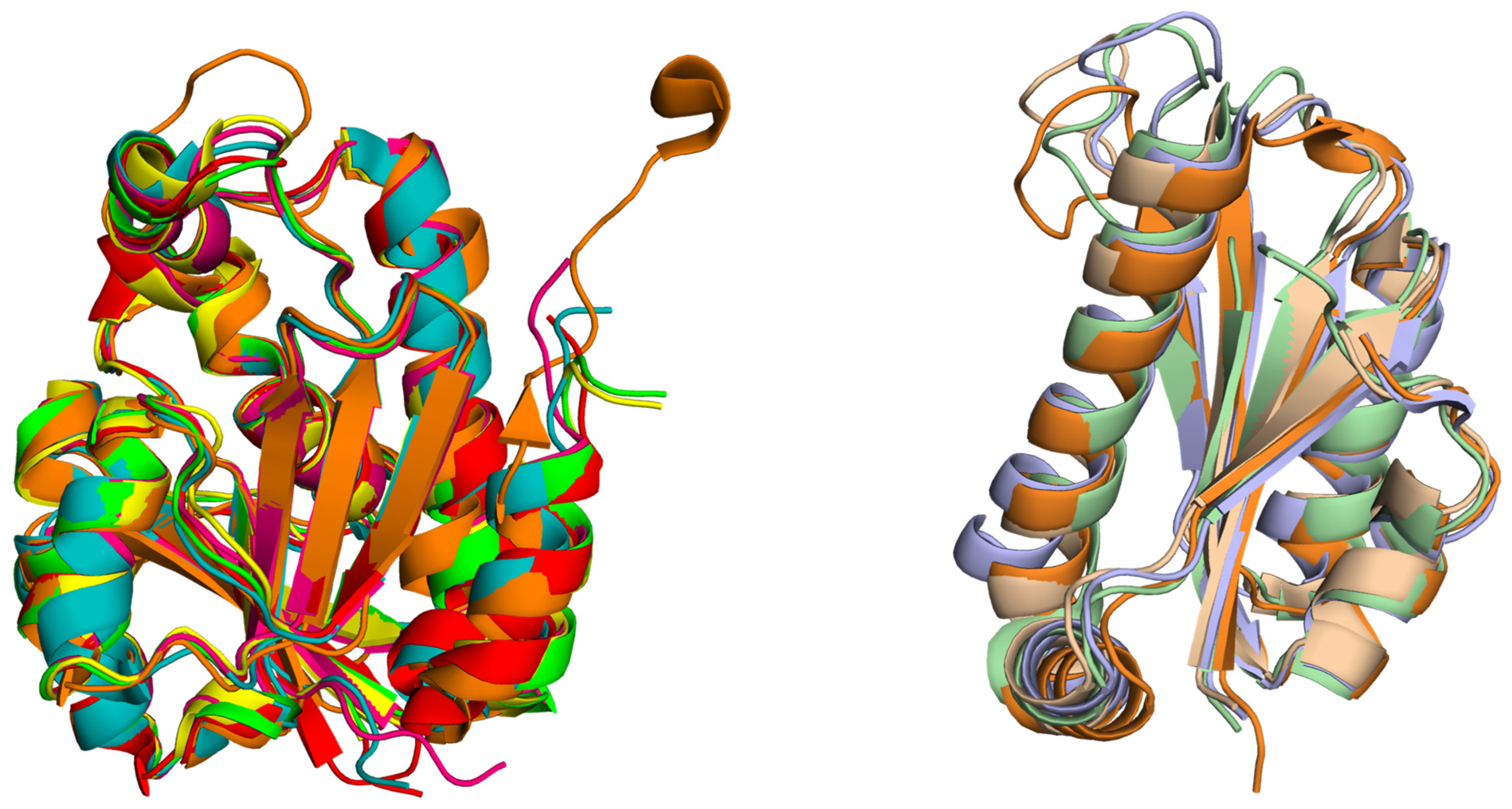
2. Structural Insights from the PDB Database
3. Structural Insights from the Pth–Substrate Interactions
- The positively charged patch of the protein and the acceptor site binding region of the substrate:
- B.
- The lid loop of the protein and CCA binding site of the tRNA:
- C.
