
Assessment of the Ferroptosis Regulators: Glutathione Peroxidase 4, Acyl-Coenzyme A Synthetase Long-Chain Family Member 4, and Transferrin Receptor 1 in Patient-Derived Endometriosis Tissue
 , , ,
, , ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Ethical Approval
2.3. Immunohistochemistry
2.4. Microscopic Evaluation
2.5. Statistical Analysis
3. Results
3.1. Patient Characteristics
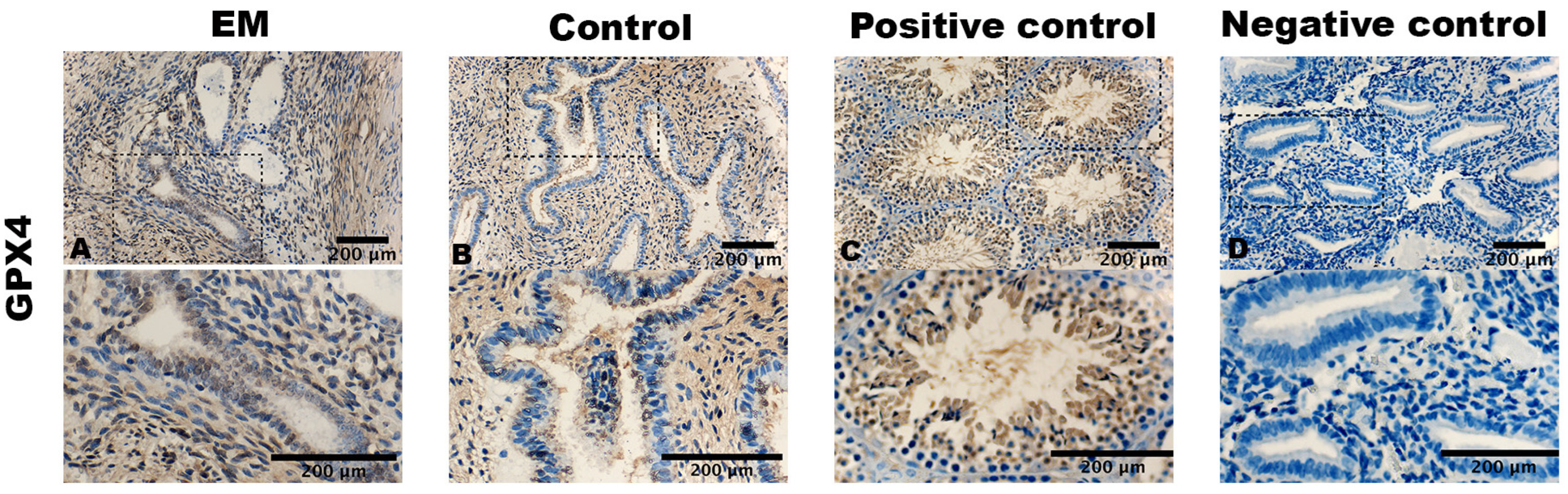
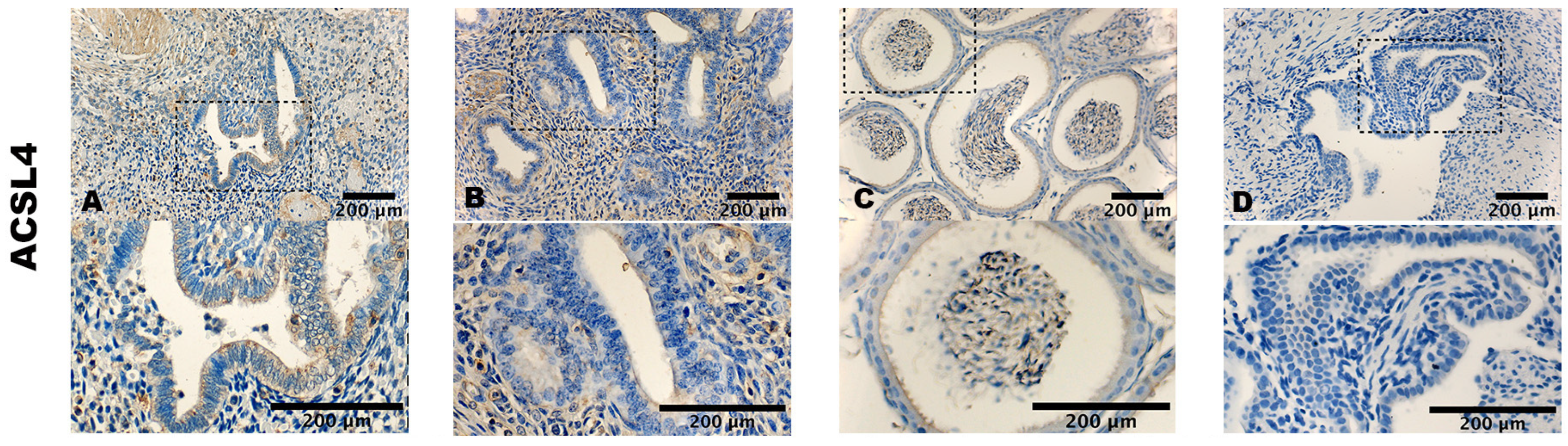
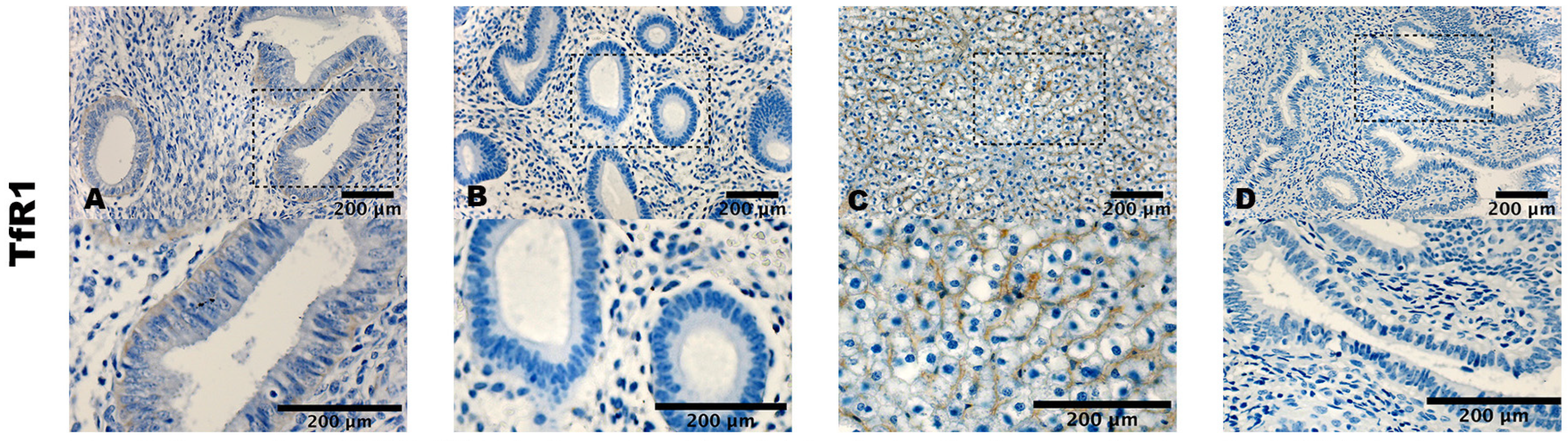
3.2. Staining Pattern of ACSL4, GPX4, and TfR1
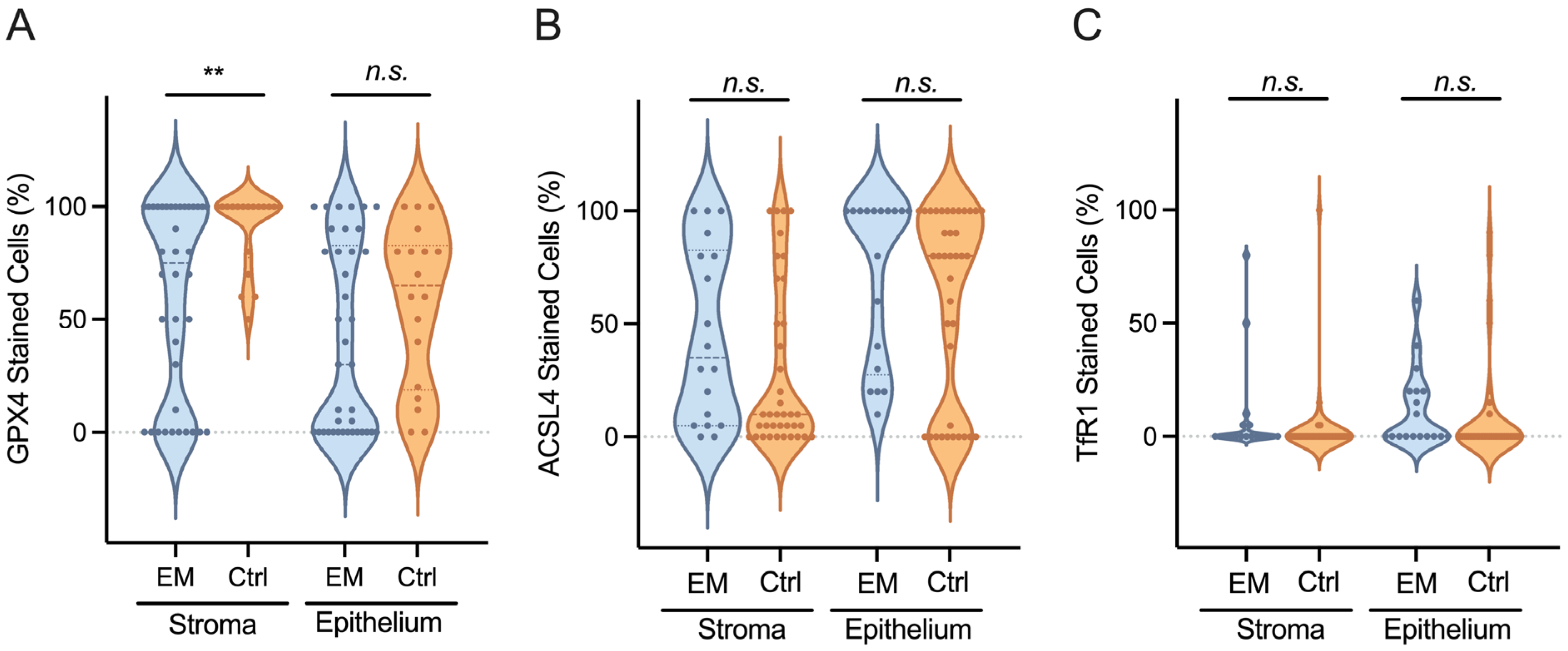
3.3. GPX4, ACSL4, and TfR1 Expression Levels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zondervan, K.T.; Becker, C.M.; Missmer, S.A. Endometriosis. N. Engl. J. Med. 2020, 382, 1244–1256. [Google Scholar] [CrossRef] [PubMed]
- Burney, R.O.; Giudice, L.C. Reprint of: Pathogenesis and pathophysiology of endometriosis. Fertil. Steril. 2019, 112 (Suppl. 1), e153–e161. [Google Scholar] [CrossRef]
- Wang, Y.; Nicholes, K.; Shih, I.M. The Origin and Pathogenesis of Endometriosis. Annu. Rev. Pathol. 2020, 15, 71–95. [Google Scholar] [CrossRef]
- Chapron, C.; Marcellin, L.; Borghese, B.; Santulli, P. Rethinking mechanisms, diagnosis and management of endometriosis. Nat. Rev. Endocrinol. 2019, 15, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Alborzi, S.; Askary, E.; Khorami, F.; Poordast, T.; Abdulwahid Hashim Alkhalidi, B.; Hamedi, M.; Alborzi, S.; Shahraki, H.R. A Detailed Study in Adenomyosis and Endometriosis: Evaluation of the Rate of Coexistence Between Uterine Adenomyosis and DIE According to Imaging and Histopathology Findings. Reprod. Sci. 2021, 28, 2387–2397. [Google Scholar] [CrossRef] [PubMed]
- Horne, A.W.; Missmer, S.A. Pathophysiology, diagnosis, and management of endometriosis. BMJ 2022, 379, e070750. [Google Scholar] [CrossRef] [PubMed]
- Leyendecker, G.; Wildt, L.; Laschke, M.W.; Mall, G. Archimetrosis: The evolution of a disease and its extant presentation: Pathogenesis and pathophysiology of archimetrosis (uterine adenomyosis and endometriosis). Arch. Gynecol. Obstet. 2023, 307, 93–112. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.A. Metastatic or Embolic Endometriosis, due to the Menstrual Dissemination of Endometrial Tissue into the Venous Circulation. Am. J. Pathol. 1927, 3, 93–110.43. [Google Scholar] [PubMed]
