
Amino-Acid Characteristics in Protein Native State Structures
 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
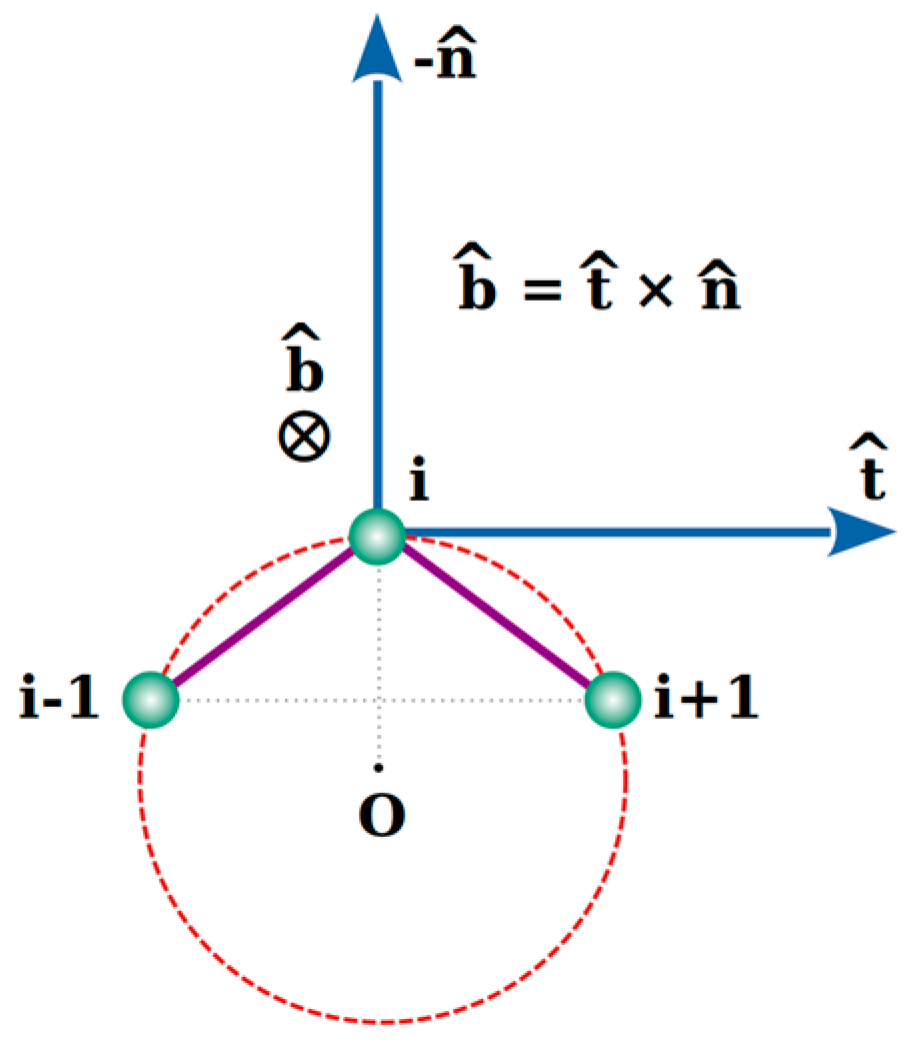
2.1. Local Frenet Coordinate System of an Amino Acid
2.2. Curation and Data Analysis
3. Results
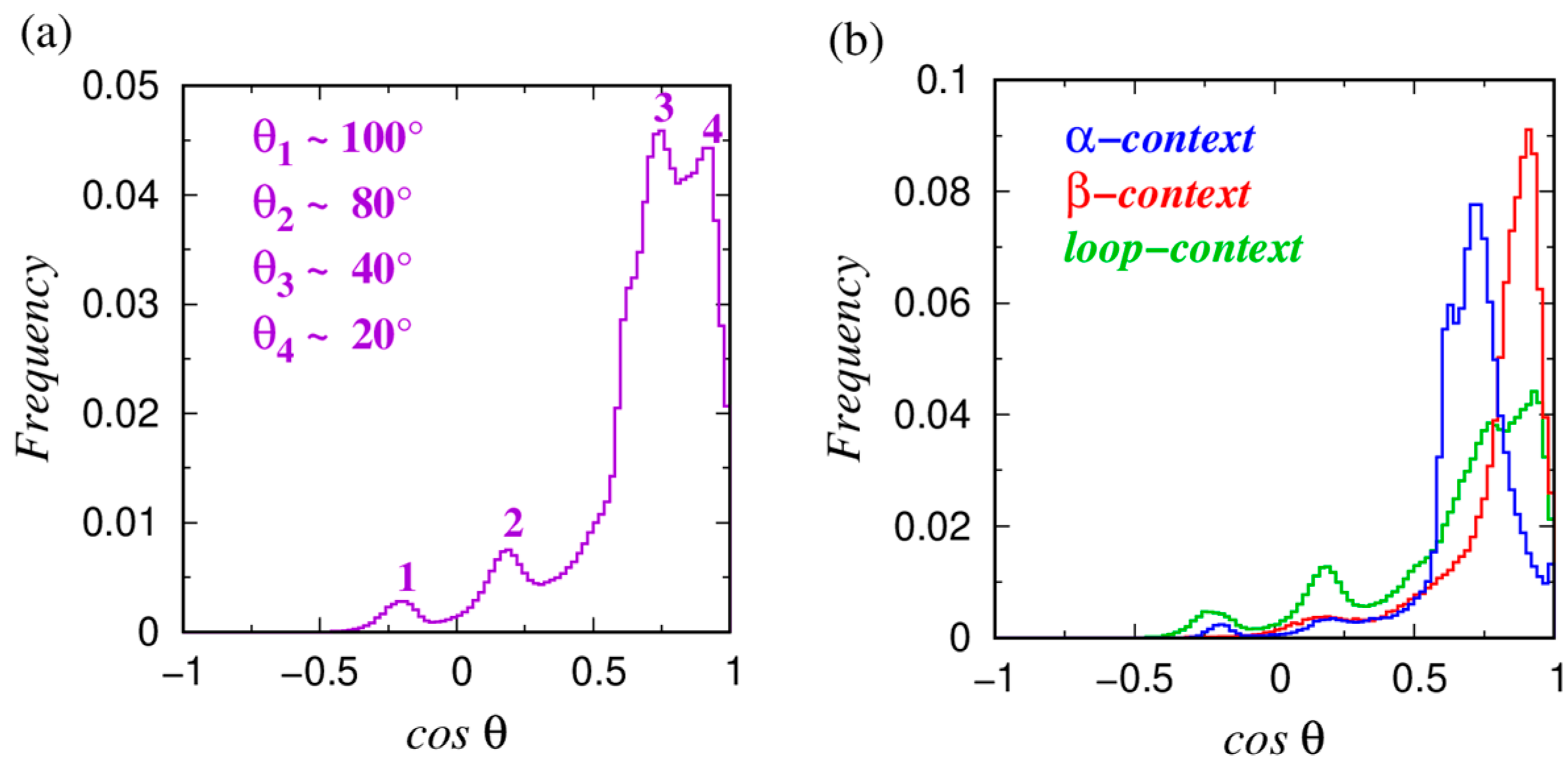
3.1. The Orientation of Amino Acids in Globular Proteins
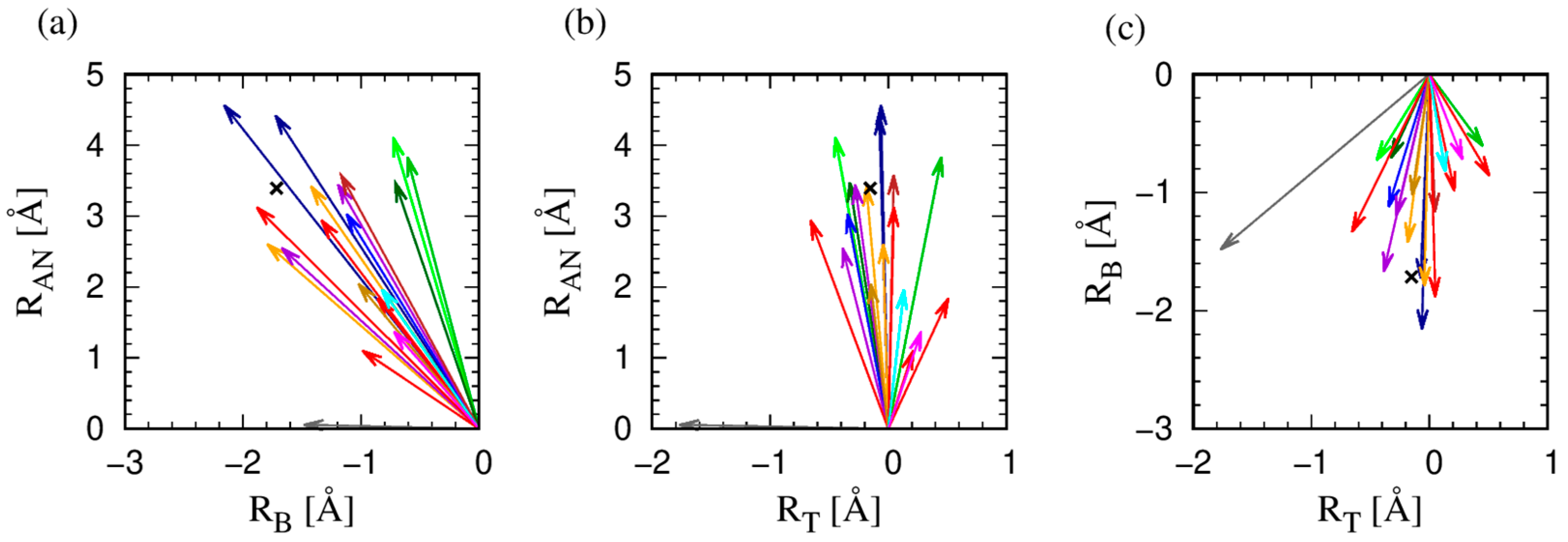
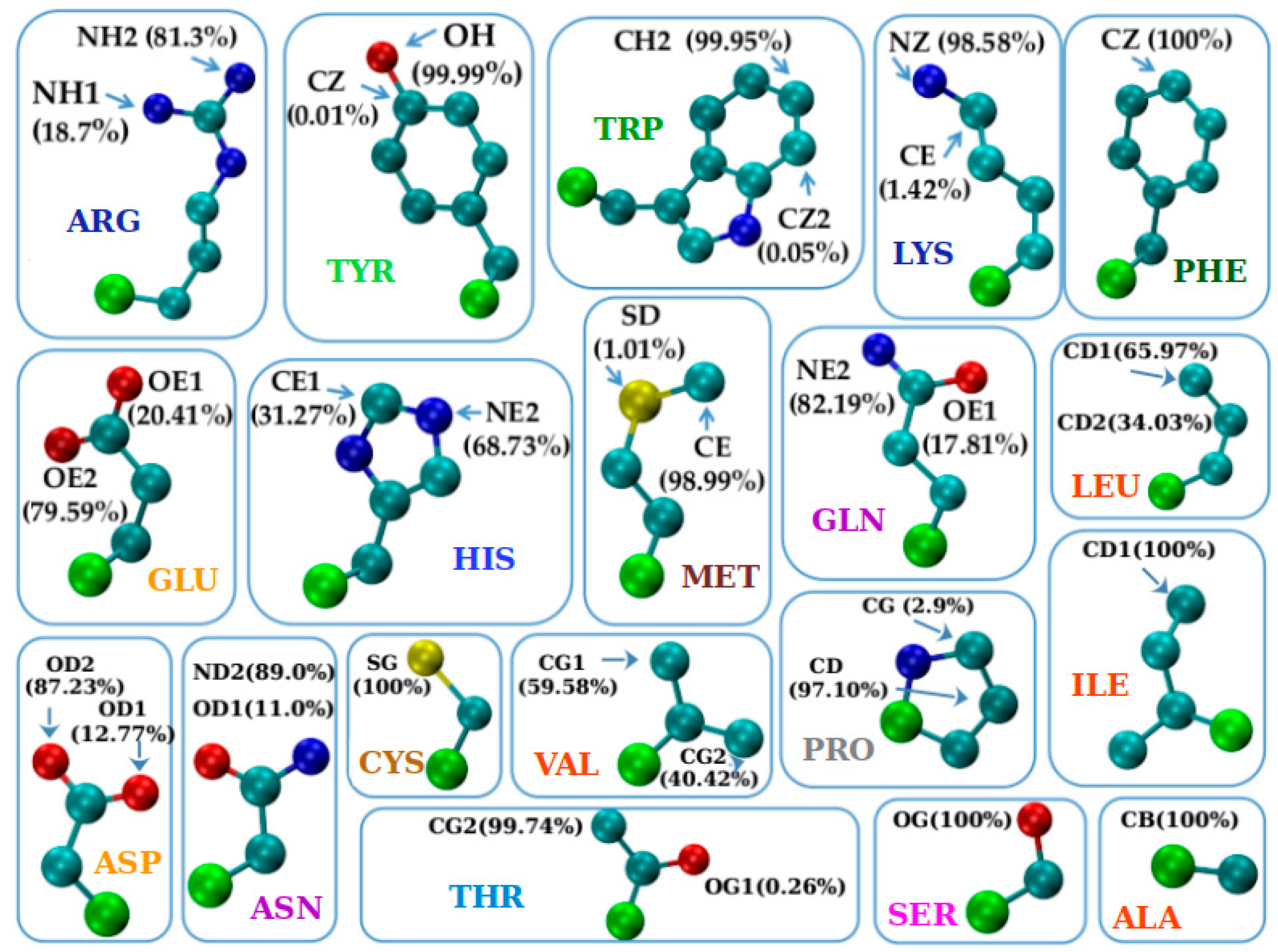
3.2. The Protruder Atom Type and Amino Acid Groupings
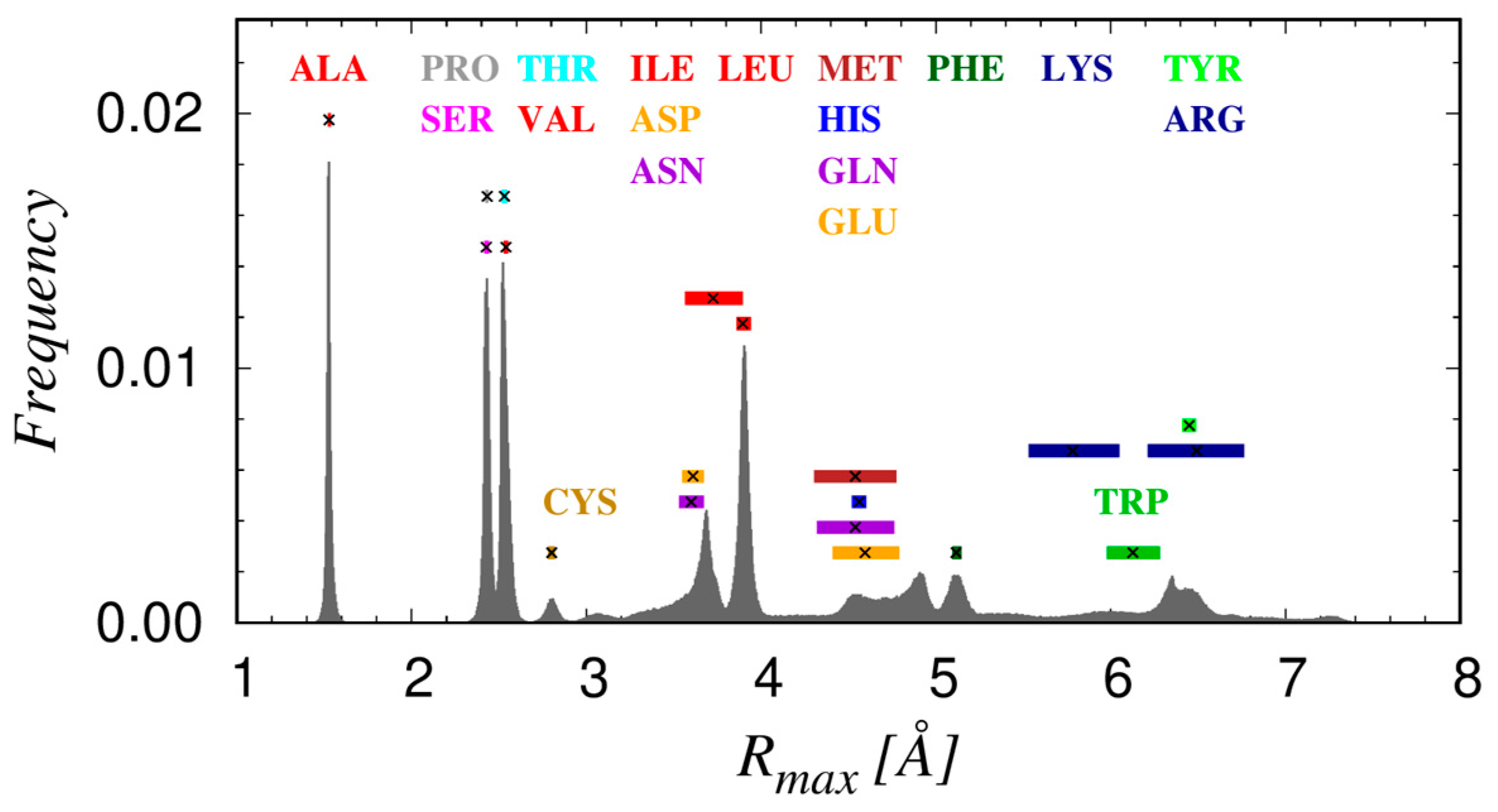
3.3. The Geometry of Amino-Acid Protrusion
- PRO due to its distinct geometry of a ring that reconnects to the protein backbone, has characteristic ɛ values that are close to or even larger than 90°. This context-independent result reflects the fact that PRO dominantly protrudes in the binormal-tangent plane unlike all the other amino acids (see Table S1 in Supplementary Information). PRO forms the singlet ‘neutral aliphatic’ group in the IMGT classification [54] and is our singlet Group I (see Table 2);