- C terminal of the protein and TψC domain of the tRNA:
4. Structural Insights from Pth and Substrate Analog Interactions
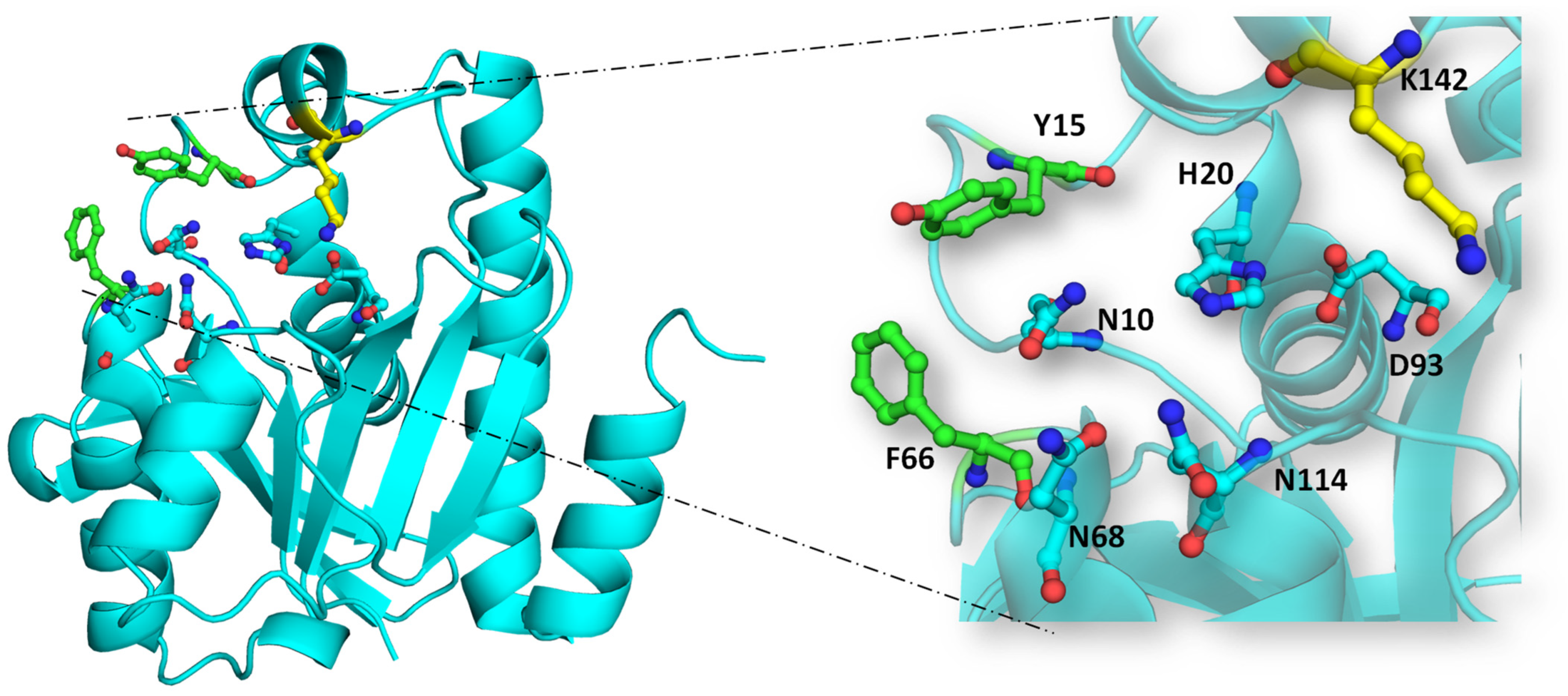
5. Peptidyl-tRNA Hydrolase: Unraveling the Mechanism of Action
- Nucleophilic attack on ester bond: The conserved histidine residue (H20 in EcPth) within the active site forms a hydrogen bond with D93, enhancing the basicity of H20. This basic histidine then interacts with a proximal water molecule situated in the active site pocket and deprotonates it. This water molecule, stabilized at the catalytic site via interaction with N114, generates a hydroxyl anion that serves as a nucleophile. This anion attacks the carbonyl group of the ester bond between the peptide and tRNA. The interplay between N10 and N114 aids in optimal substrate positioning within the catalytic pocket.
- Stabilization of tetrahedral intermediate: The interaction between the hydroxyl anion and the ester bond forms a tetrahedral intermediate, further stabilized by interactions with two key asparagine residues, N68 and N114.
- Decomposition of tetrahedral oxyanion intermediate: The tetrahedral oxyanion intermediate decomposes through general acid catalysis, where D93, polarized by H20, facilitates the production of the peptide and free tRNA [20].