- Ng, S.W.; Norwitz, S.G.; Taylor, H.S.; Norwitz, E.R. Endometriosis: The Role of Iron Overload and Ferroptosis. Reprod. Sci. 2020, 27, 1383–1390. [Google Scholar] [CrossRef]
- Li, G.; Lin, Y.; Zhang, Y.; Gu, N.; Yang, B.; Shan, S.; Liu, N.; Ouyang, J.; Yang, Y.; Sun, F.; et al. Endometrial stromal cell ferroptosis promotes angiogenesis in endometriosis. Cell Death Discov. 2022, 8, 29. [Google Scholar] [CrossRef]
- Guo, S.W. Cracking the enigma of adenomyosis: An update on its pathogenesis and pathophysiology. Reproduction 2022, 164, R101–R121. [Google Scholar] [CrossRef]
- Maruyama, S.; Imanaka, S.; Nagayasu, M.; Kimura, M.; Kobayashi, H. Relationship between adenomyosis and endometriosis; Different phenotypes of a single disease? Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 253, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Duan, H.; Wang, S.; Li, Y. Ferroptosis resistance mechanisms in endometriosis for diagnostic model establishment. Reprod. Biomed. Online 2021, 43, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R. From Oxytosis to Ferroptosis: 10 Years of Research on Oxidative Cell Death. Antioxid. Redox Signal 2023, 39, 162–165. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Yiannikourides, A.; Latunde-Dada, G.O. A Short Review of Iron Metabolism and Pathophysiology of Iron Disorders. Medicines 2019, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, M.Y.; Jiang, H.B.; Guo, C.R.; Zhu, X.D.; Yao, X.Q.; Zeng, W.W.; Zhao, Y.; Chi, L.K. Bisphenol A induces testicular oxidative stress in mice leading to ferroptosis. Asian J. Androl. 2023, 25, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Roemhild, K.; von Maltzahn, F.; Weiskirchen, R.; Knuchel, R.; von Stillfried, S.; Lammers, T. Iron metabolism: Pathophysiology and pharmacology. Trends Pharmacol. Sci. 2021, 42, 640–656. [Google Scholar] [CrossRef]
- Zhang, F.; Tao, Y.; Zhang, Z.; Guo, X.; An, P.; Shen, Y.; Wu, Q.; Yu, Y.; Wang, F. Metalloreductase Steap3 coordinates the regulation of iron homeostasis and inflammatory responses. Haematologica 2012, 97, 1826–1835. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423.e7. [Google Scholar] [CrossRef] [PubMed]