- ALA, ILE, LEU, and VAL have qualitatively similar behaviors. For both α and β contexts, one mode strongly dominates, while in the loop context, the behavior is a combination of the modes in the α and β contexts. (ALA, ILE, LEU, VAL) form the ‘hydrophobic aliphatic’ IMGT group [54] and coincides with our Group II (see Table 2);
- PHE and TYR share very similar behavior, with only one mode present in each of the contexts and all of them ~0°, meaning that these amino acids with aromatic rings protrude predominantly along the anti-normal direction. PHE is a singlet ‘hydrophobic, aromatic, with no hydrogen donor’ and TYR a singlet ‘neutral, aromatic, with both hydrogen donor and acceptor’ group in the IMGT classification [54], We denote them as singlet groups as well, Group III and Group V (see Table 2);
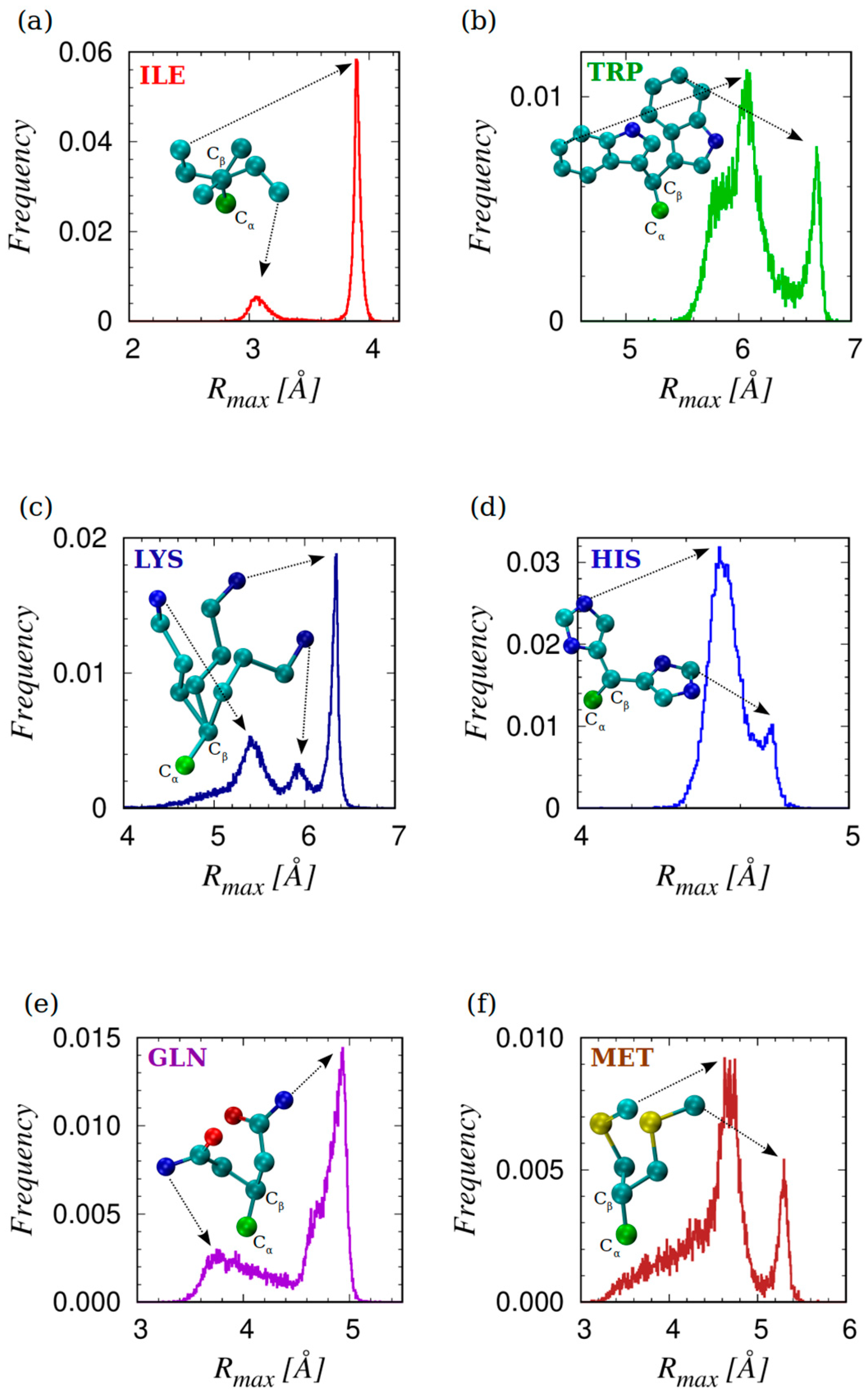
- TRP is the unique amino acid with the ‘double ring’ structure (composed of a six-atom ring and a five-atom ring, sharing one side, see Figure 5) and, contrary to all other amino acids, has an ɛ angle α-mode smaller than the ɛ angle β-mode. TRP forms the singlet ‘hydrophobic, aromatic, with hydrogen donor’ IMGT group [54] and is our singlet Group IV (see Table 2);