6. Druggability of Peptidyl-tRNA Hydrolase: Exploiting a Crucial Node in Translation Machinery
7. Discussion: Antimicrobial Resistance, Finding New Drug Targets and Inhibitor Design, and Charting the Future Course of Peptidyl-tRNA Hydrolase Research
- Selectivity: In the context of antibiotics, selectivity is paramount to avoid disrupting the beneficial microbiota while targeting pathogenic bacteria. Many traditional antibiotics have broad-spectrum activity, killing both harmful and beneficial bacteria, leading to dysbiosis and secondary infections [68,69,70,71]. Developing antibiotics with high selectivity for specific bacterial pathogens can minimize collateral damage to the microbiome and reduce the selective pressure for AMR [72,73].
- Resistance: Antibiotic resistance is a natural evolutionary response of bacteria to the selective pressure exerted by antibiotics [72]. Resistance mechanisms can arise through genetic mutations, horizontal gene transfer, or the acquisition of resistance genes from other bacteria [74,75]. Developing specific inhibitors that target essential bacterial functions or virulence factors can help minimize the emergence of resistance. Additionally, combination therapy strategies that target multiple pathways or utilize adjuvants to potentiate antibiotic activity can delay the development of resistance [76,77,78].
- Delivery Methods: Effective delivery of antibiotics to the site of infection is essential for achieving therapeutic concentrations while minimizing systemic exposure and toxicity. However, bacterial pathogens have evolved various mechanisms to evade antibiotic action, such as biofilm formation or efflux pump systems [79,80,81,82,83]. Developing innovative delivery methods, such as nanoparticles or localized drug delivery systems, can enhance the efficacy of antibiotics and overcome bacterial resistance mechanisms [84,85,86,87,88]. Targeted drug delivery strategies that exploit bacteria-specific surface markers or vulnerabilities can also improve the precision and efficiency of antibiotic treatment.
- D. Toxicity and Side Effects: Antibiotics can cause adverse effects ranging from mild gastrointestinal disturbances to life-threatening allergic reactions or organ toxicity. Developing antibiotics with improved safety profiles and reduced off-target effects is crucial for minimizing patient morbidity and mortality. Structure–activity relationship studies, pharmacokinetic optimization, and preclinical toxicity testing are essential steps in the development of safer antibiotics. Furthermore, strategies such as selective antimicrobial peptides or narrow-spectrum antibiotics can minimize the disruption of commensal microbial communities and reduce the risk of secondary infections [89,90].
Author Contributions
Funding
Conflicts of Interest
References
- Shimizu, Y. ArfA recruits RF2 into stalled ribosomes. J. Mol. Biol. 2012, 423, 624–631. [Google Scholar] [CrossRef]
- Giudice, E.; Gillet, R. The task force that rescues stalled ribosomes in bacteria. Trends Biochem. Sci. 2013, 38, 403–411. [Google Scholar] [CrossRef]
- Singh, N.S.; Varshney, U. A physiological connection between tmRNA and peptidyl-tRNA hydrolase functions in Escherichia coli. Nucleic Acids Res. 2004, 32, 6028–6037. [Google Scholar] [CrossRef]
- Cruz-Vera, L.R.; Magos-Castro, M.A.; Zamora-Romo, E.; Guarneros, G. Ribosome stalling and peptidyl-tRNA drop-off during translational delay at AGA codons. Nucleic Acids Res. 2004, 32, 4462–4468. [Google Scholar] [CrossRef]
- Caplan, A.B.; Menninger, J.R. Tests of the ribosomal editing hypothesis: Amino acid starvation differentially enhances the dissociation of peptidyl-tRNA from the ribosome. J. Mol. Biol. 1979, 134, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Nagao, A.; Nakanishi, Y.; Yamaguchi, Y.; Mishina, Y.; Karoji, M.; Toya, T.; Fujita, T.; Iwasaki, S.; Miyauchi, K.; Sakaguchi, Y.; et al. Quality control of protein synthesis in the early elongation stage. Nat. Commun. 2023, 14, 2704. [Google Scholar] [CrossRef]