- Sha, R.; Xu, Y.; Yuan, C.; Sheng, X.; Wu, Z.; Peng, J.; Wang, Y.; Lin, Y.; Zhou, L.; Xu, S.; et al. Predictive and prognostic impact of ferroptosis-related genes ACSL4 and GPX4 on breast cancer treated with neoadjuvant chemotherapy. EBioMedicine 2021, 71, 103560. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, Y.; Cheng, W.; Zhou, Z.; Ni, Z.; Yu, C. Double-edged roles of ferroptosis in endometriosis and endometriosis-related infertility. Cell Death Discov. 2023, 9, 306. [Google Scholar] [CrossRef] [PubMed]
- Keckstein, J.; Hudelist, G. Classification of deep endometriosis (DE) including bowel endometriosis: From r-ASRM to #Enzian-classification. Best. Pract. Res. Clin. Obstet. Gynaecol. 2021, 71, 27–37. [Google Scholar] [CrossRef]
- Lee, S.Y.; Koo, Y.J.; Lee, D.H. Classification of endometriosis. Yeungnam Univ. J. Med. 2021, 38, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.A.; Naz, N.; Sheikh, N.; Khan, S.; Moriconi, F.; Blaschke, M.; Ramadori, G. Comparison of changes in gene expression of transferrin receptor-1 and other iron-regulatory proteins in rat liver and brain during acute-phase response. Cell Tissue Res. 2011, 344, 299–312. [Google Scholar] [CrossRef]
- Landis, J.R.; Koch, G.G. The measurement of observer agreement for categorical data. Biometrics 1977, 33, 159–174. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Riegman, M.; Sagie, L.; Galed, C.; Levin, T.; Steinberg, N.; Dixon, S.J.; Wiesner, U.; Bradbury, M.S.; Niethammer, P.; Zaritsky, A.; et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat. Cell Biol. 2020, 22, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Polak, G.; Barczynski, B.; Wertel, I.; Kwasniewski, W.; Bednarek, W.; Derewianka-Polak, M.; Fraszczak, K.; Olajossy, M.; Kotarski, J. Disrupted iron metabolism in peritoneal fluid may induce oxidative stress in the peritoneal cavity of women with endometriosis. Ann. Agric. Environ. Med. 2018, 25, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Defrere, S.; Lousse, J.C.; Gonzalez-Ramos, R.; Colette, S.; Donnez, J.; Van Langendonckt, A. Potential involvement of iron in the pathogenesis of peritoneal endometriosis. Mol. Hum. Reprod. 2008, 14, 377–385. [Google Scholar] [CrossRef]
