
Inhibitors of SARS-CoV-2 Main Protease (Mpro) as Anti-Coronavirus Agents


Abstract
1. Introduction
2. The Structure of Mpro
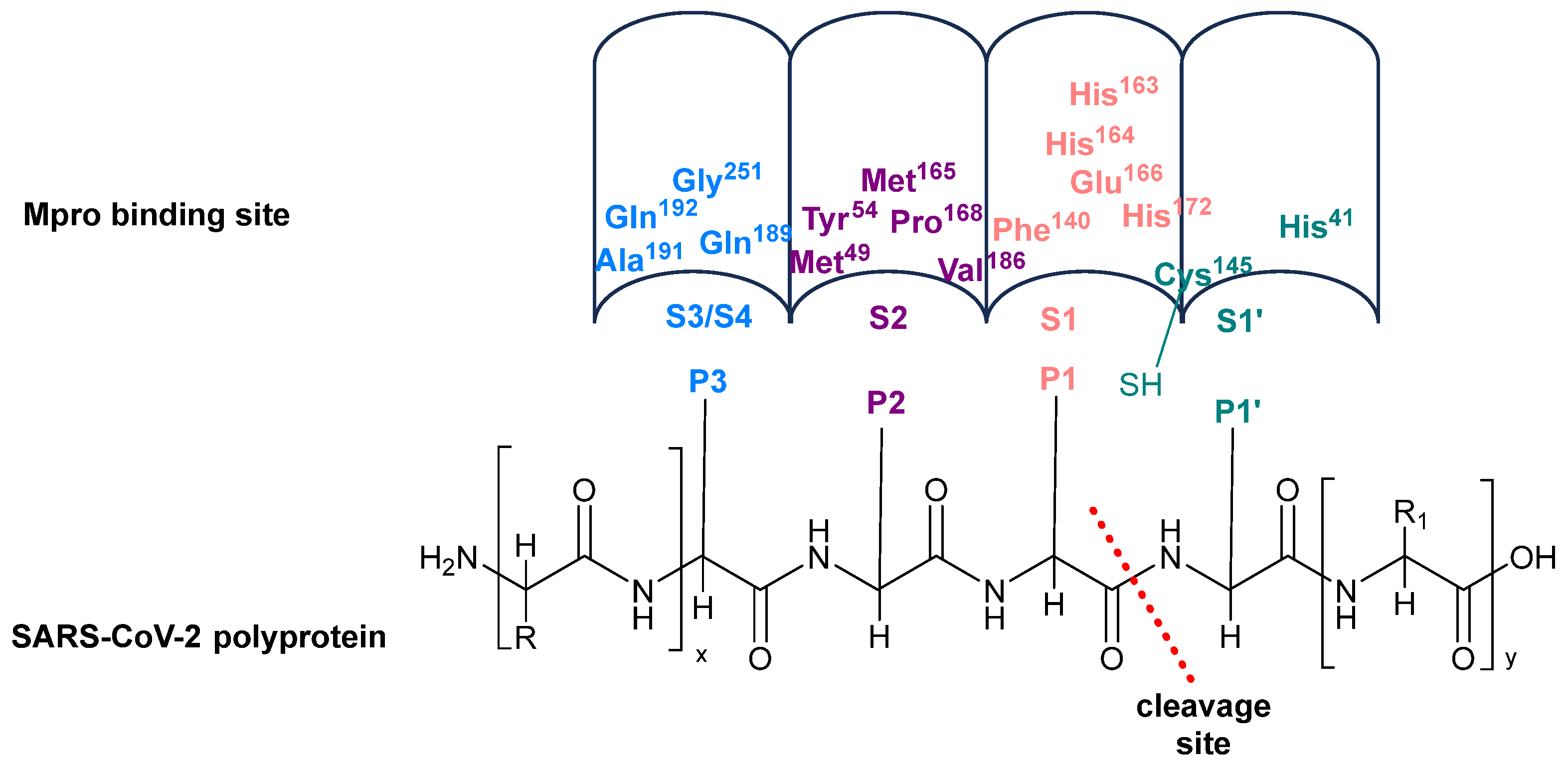
2.1. Substrate Recognition
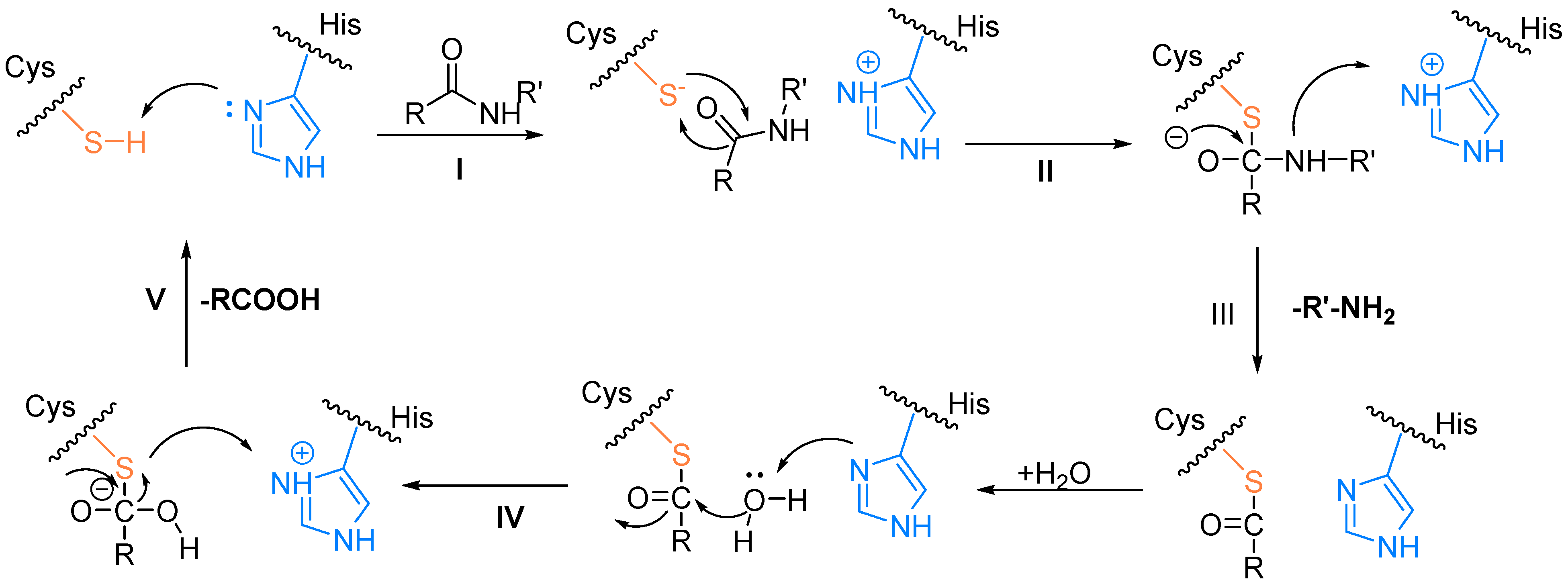
2.2. The Catalytic Mechanism for the Cleavage of Polyproteins by Mpro
3. Classification of Mpro Inhibitors
3.1. Classification of Mpro Inhibitors by Binding Kinetics
3.1.1. Non-Covalent Inhibitors of Mpro
3.1.2. Covalent Inhibitors of Mpro
3.2. Classification of Mpro Inhibitors by Origin
3.3. Classification of Mpro Inhibitors by the Chemical Structure
3.3.1. Peptidomimetics
3.3.2. Small Molecules
3.4. Classification of Mpro Inhibitors by Electrophilic Warheads
4. Clinical Trials of SARS-CoV-2 Mpro Inhibitors
4.1. Covalent Inhibitors
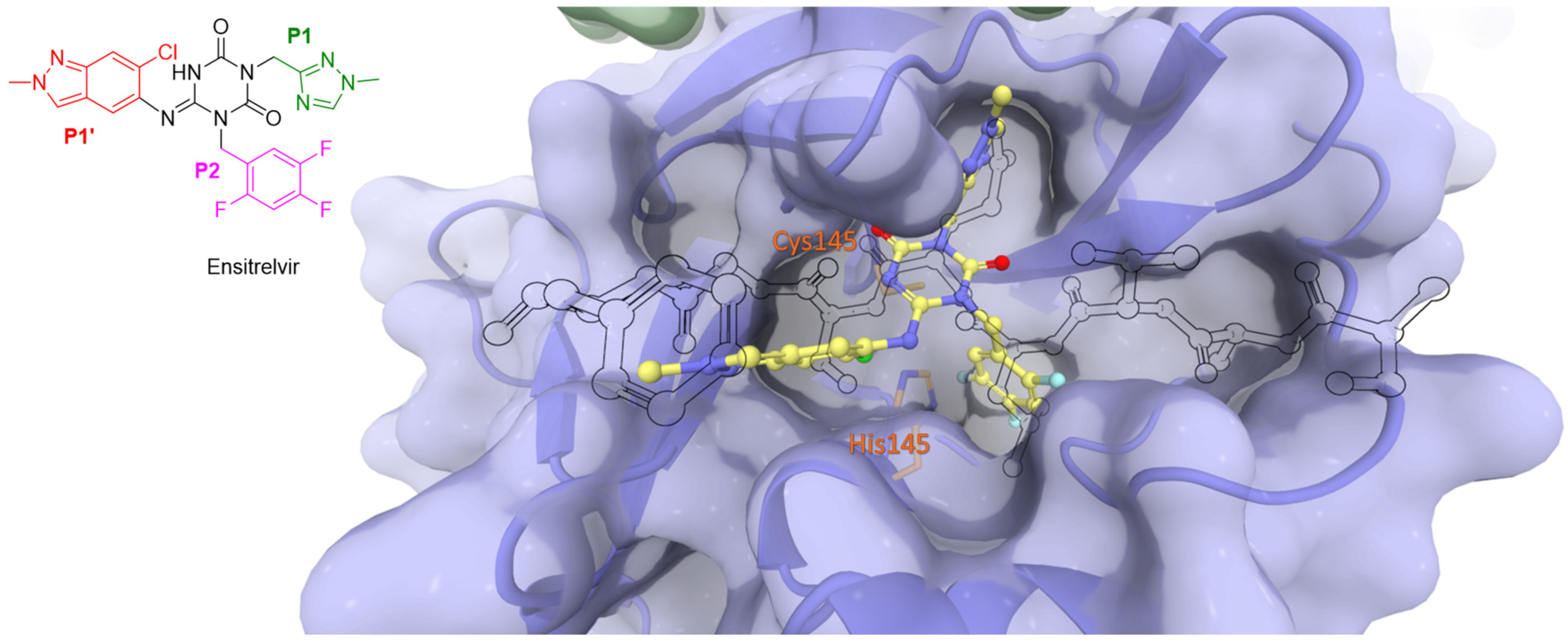
4.2. Non-Covalent Inhibitors
4.3. Dual Inhibitor of SARS-CoV-2 Mpro and Cathepsin L
5. Conclusions, Challenges, and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Naseer, S.; Khalid, S.; Parveen, S.; Abbass, K.; Song, H.; Achim, M.V. COVID-19 Outbreak: Impact on Global Economy. Front. Public Health 2022, 10, 1009393. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.J.; Cheng, S.M.S.; Leung, K.; Lee, C.K.; Hachim, A.; Tsang, L.C.H.; Yam, K.W.H.; Chaothai, S.; Kwan, K.K.H.; Chai, Z.Y.H.; et al. Real-World COVID-19 Vaccine Effectiveness against the Omicron BA.2 Variant in a SARS-CoV-2 Infection-Naive Population. Nat. Med. 2023, 29, 348–357. [Google Scholar] [CrossRef]
- Fischer, C.; Willscher, E.; Paschold, L.; Gottschick, C.; Klee, B.; Diexer, S.; Bosurgi, L.; Dutzmann, J.; Sedding, D.; Frese, T.; et al. SARS-CoV-2 Vaccination May Mitigate Dysregulation of IL-1/IL-18 and Gastrointestinal Symptoms of the Post-COVID-19 Condition. NPJ Vaccines 2024, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Beinfeld, M.; Yeung, K.; Whittington, M.D.; Mohammed, R.; Nhan, E.; Pearson, S.D. Oral Treatments for Outpatient COVID-19: Effectiveness and Value. J. Manag. Care Spec. Pharm. 2022, 28, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pan, X.; Zhang, S.; Li, M.; Ma, K.; Fan, C.; Lv, Y.; Guan, X.; Yang, Y.; Ye, X.; et al. Efficacy and Safety of Paxlovid in Severe Adult Patients with SARS-CoV-2 Infection: A Multicenter Randomized Controlled Study. Lancet Reg. Health West. Pac. 2023, 33, 100694. [Google Scholar] [CrossRef] [PubMed]
- Reis, S.; Metzendorf, M.I.; Kuehn, R.; Popp, M.; Gagyor, I.; Kranke, P.; Meybohm, P.; Skoetz, N.; Weibel, S. Nirmatrelvir Combined with Ritonavir for Preventing and Treating COVID-19. Cochrane Database Syst. Rev. 2023, 2023, CD015395. [Google Scholar] [CrossRef]
- von Delft, A.; Hall, M.D.; Kwong, A.D.; Purcell, L.A.; Saikatendu, K.S.; Schmitz, U.; Tallarico, J.A.; Lee, A.A. Accelerating Antiviral Drug Discovery: Lessons from COVID-19. Nat. Rev. Drug Discov. 2023, 22, 585–603. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.W.; Yuan, S.; Chu, H.; Sridhar, S.; Yuen, K.Y. COVID-19 Drug Discovery and Treatment Options. Nat. Rev. Microbiol. 2024, 22, 391–407. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Hilgenfeld, R.; Whitley, R.; De Clercq, E. Therapeutic Strategies for COVID-19: Progress and Lessons Learned. Nat. Rev. Drug Discov. 2023, 22, 449–475. [Google Scholar] [CrossRef]
- Amicone, M.; Borges, V.; Alves, M.J.; Isidro, J.; Zé-Zé, L.; Duarte, S.; Vieira, L.; Guiomar, R.; Gomes, J.P.; Gordo, I. Mutation Rate of SARS-CoV-2 and Emergence of Mutators during Experimental Evolution. Evol. Med. Public Health 2022, 10, 142–155. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The Evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Duan, Y.; Zhou, H.; Liu, X.; Iketani, S.; Lin, M.; Zhang, X.; Bian, Q.; Wang, H.; Sun, H.; Hong, S.J.; et al. Molecular Mechanisms of SARS-CoV-2 Resistance to Nirmatrelvir. Nature 2023, 622, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Iketani, S.; Mohri, H.; Culbertson, B.; Hong, S.J.; Duan, Y.; Luck, M.I.; Annavajhala, M.K.; Guo, Y.; Sheng, Z.; Uhlemann, A.C.; et al. Multiple Pathways for SARS-CoV-2 Resistance to Nirmatrelvir. Nature 2022, 613, 558–564. [Google Scholar] [CrossRef]
- Noske, G.D.; de Souza Silva, E.; de Godoy, M.O.; Dolci, I.; Fernandes, R.S.; Guido, R.V.C.; Sjö, P.; Oliva, G.; Godoy, A.S. Structural Basis of Nirmatrelvir and Ensitrelvir Activity against Naturally Occurring Polymorphisms of the SARS-CoV-2 Main Protease. J. Biol. Chem. 2023, 299, 103004. [Google Scholar] [CrossRef]
- Moghadasi, S.A.; Biswas, R.G.; Harki, D.A.; Harris, R.S. Rapid Resistance Profiling of SARS-CoV-2 Protease Inhibitors. NPJ Antimicrob. Resistance 2023, 1, 1–4. [Google Scholar] [CrossRef]
- Ip, J.D.; Chu, A.W.H.; Chan, W.M.; Leung, R.C.Y.; Umer Abdullah, S.M.; Sun, Y.; To, K.K.W. Global Prevalence of SARS-CoV-2 3CL Protease Mutations Associated with Nirmatrelvir or Ensitrelvir Resistance. eBioMedicine 2023, 91, 104559. [Google Scholar] [CrossRef]
- Hoang, H.D.; Naeli, P.; Alain, T.; Jafarnejad, S.M. Mechanisms of Impairment of Interferon Production by SARS-CoV-2. Biochem. Soc. Trans. 2023, 51, 1047. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The Crystal Structures of Severe Acute Respiratory Syndrome Virus Main Protease and Its Complex with an Inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef]