- ARG, LYS, and HIS, the three positively charged amino acids forming the ‘basic’ group in IMGT classification [54]. They all exhibit a ~0° β-mode, but quite different α-modes. For ARG, there are two α-modes, presumably due to the ‘double tip’ branch formed by two symmetrically placed nitrogen atoms at its end (see Figure 5). In our classification, ARG and LYS fall into Group VI, while HIS forms the singlet Group VII, due to its different topology (see Table 2);
- ASP and ASN, on one hand, and GLU and GLN, on the other, have very similar ɛ angle profiles, so they can be dubbed geometrical twins. From Figure 5, we see that this is due to the identical shape for the two corresponding pairs, with the difference that for ASP and GLU the ‘double tip’ in the amino acid ending is made up of two oxygen atoms, while for the ASN and GLN, the double tip is composed of one oxygen and one nitrogen atom. In the IMGT categorization [54], ASP and GLU constitute the ‘acidic’ group, while ASN and GLN form the ‘amide’ group. In our classification, these pairs of amino acids form Group VIII and Group IX, respectively (see Table 2);
- SER and THR constitute the ‘hydroxylic’ group in the IMGT classification [54] and have decisively different protrusion geometries, with SER most notably (and distinctively from all other amino acids) displaying the most complex ɛ profile, with three α-modes, two β-modes, as well as two loop-modes. SER is thus the champion of versatility with multiple sharp modes in all environments that is surprising because of its relatively small size. For 60% of the time, SER is found in loops. In our grouping, SER and THR form two singlet groups, Group X and Group XI, respectively (see Table 2);
- CYS and MET, placed in the ‘sulfur-containing’ group in the IMGT classification [54], have different protrusion geometries. SER has a non-zero α-mode and zero β- and loop-modes; while MET with all three zero-modes, seems more compatible geometry-wise with the aromatic duo PHE and TYR. In our grouping, CYS and MET are in two singlet groups, Group XII and Group XIII (see Table 2);
- There are three amino acids, ARG, GLN, and GLU with two dominant α-modes, that could be due to their considerable length and the ‘double tip’ shape in the amino acid ending. For GLN, this is also reflected in the double peak in the distribution of the magnitude of the maximal protrusion Rmax (see Figure 5), while for ARG, Rmax has a very broad distribution, so that no well-defined peaks could be identified.