- Wolfler, M.M.; Meinhold-Heerlein, I.M.; Henkel, C.; Rath, W.; Neulen, J.; Maass, N.; Brautigam, K. Reduced hemopexin levels in peritoneal fluid of patients with endometriosis. Fertil. Steril. 2013, 100, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, K.; Yip, Y.C. De novo formation of adhesions in endometriosis: The role of iron and free radical reactions. Fertil. Steril. 1995, 64, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, J.; Fernando, S.M.; Powell, S.G.; Hill, C.J.; Arshad, I.; Probert, C.; Ahmed, S.; Hapangama, D.K. The role of iron in the pathogenesis of endometriosis: A systematic review. Hum. Reprod. Open 2023, 2023, hoad033. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Diaz, C.P.; Nunez, M.T.; Devoto, L.; Gonzalez-Ramos, R. Endometrial expression and in vitro modulation of the iron transporter divalent metal transporter-1: Implications for endometriosis. Fertil. Steril. 2016, 106, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Yamashita, Y.; Iwase, A.; Yoshikawa, Y.; Yasui, H.; Kawai, Y.; Uchida, K.; Uno, N.; Akatsuka, S.; Takahashi, T.; et al. The ferroimmunomodulatory role of ectopic endometriotic stromal cells in ovarian endometriosis. Fertil. Steril. 2012, 98, 415–422, e411–412. [Google Scholar] [CrossRef] [PubMed]
- Akashi, K.; Nagashima, Y.; Tabata, T.; Oda, H. Immunochemical analysis of iron transporters and M2 macrophages in ovarian endometrioma and clear cell adenocarcinoma. Mol. Clin. Oncol. 2021, 15, 159. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Wu, C.H.; Liu, C.H.; Yang, S.F.; Wang, P.H.; Lin, L.Y.; Lee, T.H.; Lee, M.S. Association between the Genetic Variants of Glutathione Peroxidase 4 and Severity of Endometriosis. Int. J. Environ. Res. Public Health 2020, 17, 5089. [Google Scholar] [CrossRef]
- Wan, Y.; Gu, C.; Kong, J.; Sui, J.; Zuo, L.; Song, Y.; Chen, J. Long noncoding RNA ADAMTS9-AS1 represses ferroptosis of endometrial stromal cells by regulating the miR-6516-5p/GPX4 axis in endometriosis. Sci. Rep. 2022, 12, 2618. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, B.D.; Lukowski, S.W.; Mortlock, S.; Crawford, J.; Atluri, S.; Subramaniam, S.; Johnston, R.L.; Nirgianakis, K.; Tanaka, K.; Amoako, A.; et al. Altered differentiation of endometrial mesenchymal stromal fibroblasts is associated with endometriosis susceptibility. Commun. Biol. 2022, 5, 600. [Google Scholar] [CrossRef] [PubMed]