- Sacco, M.D.; Ma, C.; Lagarias, P.; Gao, A.; Townsend, J.A.; Meng, X.; Dube, P.; Zhang, X.; Hu, Y.; Kitamura, N.; et al. Structure and Inhibition of the SARS-CoV-2 Main Protease Reveal Strategy for Developing Dual Inhibitors against Mpro and Cathepsin L. Sci. Adv. 2020, 6, eabe0751. [Google Scholar] [CrossRef]
- MacDonald, E.A.; Frey, G.; Namchuk, M.N.; Harrison, S.C.; Hinshaw, S.M.; Windsor, I.W. Recognition of Divergent Viral Substrates by the SARS-CoV-2 Main Protease. ACS Infect. Dis. 2021, 7, 2591–2595. [Google Scholar] [CrossRef]
- Ho, B.L.; Cheng, S.C.; Shi, L.; Wang, T.Y.; Ho, K.I.; Chou, C.Y. Critical Assessment of the Important Residues Involved in the Dimerization and Catalysis of MERS Coronavirus Main Protease. PLoS ONE 2015, 10, e0144865. [Google Scholar] [CrossRef]
- Marzi, M.; Vakil, M.K.; Bahmanyar, M.; Zarenezhad, E. Paxlovid: Mechanism of Action, Synthesis, and in Silico Study. Biomed. Res. Int. 2022, 2022, 7341493. [Google Scholar] [CrossRef]
- Paul, A.S.; Islam, R.; Parves, M.R.; Mamun, A.A.; Shahriar, I.; Hossain, M.I.; Hossain, M.N.; Ali, M.A.; Halim, M.A. Cysteine Focused Covalent Inhibitors against the Main Protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 40, 1639–1658. [Google Scholar] [CrossRef]
- Kuzikov, M.; Costanzi, E.; Reinshagen, J.; Esposito, F.; Vangeel, L.; Wolf, M.; Ellinger, B.; Claussen, C.; Geisslinger, G.; Corona, A.; et al. Identification of Inhibitors of SARS-CoV-2 3CL-Pro Enzymatic Activity Using a Small Molecule in Vitro Repurposing Screen. ACS Pharmacol. Transl. Sci. 2021, 4, 1096–1110. [Google Scholar] [CrossRef]
- Ma, C.; Xia, Z.; Sacco, M.D.; Hu, Y.; Townsend, J.A.; Meng, X.; Choza, J.; Tan, H.; Jang, J.; Gongora, M.V.; et al. Discovery of Di- and Trihaloacetamides as Covalent SARS-CoV-2 Main Protease Inhibitors with High Target Specificity. J. Am. Chem. Soc. 2021, 143, 20697–20709. [Google Scholar] [CrossRef]
- Citarella, A.; Scala, A.; Piperno, A.; Micale, N. SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules 2021, 11, 607. [Google Scholar] [CrossRef]
- Qomara, W.F.; Primanissa, D.N.; Amalia, S.H.; Purwadi, F.V.; Zakiyah, N. Effectiveness of Remdesivir, Lopinavir/Ritonavir, and Favipiravir for COVID-19 Treatment: A Systematic Review. Int. J. Gen. Med. 2021, 14, 8557–8571. [Google Scholar] [CrossRef]
- De Cesco, S.; Kurian, J.; Dufresne, C.; Mittermaier, A.K.; Moitessier, N. Covalent Inhibitors Design and Discovery. Eur. J. Med. Chem. 2017, 138, 96–114. [Google Scholar] [CrossRef]
- Strelow, J.M. A Perspective on the Kinetics of Covalent and Irreversible Inhibition. SLAS Discov. 2017, 22, 3–20. [Google Scholar] [CrossRef]
- Macchiagodena, M.; Pagliai, M.; Procacci, P. Characterization of the Non-Covalent Interaction between the PF-07321332 Inhibitor and the SARS-CoV-2 Main Protease. J. Mol. Graph. Model. 2022, 110, 108042. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Raghavaiah, J.; Shahabi, D.; Yadav, M.; Anson, B.J.; Lendy, E.K.; Hattori, S.I.; Higashi-Kuwata, N.; Mitsuya, H.; Mesecar, A.D. Indole Chloropyridinyl Ester-Derived SARS-CoV-2 3CLpro Inhibitors: Enzyme Inhibition, Antiviral Efficacy, Structure-Activity Relationship, and X-ray Structural Studies. J. Med. Chem. 2021, 64, 14702–14714. [Google Scholar] [CrossRef] [PubMed]
- Manandhar, A.; Srinivasulu, V.; Hamad, M.; Tarazi, H.; Omar, H.; Colussi, D.J.; Gordon, J.; Childers, W.; Klein, M.L.; Al-Tel, T.H.; et al. Discovery of Novel Small-Molecule Inhibitors of SARS-CoV-2 Main Protease as Potential Leads for COVID-19 Treatment. J. Chem. Inf. Model. 2021, 61, 4745–4757. [Google Scholar] [CrossRef] [PubMed]
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; van Belkum, M.J.; Joyce, M.A.; et al. Feline Coronavirus Drug Inhibits the Main Protease of SARS-CoV-2 and Blocks Virus Replication. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Huang, C.; Shuai, H.; Qiao, J.; Hou, Y.; Zeng, R.; Xia, A.; Xie, L.; Fang, Z.; Li, Y.; Yoon, C.; et al. A New Generation Mpro Inhibitor with Potent Activity against SARS-CoV-2 Omicron Variants. Signal Transduct. Target. Ther. 2023, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An Oral SARS-CoV-2 Mpro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Vandyck, K.; Deval, J. Considerations for the Discovery and Development of 3-Chymotrypsin-like Cysteine Protease Inhibitors Targeting SARS-CoV-2 Infection. Curr. Opin. Virol. 2021, 49, 36–40. [Google Scholar] [CrossRef] [PubMed]
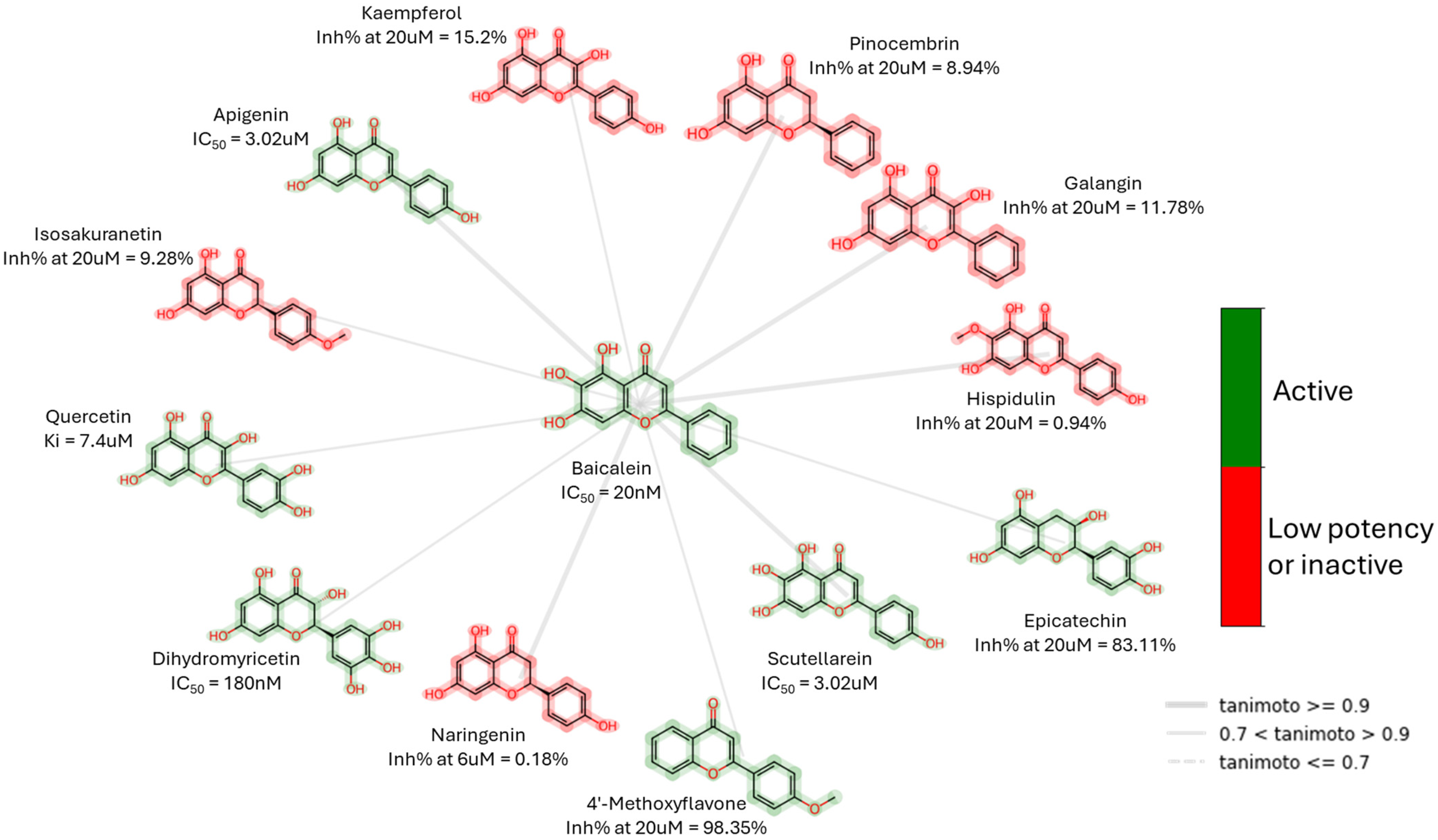
- Krüger, N.; Kronenberger, T.; Xie, H.; Rocha, C.; Pöhlmann, S.; Su, H.; Xu, Y.; Laufer, S.A.; Pillaiyar, T. Discovery of Polyphenolic Natural Products as SARS-CoV-2 Mpro Inhibitors for COVID-19. Pharmaceuticals 2023, 16, 190. [Google Scholar] [CrossRef]
- Baron, G.; Borella, S.; della Vedova, L.; Vittorio, S.; Vistoli, G.; Carini, M.; Aldini, G.; Altomare, A. An Integrated Metabolomic and Proteomic Approach for the Identification of Covalent Inhibitors of the Main Protease (Mpro) of SARS-CoV-2 from Crude Natural Extracts. Talanta 2023, 252, 123824. [Google Scholar] [CrossRef]