- Atherly, A.G. Peptidyl-transfer RNA hydrolase prevents inhibition of protein synthesis initiation. Nature 1978, 275, 769. [Google Scholar] [CrossRef] [PubMed]
- Menez, J.; Heurgué-Hamard, V.; Buckingham, R.H. Sequestration of specific tRNA species cognate to the last sense codon of an overproduced gratuitous protein. Nucleic Acids Res. 2000, 28, 4725–4732. [Google Scholar] [CrossRef] [PubMed]
- Menninger, J.R. Accumulation of peptidyl tRNA is lethal to Escherichia coli. J. Bacteriol. 1979, 137, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Cuzin, F.; Kretchmer, N.; Greenberg, R.E.; Hurwitz, R.; Chapeville, F. Enzymatic hydrolysis of N-substituted aminoacyl-tRNA. Proc. Natl. Acad. Sci. USA 1967, 58, 2079–2086. [Google Scholar] [CrossRef]
- Kössel, H.; RajBhandary, U.L. Studies on polynucleotides. LXXXVI. Enzymic hydrolysis of N-acylaminoacyl-transfer RNA. J. Mol. Biol. 1968, 35, 539–560. [Google Scholar] [CrossRef] [PubMed]
- Rodnina, M.V. Peptidyl-tRNA hydrolase as a key player in the liberation of truncated nascent chains from the ribosomal subunit. Mol. Cell 2024, 84, 614–615. [Google Scholar] [CrossRef] [PubMed]
- Das, G.; Varshney, U. Peptidyl-tRNA hydrolase and its critical role in protein biosynthesis. Microbiology 2006, 152, 2191–2195. [Google Scholar] [CrossRef] [PubMed]
- García-Villegas, M.R.; De La Vega, F.M.; Galindo, J.M.; Segura, M.; Buckingham, R.H.; Guarneros, G. Peptidyl-tRNA hydrolase is involved in lambda inhibition of host protein synthesis. EMBO J. 1991, 10, 3549–3555. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.; Mechulam, Y.; Fromant, M.; Plateau, P.; Blanquet, S. Crystal structure at 1.2 Å resolution and active site mapping of Escherichia coli peptidyl-tRNA hydrolase. EMBO J. 1997, 16, 4760–4769. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, M.; Roy, S.; Singh, N.S.; Sangeetha, R.; Varshney, U.; Vijayan, M. Structural Plasticity and Enzyme Action: Crystal Structures of Mycobacterium tuberculosis Peptidyl-tRNA Hydrolase. J. Mol. Biol. 2007, 372, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Pulavarti, S.V.S.R.K.; Jain, A.; Pathak, P.P.; Mahmood, A.; Arora, A. Solution structure and dynamics of peptidyl-tRNA hydrolase from Mycobacterium tuberculosis H37Rv. J. Mol. Biol. 2008, 378, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Fromant, M.; Ferri-Fioni, M.L.; Plateau, P.; Blanquet, S. Peptidyl-tRNA hydrolase from Sulfolobus solfataricus. Nucleic Acids Res. 2003, 31, 3227–3235. [Google Scholar] [CrossRef] [PubMed]
- De Pereda, J.M.; Waas, W.F.; Jan, Y.; Ruoslahti, E.; Schimmel, P.; Pascual, J. Crystal Structure of a Human Peptidyl-tRNA Hydrolase Reveals a New Fold and Suggests Basis for a Bifunctional Activity. J. Biol. Chem. 2004, 279, 8111–8115. [Google Scholar] [CrossRef]
- Ito, K.; Murakami, R.; Mochizuki, M.; Qi, H.; Shimizu, Y.; Miura, K.I.; Ueda, T.; Uchiumi, T. Structural basis for the substrate recognition and catalysis of peptidyl-tRNA hydrolase. Nucleic Acids Res. 2012, 40, 2849–2861. [Google Scholar] [CrossRef]
- Mao, C.; Cook, W.J.; Zhou, M.; Federov, A.A.; Almo, S.C.; Ealick, S.E. Calf spleen purine nucleoside phosphorylase complexed with substrates and substrate analogues. Biochemistry 1998, 37, 7135–7146. [Google Scholar] [CrossRef] [PubMed]
- Baugh, L.; Gallagher, L.A.; Patrapuvich, R.; Clifton, M.C.; Gardberg, A.S.; Edwards, T.E.; Armour, B.; Begley, D.W.; Dieterich, S.H.; Dranow, D.M.; et al. Combining Functional and Structural Genomics to Sample the Essential Burkholderia Structome. PLoS ONE 2013, 8, e53851. [Google Scholar] [CrossRef]