- Finally, GLY (with no heavy side chain atoms) is our singlet Group XIV and it belongs to the ‘very small, neutral aliphatic’ singlet group in the IMGT classification [54].
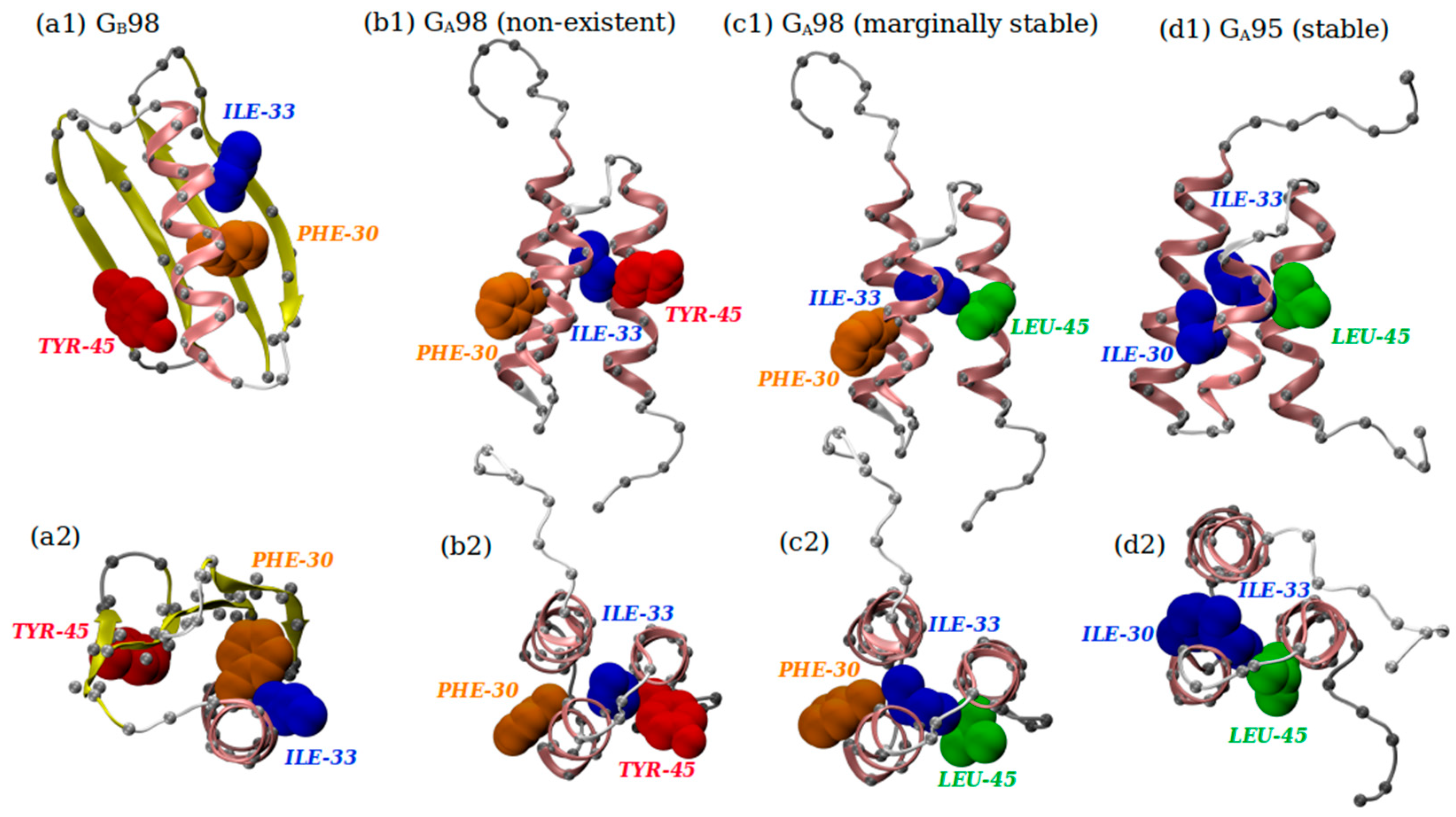
3.4. The Biology of Amino Acid Protrusion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Creighton, T.E. Proteins: Structures and Molecular Properties; W. H. Freeman: New York, NY, USA, 1993. [Google Scholar]
- Lesk, A.M. Introduction to Protein Science: Architecture, Function and Genomics; Oxford University Press: Oxford, UK, 2004. [Google Scholar]
- Bahar, I.; Jernigan, R.L.; Dill, K.A. Protein Actions; Garland Science: New York, NY, USA, 2017. [Google Scholar]
- Berg, J.M.; Tymoczko, J.L.; Gatto, G.J., Jr.; Stryer, L. Biochemistry; Macmillan Learning: New York, NY, USA, 2019. [Google Scholar]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L.; Corey, R.B.; Branson, H.R. The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 1951, 37, 205. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L.; Corey, R.B. The pleated sheet, a new layer configuration of polypeptide chains. Proc. Natl. Acad. Sci. USA 1951, 37, 251. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M.; Chothia, C. Structural patterns in globular proteins. Nature 1976, 261, 552. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C. One thousand families for the molecular biologist. Nature 1992, 357, 543. [Google Scholar] [CrossRef] [PubMed]
- Przytycka, T.; Aurora, R.; Rose, G.D. A protein taxonomy based on secondary structure. Nat. Struct. Biol. 1999, 6, 672. [Google Scholar]
- Taylor, W. A ‘periodic table’ for protein structures. Nature 2002, 416, 657. [Google Scholar] [CrossRef] [PubMed]
- Bordin, N.; Sillitoe, I.; Lees, J.G.; Orengo, C. Tracing Evolution Through Protein Structures: Nature Captured in a Few Thousand Folds. Front. Mol. Biosci. 2021, 8, 668184. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Carreno, C.; Gupta, R.J.; Petrov, A.S.; Williams, L.D. Creative destruction: New protein folds from old. Proc. Natl. Acad. Sci. USA 2022, 119, e2207897119. [Google Scholar] [CrossRef]
- Hoang, T.X.; Trovato, A.; Seno, F.; Banavar, J.R.; Maritan, A. Geometry and symmetry presculpt the free-energy landscape of proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 7960. [Google Scholar] [CrossRef]
- Banavar, J.R.; Giacometti, A.; Hoang, T.X.; Maritan, A.; Škrbić, T. A geometrical framework for thinking about proteins. Proteins 2023. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, M.; Bhat, C.R.; Vishveshwara, S. An automated approach to network features of protein structure ensembles. Protein Sci. 2013, 22, 1399. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, M.; Ghosh, S.; Vishveshwara, S. Protein Structure and Function: Looking through the Network of Side-Chain Interactions. Curr. Protein Pept. Sci. 2016, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Rose, G.D. Ramachandran maps for side chains in globular proteins. Proteins 2019, 87, 357. [Google Scholar] [CrossRef] [PubMed]
- Bryngelson, J.D.; Onuchic, J.N.; Socci, N.D.; Wolynes, P.G. Funnels, pathways, and the energy landscape of protein folding: A synthesis. Proteins 1995, 21, 167. [Google Scholar] [CrossRef] [PubMed]
- Wolynes, P.G.; Onuchic, J.N.; Thirumalai, D. Navigating the folding routes. Science 1995, 267, 1619. [Google Scholar] [CrossRef]
- Dill, K.A.; Chan, H.S. From Levinthal to pathways to funnels. Nat. Struct. Biol. 1997, 4, 10. [Google Scholar] [CrossRef]