- Zhang, Y.N.; Zhu, G.H.; Liu, W.; Chen, X.X.; Xie, Y.Y.; Xu, J.R.; Jiang, M.F.; Zhuang, X.Y.; Zhang, W.D.; Chen, H.Z.; et al. Discovery of the Covalent SARS-CoV-2 Mpro Inhibitors from Antiviral Herbs via Integrating Target-Based High-Throughput Screening and Chemoproteomic Approaches. J. Med. Virol. 2023, 95, e29208. [Google Scholar] [CrossRef]
- Shi, T.H.; Huang, Y.L.; Chen, C.C.; Pi, W.C.; Hsu, Y.L.; Lo, L.C.; Chen, W.Y.; Fu, S.L.; Lin, C.H. Andrographolide and Its Fluorescent Derivative Inhibit the Main Proteases of 2019-NCoV and SARS-CoV through Covalent Linkage. Biochem. Biophys. Res. Commun. 2020, 533, 467–473. [Google Scholar] [CrossRef]
- Khanfar, M.A.; Salaas, N.; Abumostafa, R. Discovery of Natural-Derived Mpro Inhibitors as Therapeutic Candidates for COVID-19: Structure-Based Pharmacophore Screening Combined with QSAR Analysis. Mol. Inf. Inform. 2023, 42, 2200198. [Google Scholar] [CrossRef] [PubMed]
- Samad, A.; Ajmal, A.; Mahmood, A.; Khurshid, B.; Li, P.; Jan, S.M.; Rehman, A.U.; He, P.; Abdalla, A.N.; Umair, M.; et al. Identification of Novel Inhibitors for SARS-CoV-2 as Therapeutic Options Using Machine Learning-Based Virtual Screening, Molecular Docking and MD Simulation. Front. Mol. Biosci. 2023, 10, 1060076. [Google Scholar] [CrossRef]
- Pillai U, J.; Cherian, L.; Taunk, K.; Iype, E.; Dutta, M. Identification of Antiviral Phytochemicals from Cranberry as Potential Inhibitors of SARS-CoV-2 Main Protease (Mpro). Int. J. Biol. Macromol. 2024, 261, 129655. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.A. Efficacy of Repurposed Antiviral Drugs: Lessons from COVID-19. Drug Discov. Today 2022, 27, 1954. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, F.A.; Costa, G.; Romeo, I.; Esposito, F.; Alkhatib, M.; Salpini, R.; Svicher, V.; Corona, A.; Malune, P.; Tramontano, E.; et al. Targeting SARS-CoV-2 Main Protease: A Successful Story Guided by an in Silico Drug Repurposing Approach. J. Chem. Inf. Model. 2023, 63, 3601–3613. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Jan, J.T.; Ma, S.H.; Kuo, C.J.; Juan, H.F.; Cheng, Y.S.E.; Hsu, H.H.; Huang, H.C.; Wu, D.; Brik, A.; et al. Small Molecules Targeting Severe Acute Respiratory Syndrome Human Coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 10012–10017. [Google Scholar] [CrossRef]
- El-Hddad, S.S.A.; Sobhy, M.H.; El-Morsy, A.; Shoman, N.A.; El-Adl, K. Quinazolines and Thiazolidine-2,4-Dions as SARS-CoV-2 Inhibitors: Repurposing, in Silico Molecular Docking and Dynamics Simulation. RSC Adv. 2024, 14, 13237–13250. [Google Scholar] [CrossRef]
- Xiao, B.; Wu, S.; Han, Y.; Ren, L.; Wang, J.; Luan, X.; Du, G.; She, Z. In Silico Screening of Minocycline as an Mpro Inhibitor and the Adjunctive Therapy Value for the Treatment of COVID-19. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Rothan, H.A.; Teoh, T.C. Cell-Based High-Throughput Screening Protocol for Discovering Antiviral Inhibitors Against SARS-CoV-2 Main Protease (3CLpro). Mol. Biotechnol. 2021, 63, 240–248. [Google Scholar] [CrossRef]
- Zhu, W.; Xu, M.; Chen, C.Z.; Guo, H.; Shen, M.; Hu, X.; Shinn, P.; Klumpp-Thomas, C.; Michael, S.G.; Zheng, W. Identification of SARS-CoV-2 3CL Protease Inhibitors by a Quantitative High-Throughput Screening. ACS Pharmacol. Transl. Sci. 2020, 3, 1008–1016. [Google Scholar] [CrossRef]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 Inhibitors Targeting Mpro and PLpro Using in-Cell-Protease Assay. Commun. Biol. 2022, 5, 1–17. [Google Scholar] [CrossRef]
- Zhang, J.; Yan, H.; Yan, G.; Liu, X.; Wang, Y.; Chen, Y. Protocol for High-Throughput Screening of SARS-CoV-2 Main Protease Inhibitors Using a Robust Fluorescence Polarization Assay. STAR Protoc. 2022, 3, 101794. [Google Scholar] [CrossRef]
- Zang, Y.; Su, M.; Wang, Q.; Cheng, X.; Zhang, W.; Zhao, Y.; Chen, T.; Jiang, Y.; Shen, Q.; Du, J.; et al. High-Throughput Screening of SARS-CoV-2 Main and Papain-like Protease Inhibitors. Protein Cell 2023, 14, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Davis-Gardner, M.E.; Garcia-Ordonez, R.D.; Nguyen, T.T.; Hull, M.; Chen, E.; Yu, X.; Bannister, T.D.; Baillargeon, P.; Scampavia, L.; et al. High Throughput Screening for Drugs That Inhibit 3C-like Protease in SARS-CoV-2. Slas Discov. 2023, 28, 95. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-Based Design of Antiviral Drug Candidates Targeting the SARS-CoV-2 Main Protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Lockbaum, G.J.; Reyes, A.C.; Lee, J.M.; Tilvawala, R.; Nalivaika, E.A.; Ali, A.; Yilmaz, N.K.; Thompson, P.R.; Schiffer, C.A. Crystal Structure of SARS-CoV-2 Main Protease in Complex with the Non-Covalent Inhibitor Ml188. Viruses 2021, 13, 174. [Google Scholar] [CrossRef]
- Balaramnavar, V.M.; Ahmad, K.; Saeed, M.; Ahmad, I.; Kamal, M.; Jawed, T. Pharmacophore-Based Approaches in the Rational Repurposing Technique for FDA Approved Drugs Targeting SARS-CoV-2 Mpro. RSC Adv. 2020, 10, 40264–40275. [Google Scholar] [CrossRef]