- Bonin, P.D.; Choi, G.H.; Trepod, C.M.; Mott, J.E.; Lyle, S.B.; Cialdella, J.I.; Sarver, R.W.; Marshall, V.P.; Erickson, L.A. Expression, purification, and characterization of peptidyl-tRNA hydrolase from Staphylococcus aureus. Protein Expr. Purif. 2002, 24, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.E.; Romanov, V.; Lam, R.; Gothe, S.A.; Peddi, S.R.; Razumova, E.B.; Lipman, R.S.A.; Branstrom, A.A.; Chirgadze, N.Y. Structure of francisella tularensis peptidyl-tRNA hydrolase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.C.; McFeeters, H.; Coates, L.; McFeeters, R.L. Recombinant production, crystallization and X-ray crystallographic structure determination of the peptidyl-tRNA hydrolase of Pseudomonas aeruginosa. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 1472–1476. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Singh, N.; Yamini, S.; Singh, A.; Sinha, M.; Arora, A.; Kaur, P.; Sharma, S.; Singh, T.P. The Mode of Inhibitor Binding to Peptidyl-tRNA Hydrolase: Binding Studies and Structure Determination of Unbound and Bound Peptidyl-tRNA Hydrolase from Acinetobacter baumannii. PLoS ONE 2013, 8, e67547. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, N.; Yadav, R.; Kumar, R.P.; Sharma, S.; Arora, A.; Singh, T.P. Crystal structure of peptidyl-tRNA hydrolase from mycobacterium smegmatis reveals novel features related to enzyme dynamics. Int. J. Biochem. Mol. Biol. 2012, 3, 58–69. [Google Scholar] [PubMed]
- Kabra, A.; Fatma, F.; Shahid, S.; Pathak, P.P.; Yadav, R.; Pulavarti, S.V.S.R.K.; Tripathi, S.; Jain, A.; Arora, A. Structural characterization of peptidyl-tRNA hydrolase from Mycobacterium smegmatis by NMR spectroscopy. Biochim. Biophys. Acta Proteins Proteom. 2016, 1864, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kumar, A.; Gautam, L.; Sharma, P.; Sinha, M.; Bhushan, A.; Kaur, P.; Sharma, S.; Arora, A.; Singh, T.P. Structural and binding studies of peptidyl-tRNA hydrolase from Pseudomonas aeruginosa provide a platform for the structure-based inhibitor design against peptidyl-tRNA hydrolase. Biochem. J. 2014, 463, 329–337. [Google Scholar] [CrossRef]
- Singh, A.; Gautam, L.; Sinha, M.; Bhushan, A.; Kaur, P.; Sharma, S.; Singh, T.P. Crystal structure of peptidyl-tRNA hydrolase from a Gram-positive bacterium, Streptococcus pyogenes at 2.19 Å resolution shows the closed structure of the substrate-binding cleft. FEBS Open Bio 2014, 4, 915–922. [Google Scholar] [CrossRef]
- Vandavasi, V.; Taylor-Creel, K.; McFeeters, R.L.; Coates, L.; McFeeters, H. Recombinant production, crystallization and X-ray crystallographic structure determination of peptidyl-tRNA hydrolase from Salmonella typhimurium. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2014, 70, 872–877. [Google Scholar] [CrossRef]
- Mundra, S.; Pal, R.K.; Tripathi, S.; Jain, A.; Arora, A. Structural and functional characterization of peptidyl-tRNA hydrolase from Klebsiella pneumoniae. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140554. [Google Scholar] [CrossRef] [PubMed]
- Kabra, A.; Shahid, S.; Pal, R.K.; Yadav, R.; Pulavarti, S.V.S.R.K.; Jain, A.; Tripathi, S.; Arora, A. Unraveling the stereochemical and dynamic aspects of the catalytic site of bacterial peptidyl-tRNA hydrolase. RNA 2017, 23, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Kavran, J.M.; Steitz, T.A. Structure of the Base of the L7/L12 Stalk of the Haloarcula marismortui Large Ribosomal Subunit: Analysis of L11 Movements. J. Mol. Biol. 2007, 371, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Sandoval, G.; Ambrogelly, A.; Rinehart, J.; Wei, D.; Cruz-Vera, L.R.; Graham, D.E.; Stetter, K.O.; Guarneros, G.; Söll, D. Orthologs of a novel archaeal and of the bacterial peptidyl-tRNA hydrolase are nonessential in yeast. Proc. Natl. Acad. Sci. USA 2002, 99, 16707–16712. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kaushik, S.; Sinha, M.; Kushwaha, G.S.; Singh, A.; Sikarwar, J.; Chaudhary, A.; Gupta, A.; Kaur, P.; Singh, T.P. Structural and functional insights into peptidyl-tRNA hydrolase. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Menez, J.; Buckingham, R.H.; De Zamaroczy, M.; Campelli, C.K. Peptidyl-tRNA hydrolase in Bacillus subtilis, encoded by spoVC, is essential to vegetative growth, whereas the homologous enzyme in Saccharomyces cerevisiae is dispensable. Mol. Microbiol. 2002, 45, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Sickmann, A.; Reinders, J.; Wagner, Y.; Joppich, C.; Zahedi, R.; Meyer, H.E.; Schönfisch, B.; Perschil, I.; Chacinska, A.; Guiard, B.; et al. The proteome of Saccharomyces cerevisiae mitochondria. Proc. Natl. Acad. Sci. USA 2003, 100, 13207–13212. [Google Scholar] [CrossRef] [PubMed]