- Richards, F.M. Areas, volumes, packing, and protein structure. Annu. Rev. Biophys. Bioeng. 1977, 6, 151. [Google Scholar] [CrossRef]
- Corey, R.B.; Pauling, L. Molecular models of amino acids, peptides, and proteins. Rev. Sci. Instrum. 1953, 8, 621. [Google Scholar] [CrossRef]
- Koltun, W.L. Precision space-filling atomic models. Biopolymers 1965, 3, 665. [Google Scholar] [CrossRef]
- Škrbić, T.; Hoang, T.X.; Maritan, A.; Banavar, J.R.; Giacometti, A. The elixir phase of chain molecules. Proteins 2019, 87, 176. [Google Scholar] [CrossRef] [PubMed]
- Škrbić, T.; Hoang, T.X.; Giacometti, A.; Maritan, A.; Banavar, J.R. Spontaneous dimensional reduction and ground state degeneracy in a simple chain model. Phys. Rev. E 2021, 104, L0121011. [Google Scholar] [CrossRef] [PubMed]
- Škrbić, T.; Hoang, T.X.; Giacometti, A.; Maritan, A.; Banavar, J.R. Marginally compact phase and ordered ground states in a model polymer with side spheres. Phys. Rev. E 2021, 104, L0125011. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 57, 105. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Word, J.M.; Richardson, J.S.; Richardson, D.C. The penultimate rotamer library. Proteins 2000, 40, 389. [Google Scholar] [CrossRef]
- Kuhlman, B.; Baker, D. Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. USA 2000, 97, 10383. [Google Scholar] [CrossRef] [PubMed]
- Dunbrack, R.L., Jr. Rotamer libraries in the 21st century. Curr. Opin. Struct. Biol. 2002, 12, 431. [Google Scholar] [CrossRef]
- Virrueta, A.; O’Hern, C.S.; Regan, L. Understanding the physical basis for the side chain conformational preferences of Met. Proteins 2016, 84, 900. [Google Scholar] [CrossRef]
- Gaines, J.C.; Acerbes, S.; Virrueta, A.; Butler, M.; Regan, L.; O’Hern, C.S. Comparing side chain packing in soluble proteins, protein-protein interfaces, and transmembrane proteins. Proteins 2018, 86, 581. [Google Scholar] [CrossRef]
- Huang, X.; Pearce, R.; Zhang, Y. Toward the Accuracy and Speed of Protein Side-Chain Packing: A Systematic Study on Rotamer Libraries. J. Chem. Inf. Model 2020, 60, 410. [Google Scholar] [CrossRef]
- Xu, G.; Wang, Q.; Ma, J. OPUS-Rota4: A gradient-based protein side-chain modeling framework assisted by deep learning-based predictors. Brief Bioinform. 2022, 23, bbab529. [Google Scholar]
- Jindal, A.; Kotelnikov, S.; Padhorny, D.; Kozakov, D.; Zhu, Y.; Chowdhury, R.; Vajda, S. Side-chain packing using SE(3)-transformer. Pac. Symp. Biocomput. 2022, 27, 46. [Google Scholar]
- Misiura, M.; Shroff, R.; Thyer, R.; Kolomeisky, A.B. DLPacker: Deep learning for prediction of amino acid chain conformations in proteins. Proteins 2022, 90, 1278. [Google Scholar] [CrossRef] [PubMed]
- McPartlon, M.; Xu, J. An end-to-end deep learning method for protein side-chain packing and inverse folding. Proc. Natl. Acad. Sci. USA 2023, 120, e2216438120. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Zhang, Z.; Zhong, B.; Misra, S.; Tang, J. DiffPack: A torsional diffusion model for autoregressive protein side-chain packing. arXiv 2023. [Google Scholar] [CrossRef]
- Mukhopadhay, A.; McMaster, B.; McWhirter, J.L.; Dixit, S.B. ZymePackNet: Rotamer-sampling free graph neural network method for protein sidechain prediction. BioRxiv 2023. [Google Scholar] [CrossRef]
- Yan, J.; Li, S.; Zhang, Y.; Hao, A.; Zhao, Q. ZetaDesign: An end-to-end deep learning method for protein sequence design and side-chain packing. Brief Bioinform. 2023, 24, bbad257. [Google Scholar] [CrossRef] [PubMed]