- Trivedi, G.N.; Karlekar, J.T.; Dhimmar, K.; Panchal, H. Fbdd: In-Silico Strategy to Inhibit Mpro Activity Using Drugs from Previous Outbreaks. J. Exp. Biol. Agric. Sci. 2021, 9, 472–480. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kuo, C.J.; Shin, J.S.; Jung, E.; Liang, P.H.; Jung, Y.S. Identification of Non-Covalent 3C-like Protease Inhibitors against Severe Acute Respiratory Syndrome Coronavirus-2 via Virtual Screening of a Korean Compound Library. Bioorganic Med. Chem. Lett. 2021, 42, 128067. [Google Scholar] [CrossRef]
- Luo, D.; Tong, J.B.; Zhang, X.; Xiao, X.C.; Bian, S. Computational Strategies towards Developing Novel SARS-CoV-2 Mpro Inhibitors against COVID-19. J. Mol. Struct. 2022, 1247, 131378. [Google Scholar] [CrossRef] [PubMed]
- Zhai, T.; Zhang, F.; Haider, S.; Kraut, D.; Huang, Z. An Integrated Computational and Experimental Approach to Identifying Inhibitors for SARS-CoV-2 3CL Protease. Front. Mol. Biosci. 2021, 8, 661424. [Google Scholar] [CrossRef] [PubMed]
- Mercorelli, B.; Desantis, J.; Celegato, M.; Bazzacco, A.; Siragusa, L.; Benedetti, P.; Eleuteri, M.; Croci, F.; Cruciani, G.; Goracci, L.; et al. Discovery of Novel SARS-CoV-2 Inhibitors Targeting the Main Protease Mpro by Virtual Screenings and Hit Optimization. Antivir. Res. 2022, 204, 105350. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, G.G.; Ossorio, M.A.; Rempel, S.; Kratzel, A.; Dionellis, V.S.; Barriot, S.; Tropia, L.; Gorgulla, C.; Arthanari, H.; Thiel, V.; et al. Non-Covalent SARS-CoV-2 Mpro Inhibitors Developed from in Silico Screen Hits. Sci. Rep. 2022, 12, 1–9. [Google Scholar] [CrossRef]
- Li, S.; Wang, L.; Meng, J.; Zhao, Q.; Zhang, L.; Liu, H. De Novo Design of Potential Inhibitors against SARS-CoV-2 Mpro. Comput. Biol. Med. 2022, 147, 105728. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.P.; Schultz, K.J.; Wilson, J.W.; Kruel, A.; Varikoti, R.A.; Kombala, C.J.; Kneller, D.W.; Galanie, S.; Phillips, G.; Zhang, Q.; et al. AI-Accelerated Design of Targeted Covalent Inhibitors for SARS-CoV-2. J. Chem. Inf. Model. 2023, 63, 1438–1453. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Fernandez, M.; Ban, F.; Ton, A.T.; Mslati, H.; Perez, C.F.; Leblanc, E.; Yaacoub, J.C.; Gleave, J.; Stern, A.; et al. Automated Discovery of Noncovalent Inhibitors of SARS-CoV-2 Main Protease by Consensus Deep Docking of 40 Billion Small Molecules. Chem. Sci. 2021, 12, 15960–15974. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic Peptides: Current Applications and Future Directions. Signal Transduct. Target. Ther. 2022, 7, 1–27. [Google Scholar] [CrossRef]
- Li Petri, G.; Di Martino, S.; De Rosa, M. Peptidomimetics: An Overview of Recent Medicinal Chemistry Efforts toward the Discovery of Novel Small Molecule Inhibitors. J. Med. Chem. 2022, 65, 7438–7475. [Google Scholar] [CrossRef]
- Vagner, J.; Qu, H.; Hruby, V.J. Peptidomimetics, a Synthetic Tool of Drug Discovery. Curr. Opin. Chem. Biol. 2008, 12, 292. [Google Scholar] [CrossRef] [PubMed]
- Gante, J. Peptidomimetics—Tailored Enzyme Inhibitors. Angew. Chem. Int. Ed. Engl. 1994, 33, 1699–1720. [Google Scholar] [CrossRef]
- Hirschmann, R. Medicinal Chemistry in the Golden Age of Biology: Lessons from Steroid and Peptide Research. Angew. Chem. Int. Ed. Engl. 1991, 30, 1278–1301. [Google Scholar] [CrossRef]
- Rizo, J.; Gierasch, L.M. Constrained Peptides: Models of Bioactive Peptides and Protein Substructures. Annu. Rev. Biochem. 1992, 61, 387–416. [Google Scholar] [CrossRef]
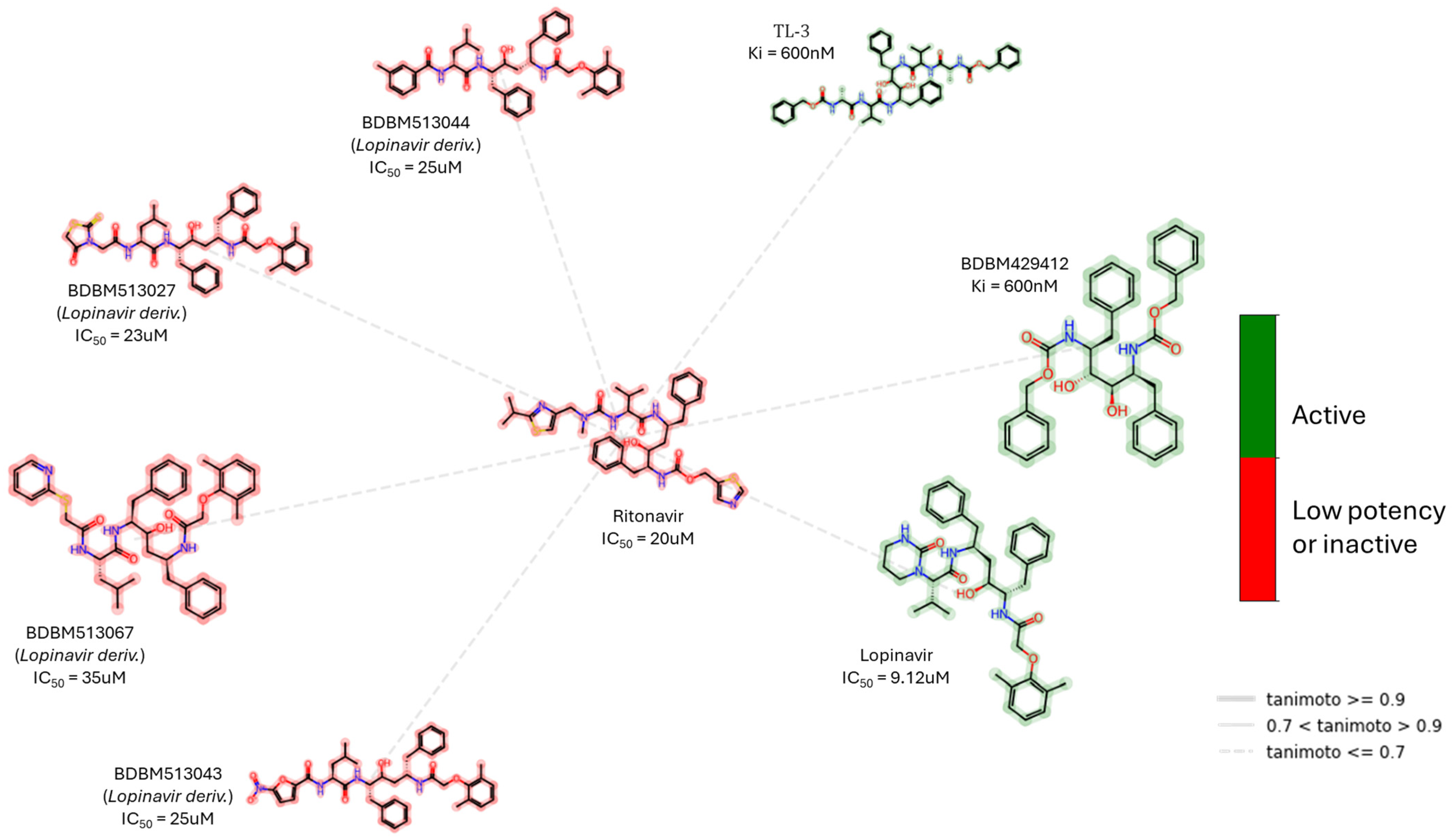
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and Calpain Inhibitors II, XII Inhibit SARS-CoV-2 Viral Replication by Targeting the Viral Main Protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Liu, Y.; Lv, S.; Peng, L.; Xie, C.; Gao, L.; Sun, H.; Lin, L.; Ding, K.; Li, Z. Development and Application of Novel Electrophilic Warheads in Target Identification and Drug Discovery. Biochem. Pharmacol. 2021, 190, 114636. [Google Scholar] [CrossRef]
- Somboon, T.; Mahalapbutr, P.; Sanachai, K.; Maitarad, P.; Lee, V.S.; Hannongbua, S.; Rungrotmongkol, T. Computational Study on Peptidomimetic Inhibitors against SARS-CoV-2 Main Protease. J. Mol. Liq. 2021, 322, 114999. [Google Scholar] [CrossRef]
- Shindo, N.; Ojida, A. Recent Progress in Covalent Warheads for in Vivo Targeting of Endogenous Proteins. Bioorganic Med. Chem. 2021, 47, 116386. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; Von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. α-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef]
- Chuck, C.P.; Chen, C.; Ke, Z.; Chi-Cheong Wan, D.; Chow, H.F.; Wong, K.B. Design, Synthesis and Crystallographic Analysis of Nitrile-Based Broad-Spectrum Peptidomimetic Inhibitors for Coronavirus 3C-like Proteases. Eur. J. Med. Chem. 2013, 59, 1–6. [Google Scholar] [CrossRef]
- Jain, R.P.; Pettersson, H.I.; Zhang, J.; Aull, K.D.; Fortin, P.D.; Huitema, C.; Eltis, L.D.; Parrish, J.C.; James, M.N.G.; Wishart, D.S.; et al. Synthesis and Evaluation of Keto-Glutamine Analogues as Potent Inhibitors of Severe Acute Respiratory Syndrome 3CLpro. J. Med. Chem. 2004, 47, 6113–6116. [Google Scholar] [CrossRef]