- Powers, R.; Mirkovic, N.; Goldsmith-Fischman, S.; Acton, T.B.; Chiang, Y.; Huang, Y.J.; Ma, L.; Rajan, P.K.; Cort, J.R.; Kennedy, M.A.; et al. Solution structure of Archaeglobus fulgidis peptidyl-tRNA hydrolase (Pth2) provides evidence for an extensive conserved family of Pth2 enzymes in archea, bacteria, and eukaryotes. Protein Sci. 2005, 14, 2849–2861. [Google Scholar] [CrossRef]
- Shimizu, K.; Kuroishi, C.; Sugahara, M.; Kunishima, N. Structure of peptidyl-tRNA hydrolase 2 from Pyrococcus horikoshii OT3: Insight into the functional role of its dimeric state. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 444–453. [Google Scholar] [CrossRef]
- Fromant, M.; Plateau, P.; Schmitt, E.; Mechulam, Y.; Blanquet, S. Receptor site for the 5′-phosphate of elongator tRNAs governs substrate selection by peptidyl-tRNA hydrolase. Biochemistry 1999, 38, 4982–4987. [Google Scholar] [CrossRef]
- Goodall, J.J.; Chen, G.J.; Page, M.G.P. Essential Role of Histidine 20 in the Catalytic Mechanism of Escherichia coli Peptidyl-tRNA Hydrolase. Biochemistry 2004, 43, 4583–4591. [Google Scholar] [CrossRef]
- Giorgi, L.; Plateau, P.; O’Mahony, G.; Aubard, C.; Fromant, M.; Thureau, A.; Grøtli, M.; Blanquet, S.; Bontems, F. NMR-based substrate analog docking to Escherichia coli peptidyl-tRNA hydrolase. J. Mol. Biol. 2011, 412, 619–633. [Google Scholar] [CrossRef]
- Giorgi, L.; Bontems, F.; Fromant, M.; Aubard, C.; Blanquet, S.; Plateau, P. RNA-binding site of Escherichia coli peptidyl-tRNA hydrolase. J. Biol. Chem. 2011, 286, 39585–39594. [Google Scholar] [CrossRef] [PubMed]
- Kulandaisamy, R.; Kushwaha, T.; Kumar, V.; De, S.; Kumar, S.; Upadhyay, S.K.; Kumar, M.; Inampudi, K.K. Characterization of active/binding site residues of peptidyl-tRNA hydrolase using biophysical and computational studies. Int. J. Biol. Macromol. 2020, 159, 877–885. [Google Scholar] [CrossRef]
- Singh, M.K.; Manoj, N. Crystal structure of Thermotoga maritima acetyl esterase complex with a substrate analog: Insights into the distinctive substrate specificity in the CE7 carbohydrate esterase family. Biochem. Biophys. Res. Commun. 2016, 476, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Shahid, S.; Kabra, A.; Mundra, S.; Pal, R.K.; Tripathi, S.; Jain, A.; Arora, A. Role of methionine 71 in substrate recognition and structural integrity of bacterial peptidyl-tRNA hydrolase. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 865–874. [Google Scholar] [CrossRef]
- Giedraitienė, A.; Vitkauskienė, A.; Naginienė, R.; Pavilonis, A. Antibiotic resistance mechanisms of clinically important bacteria. Medicina 2011, 47, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. Bacterial resistance to antibiotics: Enzymatic degradation and modification. Adv. Drug Deliv. Rev. 2005, 57, 1451–1470. [Google Scholar] [CrossRef]
- Li, H.; Rothberg, L. Colorimetric detection of DNA sequences based on electrostatic interactions with unmodified gold nanoparticles. Proc. Natl. Acad. Sci. USA 2004, 101, 14036–14039. [Google Scholar] [CrossRef]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable re-sistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Drlica, K.; Malik, M.; Kerns, R.J.; Zhao, X. Quinolone-mediated bacterial death. Antimicrob. Agents Chemother. 2008, 52, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Floss, H.G.; Yu, T.-W. Rifamycin-mode of action, resistance, and biosynthesis. Chem. Rev. 2005, 105, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Tomasz, A. The mechanism of the irreversible antimicrobial effects of penicillins: How the beta-lactam antibiotics kill and lyse bacteria. Annu. Rev. Microbiol. 1979, 33, 113–137. [Google Scholar] [CrossRef] [PubMed]