- Randolph, N.Z.; Kuhlman, B. Invariant point message passing for protein side chain packing. Proteins 2024. [Google Scholar] [CrossRef] [PubMed]
- Zhang, O.; Shubhankar, A.N.; Liu, Z.H.; Forman-Kay, J.; Head-Gordon, T. A Curated Rotamer Library for Common Post-Translational Modifications of Proteins. arXiv 2024. [Google Scholar] [CrossRef]
- Ambroggio, X.I.; Kuhlman, B. Design of protein conformational switches. Curr. Opin. Struct. Biol. 2006, 16, 525–530. [Google Scholar] [CrossRef]
- Alexander, P.A.; He, Y.; Chen, Y.; Orban, J.; Bryan, P.N. The design and characterization of two proteins with 88% sequence identity but different structure and function. Proc. Natl. Acad. Sci. USA 2007, 104, 11963. [Google Scholar] [CrossRef]
- Davidson, A.R. A folding space odyssey. Proc. Natl. Acad. Sci. USA 2008, 105, 2759–2760. [Google Scholar] [CrossRef] [PubMed]
- Alexander, P.A.; He, Y.; Chen, Y.; Orban, J.; Bryan, P.N. A minimal sequence code for switching protein structure and function. Proc. Natl. Acad. Sci. USA 2009, 106, 21149. [Google Scholar] [CrossRef] [PubMed]
- Porter, L.L.; Looger, L.L. Extant fold-switching proteins are widespread. Proc. Natl. Acad. Sci. USA 2018, 115, 5968. [Google Scholar] [CrossRef] [PubMed]
- Kamien, R.D. The geometry of soft materials: A primer. Rev. Mod. Phys. 2002, 74, 953. [Google Scholar] [CrossRef]
- Škrbić, T.; Maritan, A.; Giacometti, A.; Banavar, J.R. Local sequence-structure relationships in proteins. Protein Sci. 2021, 30, 818. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.N.; Mitra, A.K. An explanation for the rare occurrence of cis peptide units in proteins and polypeptides. J. Mol. Biol. 1976, 107, 85. [Google Scholar] [CrossRef]
- 3D Macromolecule Analysis & Kinemage Home Page at Richardson Laboratory. Available online: http://kinemage.biochem.duke.edu/databases/top8000/ (accessed on 1 January 2019).
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577. [Google Scholar] [CrossRef]
- Pommié, C.; Levadoux, S.; Sabatier, R.; Lefranc, G.; Lefranc, M.-P. IMGT (ImMunoGeneTics information system) standardized criteria for statistical analysis of immunoglobulin V-REGION amino acid properties. J. Mol. Recognit. 2004, 17, 17. [Google Scholar] [CrossRef]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009, 77, 778. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Total Number | Frequency [%] | α | β | Loop |
|---|---|---|---|---|---|
| LEU | 84,916 | 8.85 | 36,154 (~43%) | 21,387 (~25%) | 27,375 (~32%) |
| ALA | 82,208 | 8.57 | 38,896 (~47%) | 13,583 (~17%) | 29,729 (~36%) |
| GLY | 76,284 | 7.95 | 10,839 (~14%) | 10,883 (~14%) | 54,562 (~72%) |
| VAL | 69,481 | 7.24 | 20,194 (~29%) | 29,569 (~43%) | 19,718 (~28%) |
| GLU | 61,780 | 6.44 | 28,135 (~45%) | 9678 (~16%) | 23,967 (~39%) |
| ASP | 57,111 | 5.95 | 15,259 (~27%) | 6795 (~12%) | 35,057 (~61%) |
| SER | 56,318 | 5.87 | 13,965 (~25%) | 10,649 (~19%) | 31,704 (~56%) |
| ILE | 54,043 | 5.63 | 18,561 (~34%) | 20,635 (~38%) | 14,847 (~28%) |
| LYS | 53,739 | 5.60 | 20,349 (~38%) | 9605 (~18%) | 23,785 (~44%) |
| THR | 53,588 | 5.58 | 13,129 (~24%) | 14,272 (~27%) | 26,187 (~49%) |
| ARG | 46,176 | 4.81 | 18,251 (~40%) | 9217 (~20%) | 18,708 (~40%) |
| PRO | 44,397 | 4.63 | 6396 (~15%) | 4148 (~9%) | 33,853 (~76%) |
| ASN | 42,128 | 4.39 | 9757 (~23%) | 5804 (~14%) | 26,567 (~63%) |
| PHE | 38,853 | 4.05 | 12,348 (~32%) | 12,184 (~31%) | 14,321 (~37%) |
| TYR | 34,685 | 3.61 | 10,506 (~30%) | 10,825 (~31%) | 13,354 (~39%) |