- Queckborner, S.; von Grothusen, C.; Boggavarapu, N.R.; Francis, R.M.; Davies, L.C.; Gemzell-Danielsson, K. Stromal Heterogeneity in the Human Proliferative Endometrium-A Single-Cell RNA Sequencing Study. J. Pers. Med. 2021, 11, 448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, X.; Deng, M.; Xu, C.; Zhang, Y.; Wu, D.; Tang, F.; Yang, R.; Miao, J. Ferroptosis induced by iron overload promotes fibrosis in ovarian endometriosis and is related to subpopulations of endometrial stromal cells. Front. Pharmacol. 2022, 13, 930614. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Diaz, C.P.; Nunez, M.T.; Devoto, L.; Gonzalez-Ramos, R. Iron overload-modulated nuclear factor kappa-B activation in human endometrial stromal cells as a mechanism postulated in endometriosis pathogenesis. Fertil. Steril. 2015, 103, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.H.; Choi, Y.S.; Choi, J.H. Iron-Storage Protein Ferritin Is Upregulated in Endometriosis and Iron Overload Contributes to a Migratory Phenotype. Biomedicines 2020, 8, 454. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.K.; Liu, L.B.; Jin, L.P.; Meng, Y.H.; Shao, J.; Wang, Y.; Mei, J.; Li, M.Q.; Li, D.J. NME1 suppression of endometrial stromal cells promotes angiogenesis in the endometriotic milieu via stimulating the secretion of IL-8 and VEGF. Int. J. Clin. Exp. Pathol. 2013, 6, 2030–2038. [Google Scholar]
- Samimi, M.; Pourhanifeh, M.H.; Mehdizadehkashi, A.; Eftekhar, T.; Asemi, Z. The role of inflammation, oxidative stress, angiogenesis, and apoptosis in the pathophysiology of endometriosis: Basic science and new insights based on gene expression. J. Cell. Physiol. 2019, 234, 19384–19392. [Google Scholar] [CrossRef]
- Powell, S.G.; Sharma, P.; Masterson, S.; Wyatt, J.; Arshad, I.; Ahmed, S.; Lash, G.; Cross, M.; Hapangama, D.K. Vascularisation in Deep Endometriosis: A Systematic Review with Narrative Outcomes. Cells 2023, 12, 1318. [Google Scholar] [CrossRef]
- Liu, S.; Xin, X.; Hua, T.; Shi, R.; Chi, S.; Jin, Z.; Wang, H. Efficacy of Anti-VEGF/VEGFR Agents on Animal Models of Endometriosis: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0166658. [Google Scholar] [CrossRef]