- Vakulenko, S.B.; Mobashery, S. Versatility of aminoglycosides and prospects for their future. Clin. Microbiol. Rev. 2003, 16, 430–450. [Google Scholar] [CrossRef] [PubMed]
- Kohanski, M.A.; Dwyer, D.J.; Hayete, B.; Lawrence, C.A.; Collins, J.J. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 2007, 130, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, D.J.; Kohanski, M.A.; Hayete, B.; Collins, J.J. Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol. Syst. Biol. 2007, 3, 91. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, V.M.; McGrann, K.M.; Hughes, D.W.; Wright, G.D. Sampling the antibiotic resistome. Science 2006, 311, 374–377. [Google Scholar] [CrossRef]
- Fitzpatrick, M.C.; Bauch, C.T.; Townsend, J.P.; Galvani, A.P. Modelling microbial infection to address global health challenges. Nat. Microbiol. 2019, 4, 1612–1619. [Google Scholar] [CrossRef]
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.M.; Wertheim, H.F.L.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic resistance-the need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, Z.; Guo, J.; de la Fuente-Nunez, C.; Wang, J.; Han, B.; Tao, H.; Liu, J.; Wang, X. Bacterial resistance to antibacterial agents: Mechanisms, control strategies, and implications for global health. Sci. Total Environ. 2023, 860, 160461. [Google Scholar] [CrossRef] [PubMed]
- Witte, W. Antibiotic resistance. Int. J. Med. Microbiol. 2013, 303, 285–286. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.; Zembower, T.R. Antimicrobial Resistance. Gastrointest. Endosc. Clin. N. Am. 2020, 30, 619–635. [Google Scholar] [CrossRef] [PubMed]
- Christaki, E.; Marcou, M.; Tofarides, A. Antimicrobial Resistance in Bacteria: Mechanisms, Evolution, and Persistence. J. Mol. Evol. 2020, 88, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Dever, L.A.; Dermody, T.S. Mechanisms of bacterial resistance to antibiotics. Arch. Intern. Med. 1991, 151, 886–895. [Google Scholar] [CrossRef]
- Brinkac, L.; Voorhies, A.; Gomez, A.; Nelson, K.E. The Threat of Antimicrobial Resistance on the Human Microbiome. Microb. Ecol. 2017, 74, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Cechova, K.; Amlerova, J.; Hort, J. Antibiotics, gut microbiota, and Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 108. [Google Scholar] [CrossRef]
- Singh, S.B.; Young, K.; Silver, L.L. What is an “ideal” antibiotic? Discovery challenges and path forward. Biochem. Pharmacol. 2017, 133, 63–73. [Google Scholar] [CrossRef]
- Zhong, W.; Wu, K.; Long, Z.; Zhou, X.; Zhong, C.; Wang, S.; Lai, H.; Guo, Y.; Lv, D.; Lu, J.; et al. Gut dysbiosis promotes prostate cancer progression and docetaxel resistance via activating NF-κB-IL6-STAT3 axis. Microbiome 2022, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Asai, T. Role of antimicrobial selective pressure and secondary factors on antimicrobial resistance prevalence in Escherichia coli from food-producing animals in Japan. J. Biomed. Biotechnol. 2010, 2010, 180682. [Google Scholar] [CrossRef]
- Lammie, S.L.; Hughes, J.M. Antimicrobial Resistance, Food Safety, and One Health: The Need for Convergence. Annu. Rev. Food Sci. Technol. 2016, 7, 287–312. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.G.; Burgess, S.; Toombs-Ruane, L.J.; Benschop, J.; Marshall, J.C.; French, N.P. Combining mutation and horizontal gene transfer in a within-host model of antibiotic resistance. Math. Biosci. 2021, 339, 108656. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.B.I.; Rodríguez-Rojas, A.; Rolff, J.; Schreiber, F. Biocides used as material preservatives modify rates of de novo mutation and horizontal gene transfer in bacteria. J. Hazard. Mater. 2022, 437, 129280. [Google Scholar] [CrossRef]