- Zhu, L.; George, S.; Schmidt, M.F.; Al-Gharabli, S.I.; Rademann, J.; Hilgenfeld, R. Peptide Aldehyde Inhibitors Challenge the Substrate Specificity of the SARS-Coronavirus Main Protease. Antivir. Res. 2011, 92, 204–212. [Google Scholar] [CrossRef]
- Lee, T.W.; Cherney, M.M.; Huitema, C.; Liu, J.; James, K.E.; Powers, J.C.; Eltis, L.D.; James, M.N.G. Crystal Structures of the Main Peptidase from the SARS Coronavirus Inhibited by a Substrate-like Aza-Peptide Epoxide. J. Mol. Biol. 2005, 353, 1137–1151. [Google Scholar] [CrossRef]
- Wang, F.; Chen, C.; Yang, K.; Xu, Y.; Liu, X.; Gao, F.; Liu, H.; Chen, X.; Zhao, Q.; Liu, X.; et al. Michael Acceptor-Based Peptidomimetic Inhibitor of Main Protease from Porcine Epidemic Diarrhea Virus. J. Med. Chem. 2017, 60, 3212–3216. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M. Recent Discovery and Development of Inhibitors Targeting Coronaviruses. Drug Discov. Today 2020, 25, 668–688. [Google Scholar] [CrossRef]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef]
- Mahmoud, A.; Mostafa, A.; Al-Karmalawy, A.A.; Zidan, A.; Abulkhair, H.S.; Mahmoud, S.H.; Shehata, M.; Elhefnawi, M.M.; Ali, M.A. Telaprevir Is a Potential Drug for Repurposing against SARS-CoV-2: Computational and in Vitro Studies. Heliyon 2021, 7, e07962. [Google Scholar] [CrossRef]
- Arafet, K.; Serrano-Aparicio, N.; Lodola, A.; Mulholland, A.J.; González, F.V.; Świderek, K.; Moliner, V. Mechanism of Inhibition of SARS-CoV-2 Mpro by N3 Peptidyl Michael Acceptor Explained by QM/MM Simulations and Design of New Derivatives with Tunable Chemical Reactivity. Chem. Sci. 2021, 12, 1433–1444. [Google Scholar] [CrossRef]
- Martí, S.; Arafet, K.; Lodola, A.; Mulholland, A.J.; Świderek, K.; Moliner, V. Impact of Warhead Modulations on the Covalent Inhibition of SARS-CoV-2 Mpro Explored by QM/MM Simulations. ACS Catal. 2022, 12, 698–708. [Google Scholar] [CrossRef]
- Zaidman, D.; Gehrtz, P.; Filep, M.; Fearon, D.; Gabizon, R.; Douangamath, A.; Prilusky, J.; Duberstein, S.; Cohen, G.; Owen, C.D.; et al. An Automatic Pipeline for the Design of Irreversible Derivatives Identifies a Potent SARS-CoV-2 Mpro Inhibitor. Cell Chem. Biol. 2021, 28, 1795–1806.e5. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Sacco, M.; Tan, H.; Li, K.; Joyce, R.; Zhang, X.; Chen, Y.; Wang, J. Exploring Diverse Reactive Warheads for the Design of SARS-CoV-2 Main Protease Inhibitors. Eur. J. Med. Chem. 2023, 259, 115667. [Google Scholar] [CrossRef]
- Singh, R.S.P.; Toussi, S.S.; Hackman, F.; Chan, P.L.; Rao, R.; Allen, R.; Van Eyck, L.; Pawlak, S.; Kadar, E.P.; Clark, F.; et al. Innovative Randomized Phase I Study and Dosing Regimen Selection to Accelerate and Inform Pivotal COVID-19 Trial of Nirmatrelvir. Clin. Pharmacol. Ther. 2022, 112, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simón-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with COVID-19. N. Engl. J. Med. 2022, 386, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Pawlak, S.; Toussi, S.S.; Hackman, F.; Thompson, K.; Song, W.; Salageanu, J.; Winter, E.; Shi, H.; Winton, J.; et al. Safety, Tolerability, and Pharmacokinetics of Intravenous Doses of PF-07304814, a Phosphate Prodrug Protease Inhibitor for the Treatment of SARS-CoV-2, in Healthy Adult Participants. Clin. Pharmacol. Drug Dev. 2022, 11, 1382–1393. [Google Scholar] [CrossRef]
- Robinson, P.; Toussi, S.S.; Aggarwal, S.; Bergman, A.; Zhu, T.; Hackman, F.; Sathish, J.G.; Updyke, L.; Loudon, P.; Krishna, G.; et al. Safety, Tolerability, and Pharmacokinetics of Single and Multiple Ascending Intravenous Infusions of PF-07304814 (Lufotrelvir) in Participants Hospitalized With COVID-19. Open Forum Infect. Dis. 2023, 10, ofad355. [Google Scholar] [CrossRef]
- Allerton, C.M.N.; Arcari, J.T.; Aschenbrenner, L.M.; Avery, M.; Bechle, B.M.; Behzadi, M.A.; Boras, B.; Buzon, L.M.; Cardin, R.D.; Catlin, N.R.; et al. A Second-Generation Oral SARS-CoV-2 Main Protease Inhibitor Clinical Candidate for the Treatment of COVID-19. J. Med. Chem. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, J.; Chen, H.; Mao, J.; Tang, Y.; Yan, W.; Zhang, T.; Li, C.; Chen, S.; Li, G.; et al. Phase I Study, and Dosing Regimen Selection for a Pivotal COVID-19 Trial of GST-HG171. Antimicrob. Agents Chemother. 2024, 68, e01115-23. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhang, G.; Mao, J.; Chen, X.; Zhan, Y.; Lin, L.; Zhang, T.; Tang, Y.; Lin, F.; Zhu, F.; et al. Efficacy and Safety of GST-HG171 in Adult Patients with Mild to Moderate COVID-19: A Randomised, Double-Blind, Placebo-Controlled Phase 2/3 Trial. eClinicalMedicine 2024, 71, 102582. [Google Scholar] [CrossRef]
- Wang, F.; Xiao, W.; Tang, Y.; Cao, M.; Shu, D.; Asakawa, T.; Xu, Y.; Jiang, X.; Zhang, L.; Wang, W.; et al. Efficacy and Safety of SIM0417 (SSD8432) plus Ritonavir for COVID-19 Treatment: A Randomised, Double-Blind, Placebo-Controlled, Phase 1b Trial. Lancet Reg. Health West. Pac. 2023, 38, 100835. [Google Scholar] [CrossRef]