- Cacciottola, L.; Donnez, J.; Dolmans, M.M. Can Endometriosis-Related Oxidative Stress Pave the Way for New Treatment Targets? Int. J. Mol. Sci. 2021, 22, 7138. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; An, P.; Xie, E.; Wu, Q.; Fang, X.; Gao, H.; Zhang, Z.; Li, Y.; Wang, X.; Zhang, J.; et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017, 66, 449–465. [Google Scholar] [CrossRef]
- Liang, D.; Feng, Y.; Zandkarimi, F.; Wang, H.; Zhang, Z.; Kim, J.; Cai, Y.; Gu, W.; Stockwell, B.R.; Jiang, X. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell 2023, 186, 2748–2764.e22. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, S.; Li, Q.; Sun, H.; Wang, H. Pharmacological Inhibition of Ferroptosis as a Therapeutic Target for Neurodegenerative Diseases and Strokes. Adv. Sci. 2023, 10, e2300325. [Google Scholar] [CrossRef] [PubMed]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Meresman, G.F.; Gotte, M.; Laschke, M.W. Plants as source of new therapies for endometriosis: A review of preclinical and clinical studies. Hum. Reprod. Update 2021, 27, 367–392. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Women N = 56 Median (25th–75th Percentile) | Endometriosis n = 38 Median (25th–75th Percentile) | Controls n = 18 Median (25th–75th Percentile) | p-Value | |
|---|---|---|---|---|
| Age | 30.5 (26.0–35.0) | 29.8 (25.0–37.0) | 32.1 (30.5–36.3) | n.s. |
| BMI (kg/m2) | 24.9 (21.0–26.8) | 24.7 (21.0–31.9) | 25.4 (21.0–29.5) | n.s. |
| Hb (g/dL) | 12.7 (12.2–13.5) | 12.8 (12.2–13.5) | 12.5 (12.2–13.4) | n.s. |
| All Women N = 56 | Endometriosis n = 38 | Controls n = 18 | p-Value | |
|---|---|---|---|---|
| Past hormone therapy | 39 | 30 | 9 | 0.028 |
| Current hormone therapy | 17 | 16 | 1 | 0.005 |
| Dysmenorrhea | 35 | 35 | 0 | - |
| Infertility | 17 | 5 | 12 | 0.000 |
| Irregular cycle | 22 | 20 | 2 | 0.003 |
| Bleeding disorder | 25 | 16 | 9 | n.s. |
| Analgesics | 26 | 26 | 0 | - |
| rASRM I n = 10 | rASRM II n = 9 | rASRM III n = 9 | rASRM IV n = 10 | |
|---|---|---|---|---|
| SUP | 10 | 9 | 8 | 9 |
| DIE | 2 | 5 | 9 | 10 |
| P | 8 | 9 | 9 | 10 |
| O | 0 | 3 | 6 | 5 |
| T | 0 | 2 | 5 | 8 |
| A | 1 | 1 | 5 | 9 |
| B | 0 | 4 | 7 | 10 |
| C | 1 | 1 | 4 | 9 |
| FA | 9 | 8 | 9 | 10 |
| FB | 0 | 1 | 2 | 1 |