- Hernandez-Rodriguez, P.; Baquero, L.P. Combination Therapy as a Strategy to Control Infections Caused by Multi-resistant Bacteria: Current Review. Curr. Drug Targets 2022, 23, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Coates, A.R.M.; Hu, Y.; Holt, J.; Yeh, P. Antibiotic combination therapy against resistant bacterial infections: Synergy, rejuvenation and resistance reduction. Expert Rev. Anti Infect. Ther. 2020, 18, 5–15. [Google Scholar] [CrossRef]
- Tarín-Pelló, A.; Suay-García, B.; Pérez-Gracia, M.-T. Antibiotic resistant bacteria: Current situation and treatment options to accelerate the development of a new antimicrobial arsenal. Expert Rev. Anti Infect. Ther. 2022, 20, 1095–1108. [Google Scholar] [CrossRef]
- Panjla, A.; Kaul, G.; Chopra, S.; Titz, A.; Verma, S. Short Peptides and Their Mimetics as Potent Antibacterial Agents and Antibiotic Adjuvants. ACS Chem. Biol. 2021, 16, 2731–2745. [Google Scholar] [CrossRef]
- Roy, R.; Tiwari, M.; Donelli, G.; Tiwari, V. Strategies for combating bacterial biofilms: A focus on anti-biofilm agents and their mechanisms of action. Virulence 2018, 9, 522–554. [Google Scholar] [CrossRef]
- O’Toole, G.; Kaplan, H.B.; Kolter, R. Biofilm formation as microbial development. Annu. Rev. Microbiol. 2000, 54, 49–79. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.M.A.; Richmond, G.E.; Piddock, L.J.V. Multidrug efflux pumps in Gram-negative bacteria and their role in antibiotic resistance. Future Microbiol. 2014, 9, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Gil-Gil, T.; Laborda, P.; Ochoa-Sánchez, L.E.; Martínez, J.L.; Hernando-Amado, S. Efflux in Gram-negative bacteria: What are the latest opportunities for drug discovery? Expert Opin. Drug Discov. 2023, 18, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhuang, J.; Li, Z.; Gong, H.; de Ávila, B.E.-F.; Duan, Y.; Zhang, Q.; Zhou, J.; Yin, L.; Karshalev, E.; et al. Nanoparticle-modified microrobots for in vivo antibiotic delivery to treat acute bacterial pneumonia. Nat. Mater. 2022, 21, 1324–1332. [Google Scholar] [CrossRef]
- Pelgrift, R.Y.; Friedman, A.J. Nanotechnology as a therapeutic tool to combat microbial resistance. Adv. Drug Deliv. Rev. 2013, 65, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Rashki, S.; Asgarpour, K.; Tarrahimofrad, H.; Hashemipour, M.; Ebrahimi, M.S.; Fathizadeh, H.; Khorshidi, A.; Khan, H.; Marzhoseyni, Z.; Salavati-Niasari, M.; et al. Chitosan-based nanoparticles against bacterial infections. Carbohydr. Polym. 2021, 251, 117108. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Thamphiwatana, S.; Angsantikul, P.; Zhang, L. Nanoparticle approaches against bacterial infections. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2014, 6, 532–547. [Google Scholar] [CrossRef] [PubMed]
- Bruna, T.; Maldonado-Bravo, F.; Jara, P.; Caro, N. Silver Nanoparticles and Their Antibacterial Applications. Int. J. Mol. Sci. 2021, 22, 7202. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef]
- Boullet, H.; Bentot, F.; Hequet, A.; Ganem-Elbaz, C.; Bechara, C.; Pacreau, E.; Launay, P.; Sagan, S.; Jolivalt, C.; Lacombe, C.; et al. Small AntiMicrobial Peptide with In Vivo Activity against Sepsis. Molecules 2019, 24, 1702. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mundra, S.; Kabra, A. Unveiling the Druggable Landscape of Bacterial Peptidyl tRNA Hydrolase: Insights into Structure, Function, and Therapeutic Potential. Biomolecules 2024, 14, 668. https://doi.org/10.3390/biom14060668
Mundra S, Kabra A. Unveiling the Druggable Landscape of Bacterial Peptidyl tRNA Hydrolase: Insights into Structure, Function, and Therapeutic Potential. Biomolecules. 2024; 14(6):668. https://doi.org/10.3390/biom14060668
Chicago/Turabian StyleMundra, Surbhi, and Ashish Kabra. 2024. "Unveiling the Druggable Landscape of Bacterial Peptidyl tRNA Hydrolase: Insights into Structure, Function, and Therapeutic Potential" Biomolecules 14, no. 6: 668. https://doi.org/10.3390/biom14060668