- Yang, X.M.; Yang, Y.; Yao, B.F.; Ye, P.P.; Xu, Y.; Peng, S.P.; Yang, Y.M.; Shu, P.; Li, P.J.; Li, S.; et al. A First-in-Human Phase 1 Study of Simnotrelvir, a 3CL-like Protease Inhibitor for Treatment of COVID-19, in Healthy Adult Subjects. Eur. J. Pharm. Sci. 2023, 191, 106598. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Lu, H.; Huang, C.; Yang, Y.; Shang, L.; Chen, Z.; Jiang, R.; Liu, Y.; Lin, L.; et al. Oral Simnotrelvir for Adult Patients with Mild-to-Moderate COVID-19. N. Engl. J. Med. 2024, 390, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Lin, Z.; Liang, J.; Sun, R.; Li, Y.; Lin, B.; Ge, F.; Lin, L.; Lu, H.; Su, L.; et al. Leritrelvir for the Treatment of Mild or Moderate COVID-19 without Co-Administered Ritonavir: A Multicentre Randomised, Double-Blind, Placebo-Controlled Phase 3 Trial. eClinicalMedicine 2023, 67, 102359. [Google Scholar] [CrossRef] [PubMed]
- Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.; Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.; et al. Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19. J. Med. Chem. 2022, 65, 6499–6512. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, R.; Sonoyama, T.; Fukuhara, T.; Kuwata, A.; Matsuo, Y.; Kubota, R. A Phase 1 Study of Ensitrelvir Fumaric Acid Tablets Evaluating the Safety, Pharmacokinetics and Food Effect in Healthy Adult Populations. Clin. Drug Investig. 2023, 43, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Yamato, M.; Bac, N.H.; Cha, B.K.; Imamura, T.; Sonoyama, T.; Ichihashi, G.; Sanaki, T.; et al. Efficacy and Safety of 5-Day Oral Ensitrelvir for Patients With Mild to Moderate COVID-19: The SCORPIO-SR Randomized Clinical Trial. JAMA Netw. Open 2024, 7, E2354991. [Google Scholar] [CrossRef] [PubMed]
- She, Z.; Yao, Y.; Wang, C.; Li, Y.; Xiong, X.; Liu, Y. Mpro-Targeted Anti-SARS-CoV-2 Inhibitor-Based Drugs. J. Chem. Res. 2023, 47, 17475198231184799. [Google Scholar] [CrossRef] [PubMed]
- Hou, N.; Shuai, L.; Zhang, L.; Xie, X.; Tang, K.; Zhu, Y.; Yu, Y.; Zhang, W.; Tan, Q.; Zhong, G.; et al. Development of Highly Potent Noncovalent Inhibitors of SARS-CoV-2 3CLpro. ACS Cent. Sci. 2022, 9, 217–227. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Z. Bench-to-Bedside: Innovation of Small Molecule Anti-SARS-CoV-2 Drugs in China. Eur. J. Med. Chem. 2023, 257, 115503. [Google Scholar] [CrossRef]
- Mao, L.; Shaabani, N.; Zhang, X.; Jin, C.; Xu, W.; Argent, C.; Kushnareva, Y.; Powers, C.; Stegman, K.; Liu, J.; et al. Olgotrelvir, a Dual Inhibitor of SARS-CoV-2 Mpro and Cathepsin L, as a Standalone Antiviral Oral Intervention Candidate for COVID-19. Med 2024, 5, 42–61.e23. [Google Scholar] [CrossRef]
- Sorrento Announces Phase 3 Trial Met Primary Endpoint and Key Secondary Endpoint in Mild or Moderate COVID-19 Adult Patients Treated with Ovydso (Olgotrelvir), an Oral Mpro Inhibitor as a Standalone Treatment for COVID-19|BioSpace. Available online: https://www.biospace.com/article/releases/sorrento-announces-phase-3-trial-met-primary-endpoint-and-key-secondary-endpoint-in-mild-or-moderate-covid-19-adult-patients-treated-with-ovydso-olgotrelvir-an-oral-mpro-inhibitor-as-a-standalone-treatment-for-covid-19/ (accessed on 24 June 2024).
- Anderson, A.S.; Caubel, P.; Rusnak, J.M. Nirmatrelvir–Ritonavir and Viral Load Rebound in COVID-19. N. Engl. J. Med. 2022, 387, 1047–1049. [Google Scholar] [CrossRef]
- Lübbert, C.; Dykukha, I.; Pelz, J.P.; Yearley, H.; Junker, W.; Gruber, N.; Escher, S.; Biereth, K.; Melnik, S.; Puschmann, J. Individuals at Risk for Severe COVID-19 in Whom Ritonavir-Containing Therapies Are Contraindicated or May Lead to Interactions with Concomitant Medications: A Retrospective Analysis of German Health Insurance Claims Data. Drugs Context 2023, 12, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Gao, S.; Ye, B.; Yang, M.; Cheng, Y.; Kang, D.; Yi, F.; Sun, J.P.; Menéndez-Arias, L.; Neyts, J.; et al. Medicinal Chemistry Strategies towards the Development of Non-Covalent SARS-CoV-2 Mpro Inhibitors. Acta Pharm. Sin. B 2024, 14, 87–109. [Google Scholar] [CrossRef] [PubMed]
- Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura, T.; Sonoyama, T.; Ichihashi, G.; Sanaki, T.; Tsuge, Y.; Uehara, T.; Mukae, H. A Phase 2/3 Study of S-217622 in Participants with SARS-CoV-2 Infection (Phase 3 Part). Medicine 2023, 102, E33024. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Han, B.; Xu, W.; Zhang, X.; Peng, C.; Dang, Q.; Sun, W.; Lin, L.; Lin, Y.; Fan, L.; et al. Olgotrelvir as a Single-Agent Treatment of Nonhospitalized Patients with COVID-19. NEJM Evid. 2024, 3, EVIDoa2400026. [Google Scholar] [CrossRef]




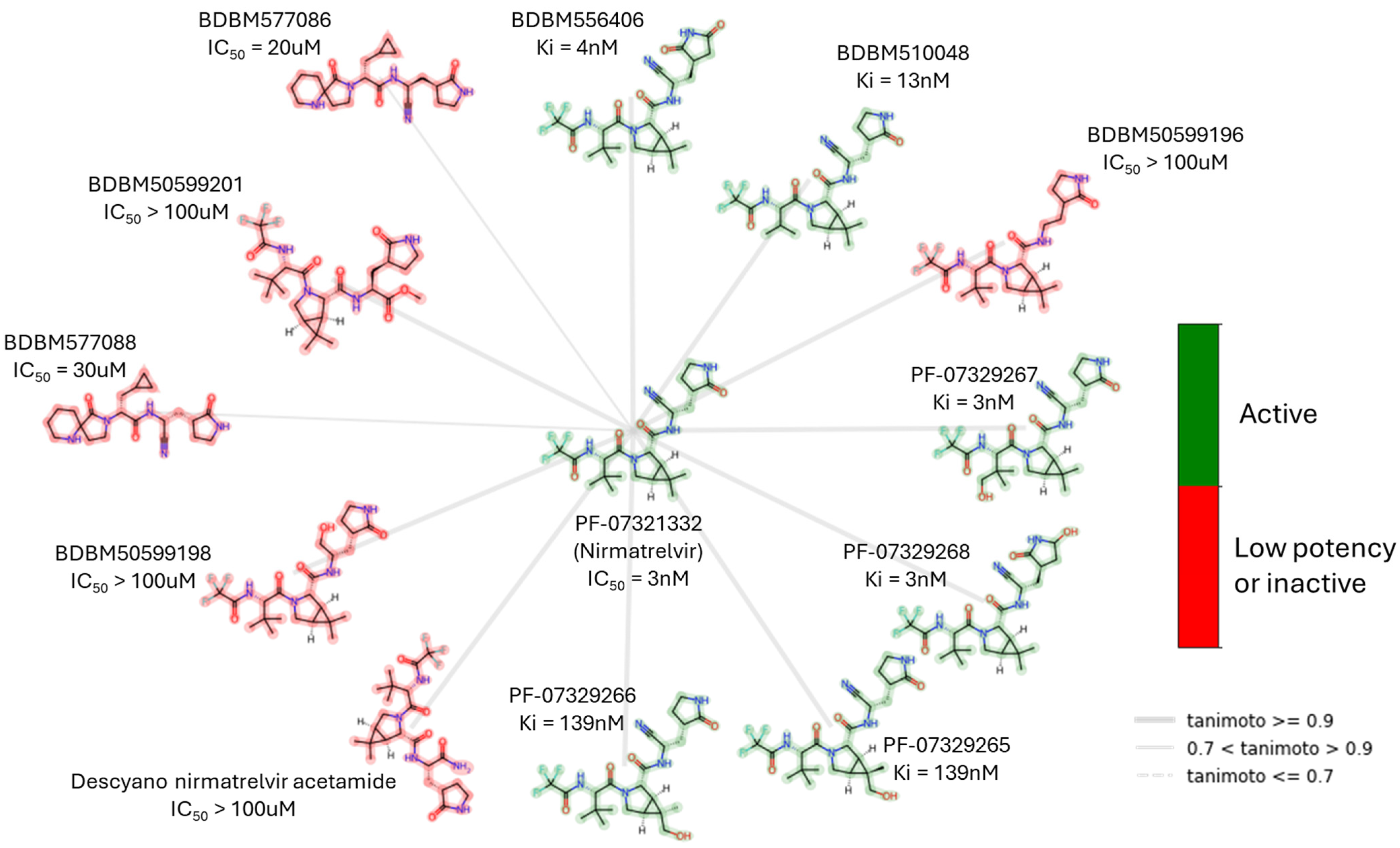
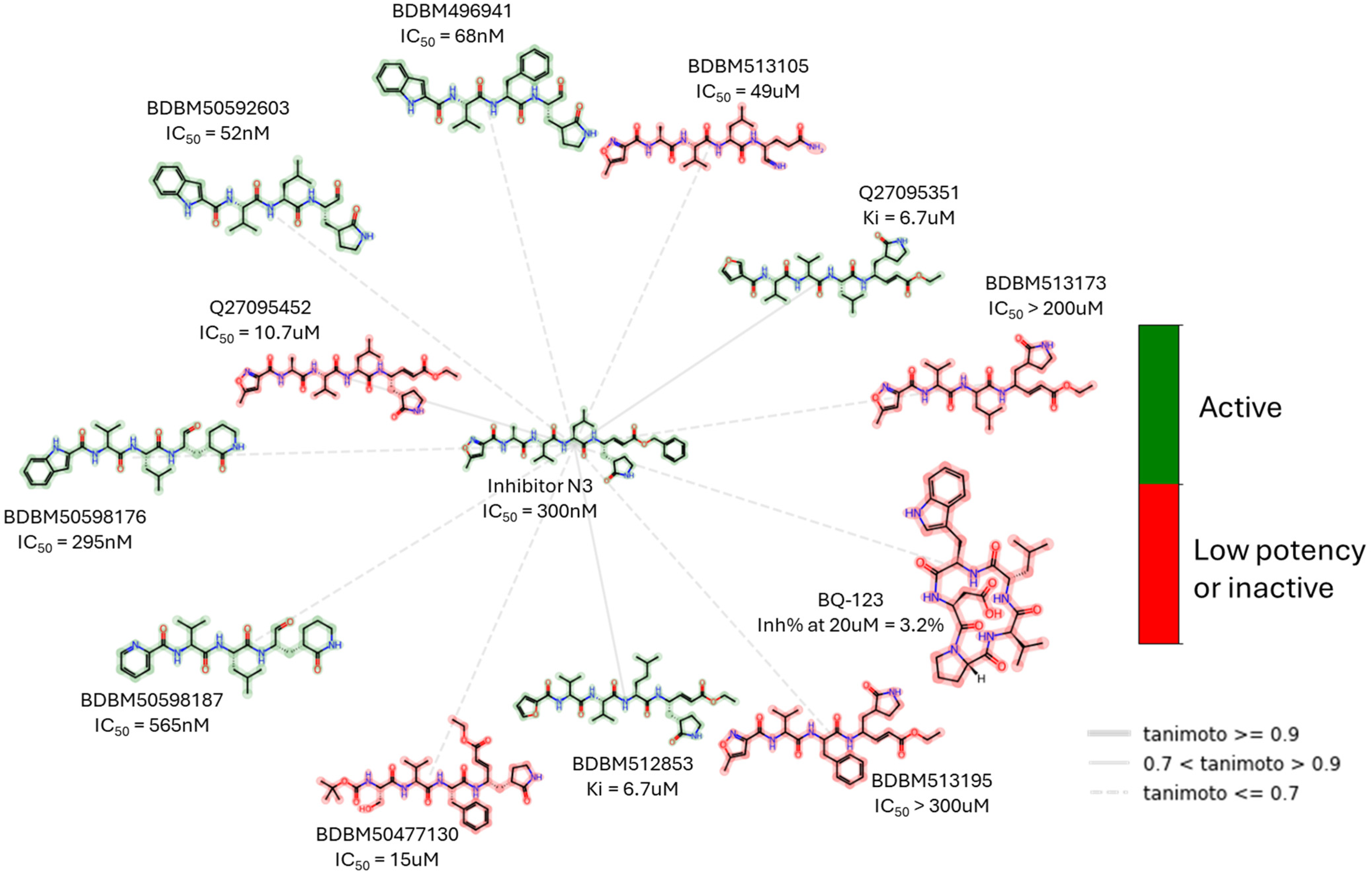

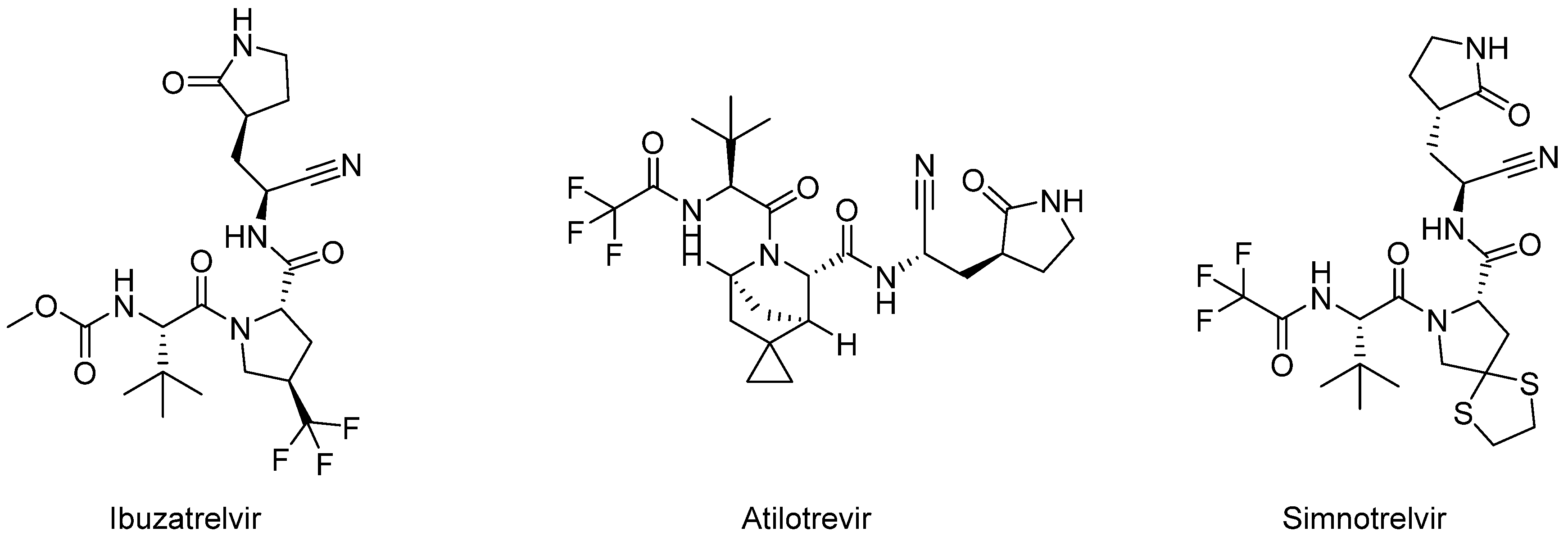
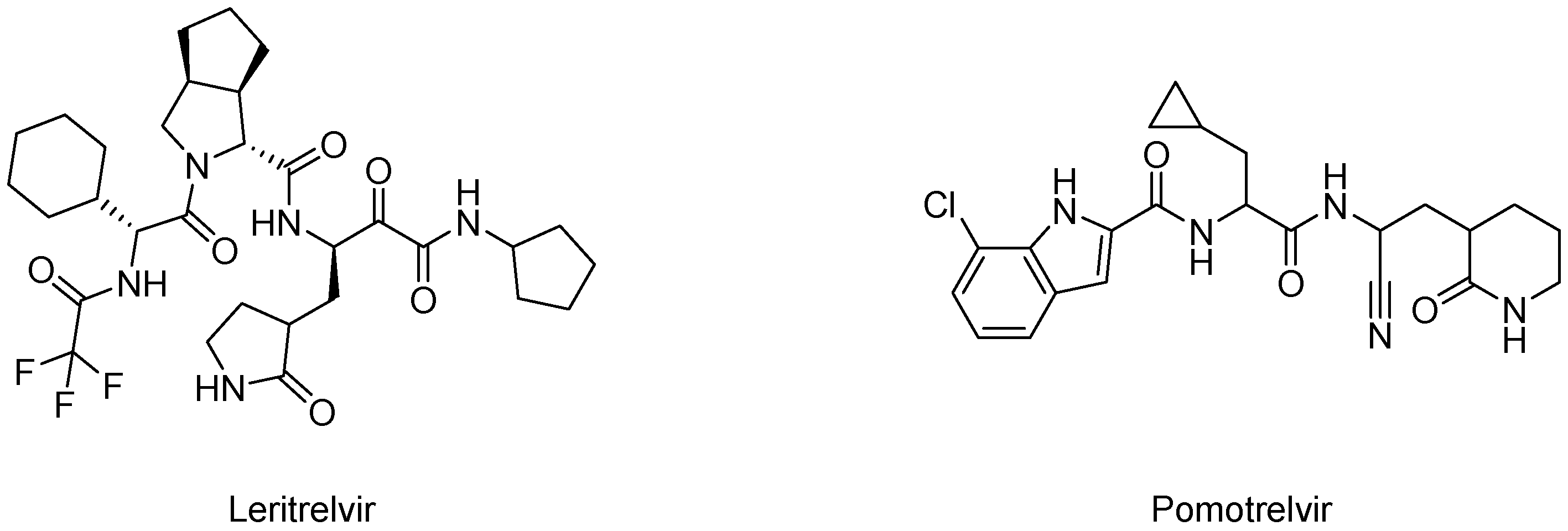

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptidomimetics | Small Molecules |
|---|---|
| Advantages | |
| Mimic the complex interactions of natural peptides with their receptors or other targets. | Can interact with a wide range of biological targets. Versatile tools in drug discovery |
| Like natural peptides, showed high specificity and selectivity for targets, fewer off-target effects, and reduced toxicity. | Better membrane permeability, easily cross the cell membranes, reaching intracellular targets. |