| GLN | 34,361 | 3.58 | 14,372 (~42%) | 5870 (~17%) | 14,119 (~41%) |
| HIS | 22,392 | 2.33 | 6261 (~28%) | 4897 (~22%) | 11,234 (~50%) |
| MET | 19,524 | 2.03 | 8273 (~42%) | 4513 (~23%) | 6738 (~35%) |
| TRP | 14,579 | 1.52 | 4698 (~32%) | 4205 (~29%) | 5676 (~39%) |
| CYS | 13,128 | 1.37 | 3469 (~26%) | 3656 (~28%) | 6003 (~46%) |
| Group I | PRO | Ring connects back to the backbone |
| Group II | ALA, ILE, LEU, VAL | Linear (C); C: max |
| Group III | PHE | Ring (C); C: max |
| Group IV | TRP | Ring (C, N); C: max |
| Group V | TYR | Ring (C, O); O: max |
| Group VI | ARG, LYS | Linear (C, N); N: max |
| Group VII | HIS | Ring (C, N); N: max |
| Group VIII | ASP, GLU | Linear (C, O, O); O: max |
| Group IX | ASN, GLN | Linear (C, N, O); N: max |
| Group X | SER | Linear (C, O); O: max |
| Group XI | THR | Linear (C, O); C: max |
| Group XII | CYS | Linear (C, S); S: max |
| Group XIII | MET | Linear (C, S); C: max |
| Group XIV | GLY | No heavy atoms |
| Type | ɛα Mode [°] | ɛα Mean [°] | ɛβ Mode [°] | ɛβ Mean [°] | ɛloop Mode [°] | ɛloop Mean [°] |
|---|---|---|---|---|---|---|
| PRO | 105 | 104.9 ± 5.5 | 77 | 74.8 ± 13.1 | 73, 108 | 83.2 ± 21.1 |
| ALA | 50 | 50.0 ± 2.3 | 25 | 28.2 ± 7.3 | 30, 48 | 37.7 ± 10.4 |
| ILE | 45 | 37.2 ±15.9 | 12 | 20.0 ± 14.6 | 12, 53 | 29.3 ± 20.3 |
| LEU | 43 | 40.8 ± 5.8 | 16 | 19.4 ± 9.7 | 18, 38 | 27.9 ± 12.3 |
| VAL | 24 | 32.7 ± 15.5 | 5 | 16.3 ± 20.7 | 7, 23 | 29.8 ± 26.3 |
| PHE | 3 | 14.1 ± 14.6 | 3 | 24.5 ± 28.8 | 3 | 21.1 ± 25.2 |
| TRP | 18 | 30.5 ± 24.4 | 32 | 36.7 ± 23.4 | 30 | 39.9 ± 30.2 |
| TYR | 0 | 14.1 ± 17.1 | 4 | 25.6 ± 28.6 | 4 | 24.1 ± 27.6 |
| ARG | 30, 70 | 38.6 ± 24.1 | 2 | 23.9 ± 20.5 | 3 | 29.9 ± 23.5 |
| LYS | 38 | 31.1 ± 16.5 | 7 | 20.3 ± 16.2 | 12 | 27.8 ± 19.8 |
| HIS | 14 | 24.8 ± 18.4 | 5 | 19.6 ± 24.3 | 0 | 26.9 ± 27.7 |
| ASP | 42 | 42.2 ± 10.5 | 14 | 19.8 ± 16.2 | 10, 40, 60 | 34.9 ± 21.0 |
| GLU | 3, 35 | 29.3 ± 17.4 | 0 | 18.8 ± 17.4 | 3 | 29.9 ± 23.3 |
| ASN | 43 | 40.4 ± 11.0 | 15 | 22.3 ± 16.5 | 18, 37, 57 | 34.5 ± 19.5 |
| GLN | 0, 29 | 28.4 ± 17.1 | 0 | 20.7 ± 17.6 | 0 | 27.7 ± 20.9 |
| SER | 25, 38, 78 | 49.6 ± 22.9 | 3, 58 | 30.5 ± 27.0 | 10, 77 | 48.6 ± 28.3 |
| THR | 23 | 26.9 ± 9.3 | 5 | 16.4 ± 20.2 | 17 | 24.6 ± 16.8 |
| CYS | 32 | 32.9 ± 13.6 | 0 | 18.7 ± 24.8 | 3 | 29.2 ± 27.0 |
| MET | 0 | 28.7 ± 21.1 | 0 | 24.9 ± 17.7 | 0 | 24.5 ± 19.3 |
| Position | 1 10 20 30 40 50 |
|---|---|
| GB98 | TTYKLILNLKQAKEEAIKELVDAGTAEKYFKLIANAKTVEGVWTYKDEIKTFTVTE |
| GA98 | TTYKLILNLKQAKEEAIKELVDAGTAEKYFKLIANAKTVEGVWTLKDEIKTFTVTE |
| GA95 | TTYKLILNLKQAKEEAIKELVDAGTAEKYIKLIANAKTVEGVWTLKDEIKTFTVTE |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Škrbić, T.; Giacometti, A.; Hoang, T.X.; Maritan, A.; Banavar, J.R. Amino-Acid Characteristics in Protein Native State Structures. Biomolecules 2024, 14, 805. https://doi.org/10.3390/biom14070805
Škrbić T, Giacometti A, Hoang TX, Maritan A, Banavar JR. Amino-Acid Characteristics in Protein Native State Structures. Biomolecules. 2024; 14(7):805. https://doi.org/10.3390/biom14070805
Chicago/Turabian StyleŠkrbić, Tatjana, Achille Giacometti, Trinh X. Hoang, Amos Maritan, and Jayanth R. Banavar. 2024. "Amino-Acid Characteristics in Protein Native State Structures" Biomolecules 14, no. 7: 805. https://doi.org/10.3390/biom14070805
APA StyleŠkrbić, T., Giacometti, A., Hoang, T. X., Maritan, A., & Banavar, J. R. (2024). Amino-Acid Characteristics in Protein Native State Structures. Biomolecules, 14(7), 805. https://doi.org/10.3390/biom14070805