| FI | 0 | 0 | 1 | 2 |
| FU | 0 | 0 | 0 | 1 |
| F | 1 | 1 | 1 | 0 |
| Endometriosis n = 38 | Controls n = 18 | p-Value | |
|---|---|---|---|
| GPX4 stromal cells | 1.0 ± 0.7 | 1.4 ± 0.6 | 0.031 |
| GPX4 epithelial cells | 1.2 ± 1.1 | 1.3 ± 0.7 | n.s |
| ACSL4 stromal cells | 1.1 ± 0.8 | 1.2 ± 0.4 | n.s |
| ACSL4 epithelial cells | 1.1 ± 0.9 | 1.1 ± 0.6 | n.s |
| TfR1 stromal cells | 0.2 ± 0.4 | 0.7 ± 1.2 | 0.014 |
| TfR1 epithelial cells | 0.1 ± 0.3 | 0.3 ± 0.5 | n.s |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mielke Cabello, L.A.; Meresman, G.; Darici, D.; Carnovale, N.; Heitkötter, B.; Schulte, M.; Espinoza-Sánchez, N.A.; Le, Q.-K.; Kiesel, L.; Schäfer, S.D.; et al. Assessment of the Ferroptosis Regulators: Glutathione Peroxidase 4, Acyl-Coenzyme A Synthetase Long-Chain Family Member 4, and Transferrin Receptor 1 in Patient-Derived Endometriosis Tissue. Biomolecules 2024, 14, 876. https://doi.org/10.3390/biom14070876
Mielke Cabello LA, Meresman G, Darici D, Carnovale N, Heitkötter B, Schulte M, Espinoza-Sánchez NA, Le Q-K, Kiesel L, Schäfer SD, et al. Assessment of the Ferroptosis Regulators: Glutathione Peroxidase 4, Acyl-Coenzyme A Synthetase Long-Chain Family Member 4, and Transferrin Receptor 1 in Patient-Derived Endometriosis Tissue. Biomolecules. 2024; 14(7):876. https://doi.org/10.3390/biom14070876
Chicago/Turabian StyleMielke Cabello, Lidia A., Gabriela Meresman, Dogus Darici, Noelia Carnovale, Birthe Heitkötter, Miriam Schulte, Nancy A. Espinoza-Sánchez, Quang-Khoi Le, Ludwig Kiesel, Sebastian D. Schäfer, and et al. 2024. "Assessment of the Ferroptosis Regulators: Glutathione Peroxidase 4, Acyl-Coenzyme A Synthetase Long-Chain Family Member 4, and Transferrin Receptor 1 in Patient-Derived Endometriosis Tissue" Biomolecules 14, no. 7: 876. https://doi.org/10.3390/biom14070876
APA StyleMielke Cabello, L. A., Meresman, G., Darici, D., Carnovale, N., Heitkötter, B., Schulte, M., Espinoza-Sánchez, N. A., Le, Q.-K., Kiesel, L., Schäfer, S. D., & Götte, M. (2024). Assessment of the Ferroptosis Regulators: Glutathione Peroxidase 4, Acyl-Coenzyme A Synthetase Long-Chain Family Member 4, and Transferrin Receptor 1 in Patient-Derived Endometriosis Tissue. Biomolecules, 14(7), 876. https://doi.org/10.3390/biom14070876