| Improved metabolic stability compared to natural peptides | Good oral bioavailability, more convenient for patients. |
| Disadvantages | |
| Many peptidomimetics still suffer from poor oral bioavailability and may require parenteral administration. | Small molecules can be rapidly metabolized and excreted, sometimes requiring frequent dosing or leading to short half-lives. |
| Compared to small molecules, inadequate membrane permeability | High potential for off-target effects and toxicity. |
| Complex and expensive synthetic processes are barriers to large-scale production. | Limited capability to mimic complex and specific interactions of larger biomolecules like peptides. |
| They can sometimes elicit immune responses. | More prone to the development of drug resistance. |
| Warhead | Mechanism of Action | Example |
|---|---|---|
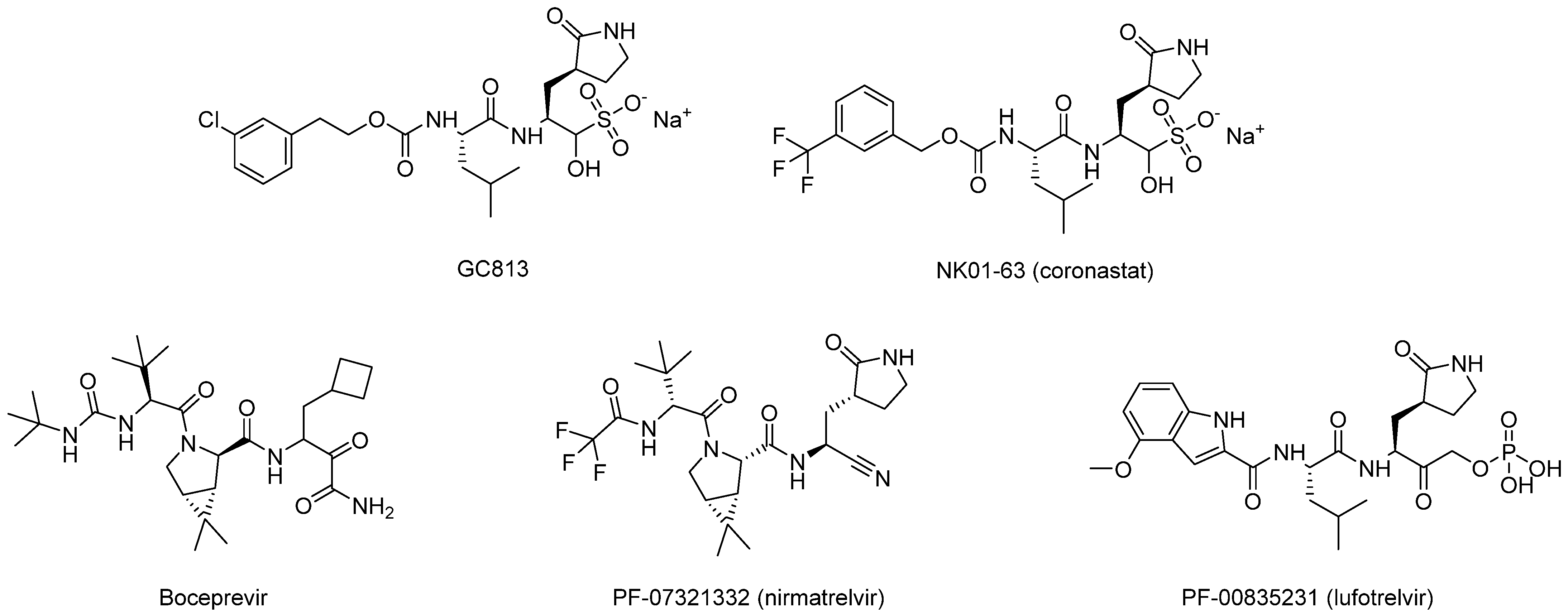
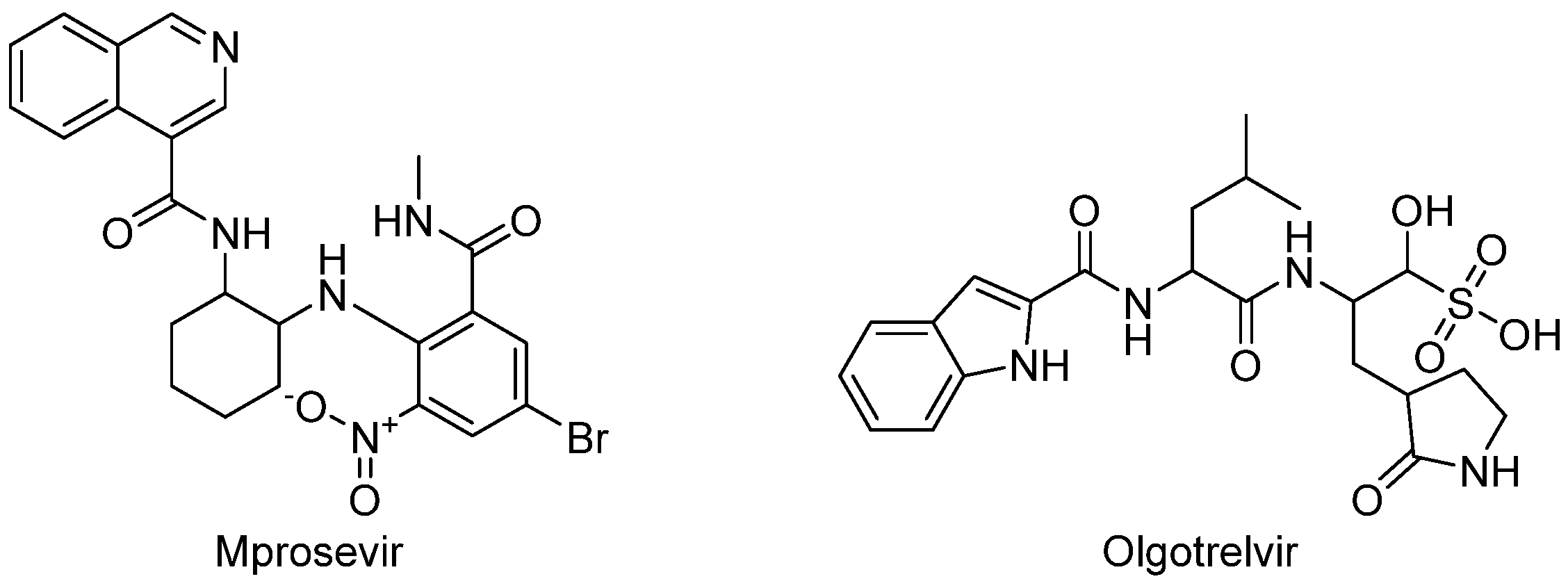
Carbonyl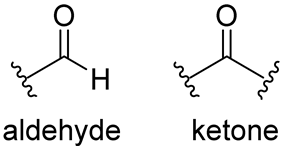 | The nucleophilic addition of the cysteine -SH leads to the formation of a reversible hemithioacetal adduct.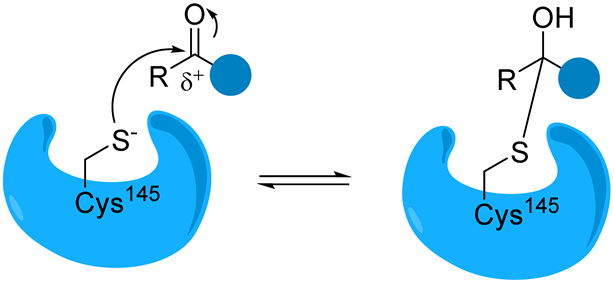 | (a) aldehyde GC373 GC376 [86] NK01-63 (coronastat) (b) ketone PF-00835231 PF-07304814 [87] |
α-Ketonamide | The α-ketoamide moiety forms additional non-covalent H bonds with the amino acids of the active site via the carbonyl oxygen and the -OH of the hemithioacetal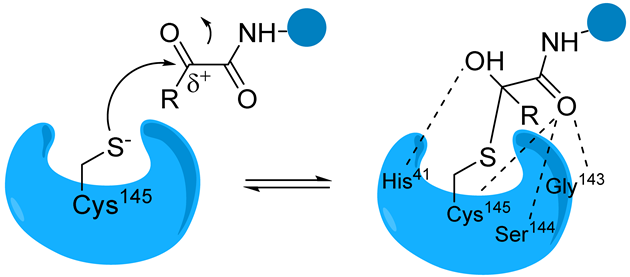 | Boceprevir [76] Telaprevir [88] UAWJ246 UAWJ248 [20] |
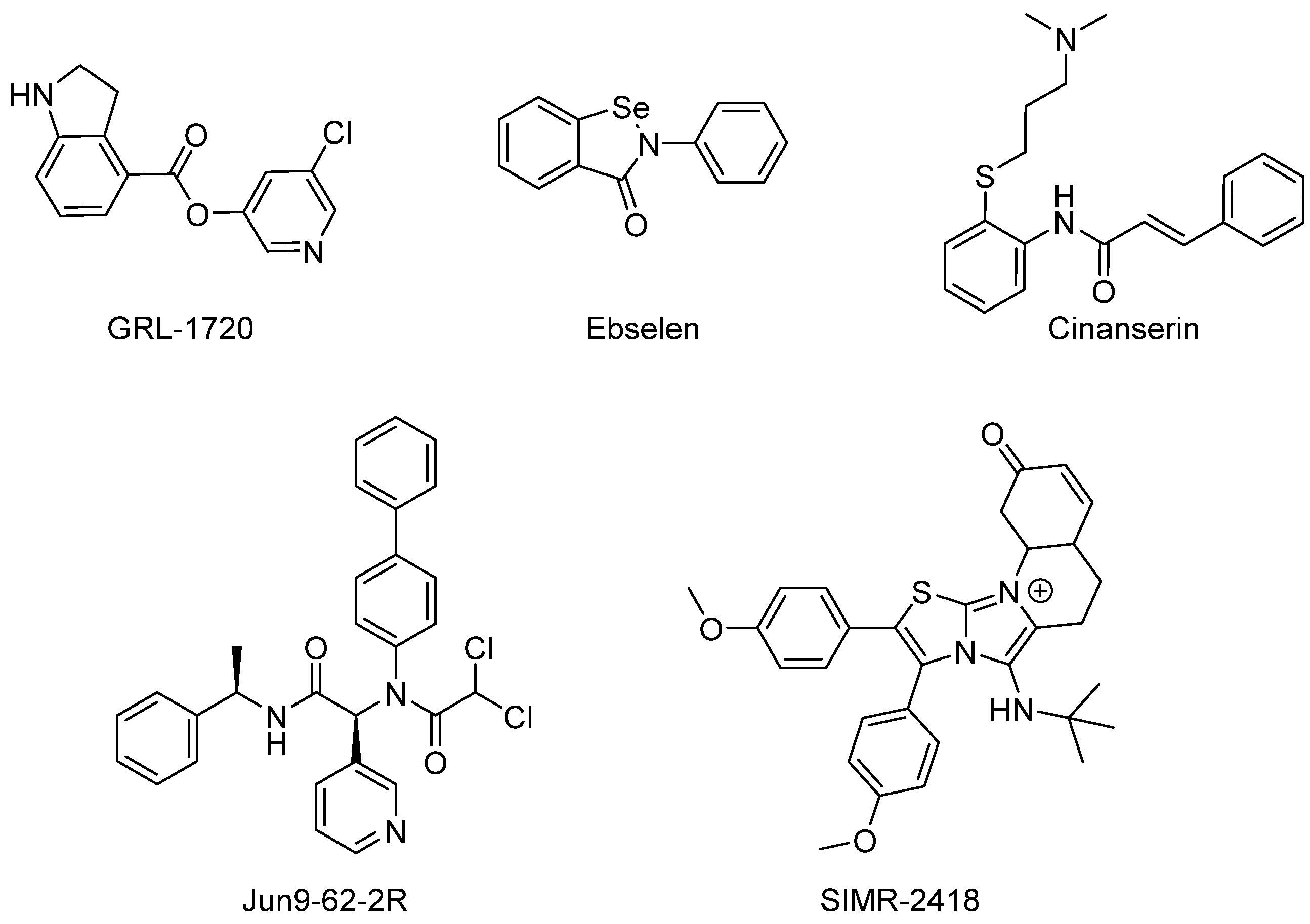
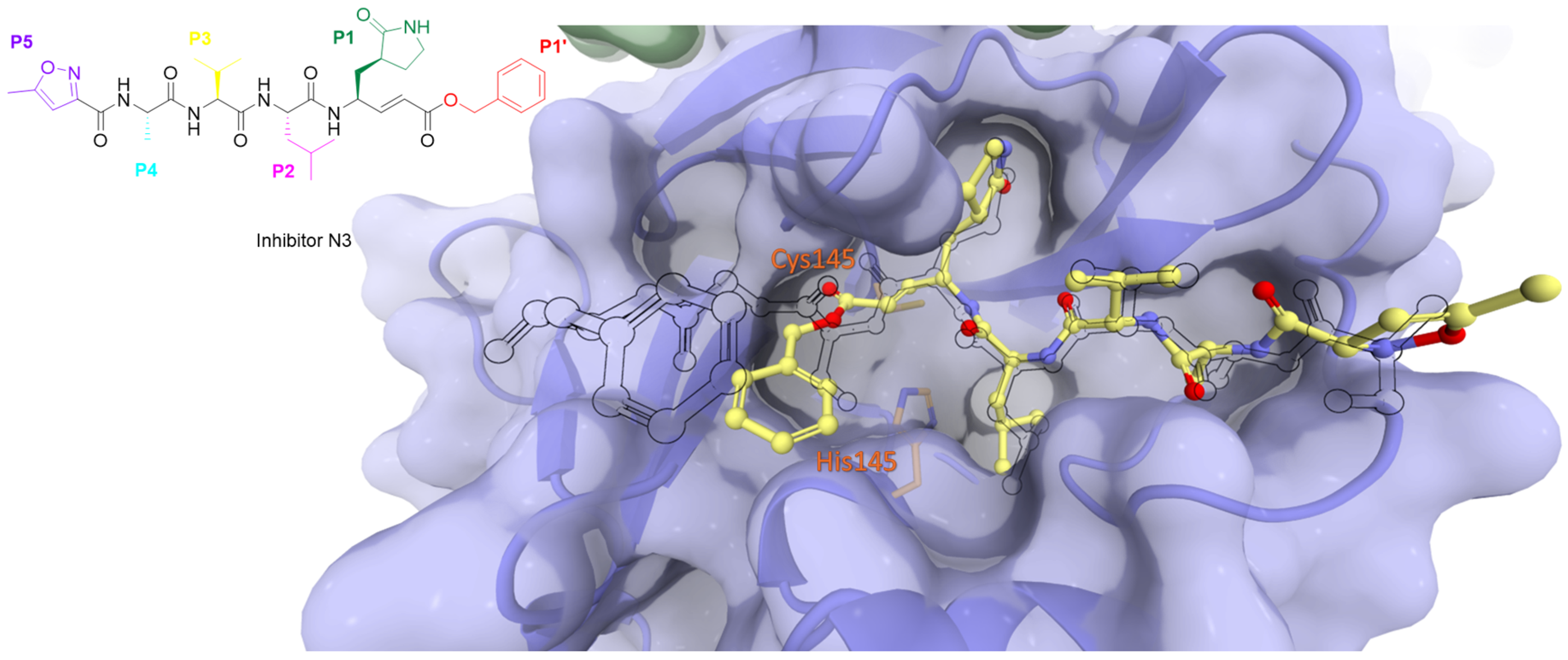
Michael acceptor group | Michael acceptor groups inhibit the enzymes via conjugate addition of the nucleophilic cysteinyl -SH to the electrophilic Cβ of the unsaturated system, producing a nearly irreversible and longer-lasting adduct.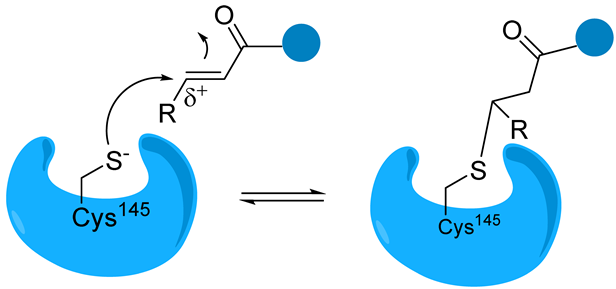 | Compound N3 [89,90] SIMR-2418 [34] Cinanserin [32] ML188 [91] |
Nitrile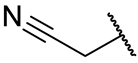 | Due to the difference in electronegativity with the nitrogen atom, the nitrile carbon is vulnerable to nucleophilic addition by the protease’s catalytic cysteine, leading to the formation of a reversible thioimidate covalent adduct. | PF-07321332 (nirmatrelvir) [37] PF-07817883 [92] GST-HG171 (atilotrevir) [36] |
| Active Compound (Sponsor) | ClinicalTrials.gov Identifier | Phase | Summary of the Study |
|---|---|---|---|
| Nilmatrelvir (PF-07321332)/ Ritonavir (Pfizer, New York, NY, USA) | NCT04756531 | 1 | Study of PF-07321332 in healthy participants. |
| NCT04960202 | 2/3 | EPIC-HR: Study of Oral PF-07321332/Ritonavir Compared with Placebo in Nonhospitalized High Risk Adults With COVID-19. | |
| NCT05047601 | 2/3 | A Study of a Potential Oral Treatment to Prevent COVID-19 in Adults Who Are Exposed to Household Member(s) With a Confirmed Symptomatic COVID-19 Infection. | |
| NCT05129475 | 1 | Food Effect Study to Evaluate the Effect of High-Fat Meal on the Relative Bioavailability of PF-07321332 Boosted with Ritonavir in Healthy Adult Participants. | |
| NCT05263895 | 1 | Relative Bioavailability Study of 4 Different Formulations of PF-07321332 Relative to the Commercial Tablet Formulation. | |
| NCT05339334 | 1 | A Study to Learn About the Study Medicine PF-07321332 and Ritonavir in Adult Healthy Chinese Participants. | |
| NCT05441215 | 1 | A Study to Learn About the Medicine (PF-07321332 or Nirmatrelvir/Ritonavir) in Healthy Lactating Women. | |
| NCT05263921 | 1 | Relative Bioavailability Study of PF-07321332/Ritonavir Oral Powder Relative to the Commercial Tablets in Healthy Participants. | |
| Lufotrelvir (PF-07304814)/ Ritonavir (Pfizer, New York, NY, USA) | NCT04535167 | 1 | First-In-Human Study to Evaluate Safety, Tolerability, And Pharmacokinetics Following Single Ascending and Multiple Ascending Doses of PF-07304814 In Hospitalized Participants With COVID-19. |
| NCT04627532 | 1 | Single Ascending Dose Study of Intravenous Infusion of PF 07304814 in Healthy Adult Participants. | |
| NCT05050682 | 1 | Study to Investigate the Mass Balance, Metabolism, and Excretion of [14C]-PF-07304814 in Healthy Participants. | |
| Ibuzatrelvir (PF- 07817883)/ Ritonavir (Pfizer, New York, NY, USA) | NCT05580003 | 1 | A Study to Learn Safety and Blood Levels of PF-07817883 in Healthy People. |
| NCT05799495 | 2 | A Study to Understand the Effect and Safety of the Study Medicine PF-07817883 in Adults Who Have Symptoms of COVID-19 But Are Not Hospitalized. | |
| NCT06122194 | 1 | A Study to Learn PF-07817883 Blood Levels After Administration of Tablets of Study Drug to Healthy Adult Volunteers. | |
| Atilotrelvir (GST-HG171)/ Ritonavir (Fujian Akeylink Biotechnology Co., Ltd., Ningde, Fujian, China) | NCT05668897 | 1 | Safety, Tolerability and Pharmacokinetic Characteristics Evaluation on GST-HG171 Tablets. |
| NCT05656443 | 2/3 | Study of GST-HG171/Ritonavir Compared with Placebo in Patients with Mild to Moderate COVID-19. | |
| NCT06106113 | 1 | Pharmacokinetics and Safety of GST-HG171 Tablets in Subjects with Impaired and Normal Liver Function. | |
| NCT06084507 | 1 | Food Effects of GST-HG171 Tablets Combined with Ritonavir in Healthy Chinese Participants. | |
| NCT06087055 | 1 | Drug-Drug Interaction Study of Itraconazole With GST-HG171/Ritonavir in Healthy Participants. | |
| Simnotrelvir (SIM-0417, SSD8432)/ Ritonavir (Jiangsu Simcere Pharmaceutical Co., Ltd., Nanjing, China) | NCT05339646 | 1 | A Phase 1 Clinical Study of SSD8432 in Healthy Adult Subjects. |
| NCT05369676 | 1/2 | To Evaluate SSD8432/Ritonavir in Adults With COVID-19. | |
| NCT05475834 | 1 | Study to Investigate the Mass Balance and Biotransformation of SIM0417 in Healthy Adult Chinese Male Participants. | |
| NCT05506176 | 2/3 | A Clinical Study to Evaluate the Efficacy and Safety of SIM0417 Orally Co-Administered with Ritonavir in Symptomatic Adult Participants with Mild to Moderate COVID-19. | |
| NCT05665647 | 1 | Drug-Drug Interaction Study of Itraconazole, Rifampicin and Midazolam with SIM0417/Ritonavir in Healthy Participants. | |
| Leritrelwir (RAY1216) (Guangdong Raynovent Biotech Co., Ltd., Guangzhou, China) | NCT05829551 | 1 | The Safety, Tolerability and Pharmacokinetics Study of RAY1216 in Healthy Adult Participants. |
| NCT05620160 | 3 | Study of RAY1216 Tablets Compared with Placebo in Patients with Mild to Moderate COVID-19. | |
| NCT06031454 | 1 | Drug-durg Interaction of Leritrelvir (RAY1216) With Midazolam, Omeprazole, Rosuvastatin, Verapamil, and Rifampin. | |
| NCT06169085 | 1 | Pharmacokinetics of Leritrelvir (Ray1216) in Elder Participants. | |
| NCT06160622 | 1 | Pharmacokinetics of Leritrelvir (RAY1216) in Participants with Severe Kidney Disease. | |
| NCT06161259 | 1 | Pharmacokinetics of Leritrelvir (RAY1216) in Participants with Hepatic Impairment. | |
| NCT06362460 | 1 | Mass Balance Study of [14C] RAY1216 in Healthy Adult Male Subjects in China. | |
| Pomotrelvir (PBI-0451) (Pardes Biosciences, Inc., Carlsbad, CA, USA) | NCT05011812 | 1 | Study of PBI-0451 in Healthy Subjects. |
| NCT05543707 | 2 | PBI-0451 (Pomotrelvir) Phase 2 Study in Nonhospitalized Symptomatic Adults With COVID-19. | |
| Ensitrelvir (S-217622) | jRCT2031210202 * | 1 | A Phase 1 study of S-217622. |
| (Nagata Tsute, Osaka, Japan) | |||
| (Gomez JuanCarlos, Osaka, Japan) | jRCT2031210350 * | 2/3 | A Phase 2/3 study of S-217622. |
| (Timothy Henrich, San Francisco, CA, USA) | NCT06161688 | 2 | Ensitrelvir for Viral Persistence and Inflammation in People Experiencing Long COVID (PREVAIL-LC). |
| (Shionogi Inc., Osaka, Japan) | NCT05897541 | 3 | Phase 3 Study of S-217622 in Prevention of Symptomatic SARS-CoV-2 Infection (SCORPIO-PEP). |
| NCT05305547 | 3 | A Study to Compare S-217622 With Placebo in Non-Hospitalized Participants With COVID-19 (SCORPIO-HR). | |
| (University of Minnesota, Minneapolis, MN, USA) | NCT05605093 | 3 | Strategies and Treatments for Respiratory Infections & Viral Emergencies (STRIVE): Shionogi Protease Inhibitor (Ensitrelvir). |
| Mprosevir (WPV01, WU-04)/Ritonavir (Westlake Pharmaceuticals (Hangzhou) Co., Ltd., Hangzhou, China) | NCT06205329 | 1 | Study of WPV01 in Healthy Subjects. |
| NCT05752175 | 2 | Study of WPV01 Compared with Placebo in Patients with Mild/Moderate COVID-19 Infection | |
| NCT06197217 | 3 | Phase 3 Clinical Study Evaluating the Efficacy and Safety of WPV01 in Patients with Mild/Moderate COVID-19. | |
| Olgotrelvir (STI-1558) | NCT05364840 | 1 | Study to Assess the Safety, Tolerability and Pharmacokinetics of STI-1558 in Healthy Volunteers. |
| (Sorrento Therapeutics, Inc., San Diego, CA, USA) | |||
| (Zhejiang ACEA Pharmaceutical Co., Ltd., Hangzhou, China) | NCT05523739 | 1 | Safety and Efficacy Study of STI-1558 in Healthy Adults and SARS-CoV-2-Positive Patients. |
| (Zhejiang ACEA Pharmaceutical Co., Ltd., Hangzhou, China) | NCT05716425 | 3 | Study to Assess the Efficacy and Safety of STI-1558 in Adult Subjects with Mild or Moderate. |
| (Zhejiang ACEA Pharmaceutical Co., Ltd., Hangzhou, China) | NCT05754411 | 1 | Study on Mass Balance and Biotransformation of STI-1558 in Healthy Chinese Adult Male Subjects. |
| (Zhejiang ACEA Pharmaceutical Co., Ltd., Hangzhou, China) | NCT06044233 | 1 | A Bioequivalence Trial of Fasting Single Oral STI-1558 Capsule in Healthy Chinese Subjects. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zagórska, A.; Czopek, A.; Fryc, M.; Jończyk, J. Inhibitors of SARS-CoV-2 Main Protease (Mpro) as Anti-Coronavirus Agents. Biomolecules 2024, 14, 797. https://doi.org/10.3390/biom14070797
Zagórska A, Czopek A, Fryc M, Jończyk J. Inhibitors of SARS-CoV-2 Main Protease (Mpro) as Anti-Coronavirus Agents. Biomolecules. 2024; 14(7):797. https://doi.org/10.3390/biom14070797
Chicago/Turabian StyleZagórska, Agnieszka, Anna Czopek, Monika Fryc, and Jakub Jończyk. 2024. "Inhibitors of SARS-CoV-2 Main Protease (Mpro) as Anti-Coronavirus Agents" Biomolecules 14, no. 7: 797. https://doi.org/10.3390/biom14070797
APA StyleZagórska, A., Czopek, A., Fryc, M., & Jończyk, J. (2024). Inhibitors of SARS-CoV-2 Main Protease (Mpro) as Anti-Coronavirus Agents. Biomolecules, 14(7), 797. https://doi.org/10.3390/biom14070797