

Are Women with Polycystic Ovary Syndrome at Increased Risk of Alzheimer Disease? Lessons from Insulin Resistance, Tryptophan and Gonadotropin Disturbances and Their Link with Amyloid-Beta Aggregation
 and
and
Abstract
:1. Introduction
2. Outline of the Pathophysiology of Alzheimer Disease
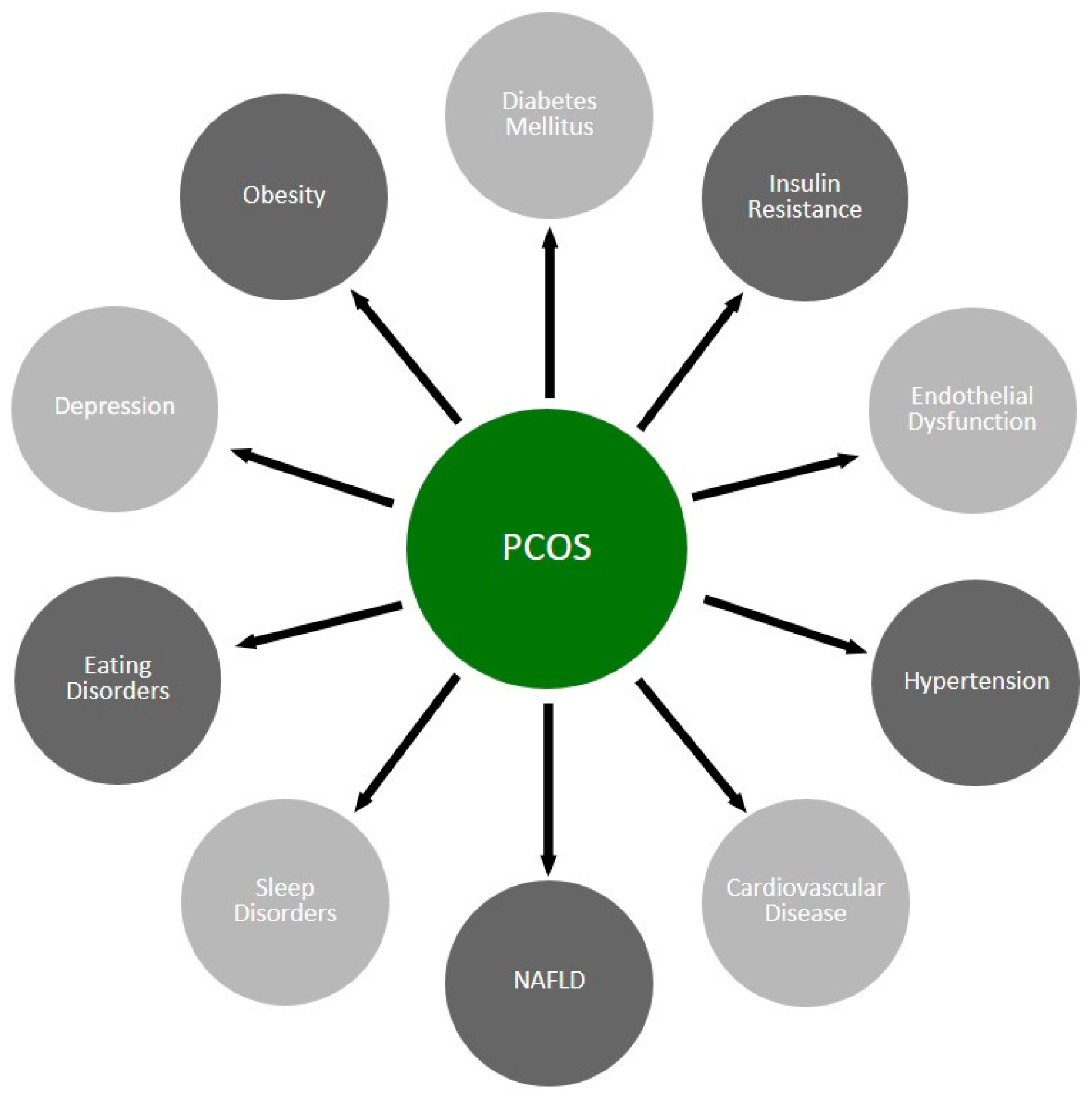
3. Outline of the Pathophysiology of PCOS
4. Physiology of Tryptophan Metabolism
4.1. Production of Serotonin and Biogenic Amines
4.2. Indole Derivatives
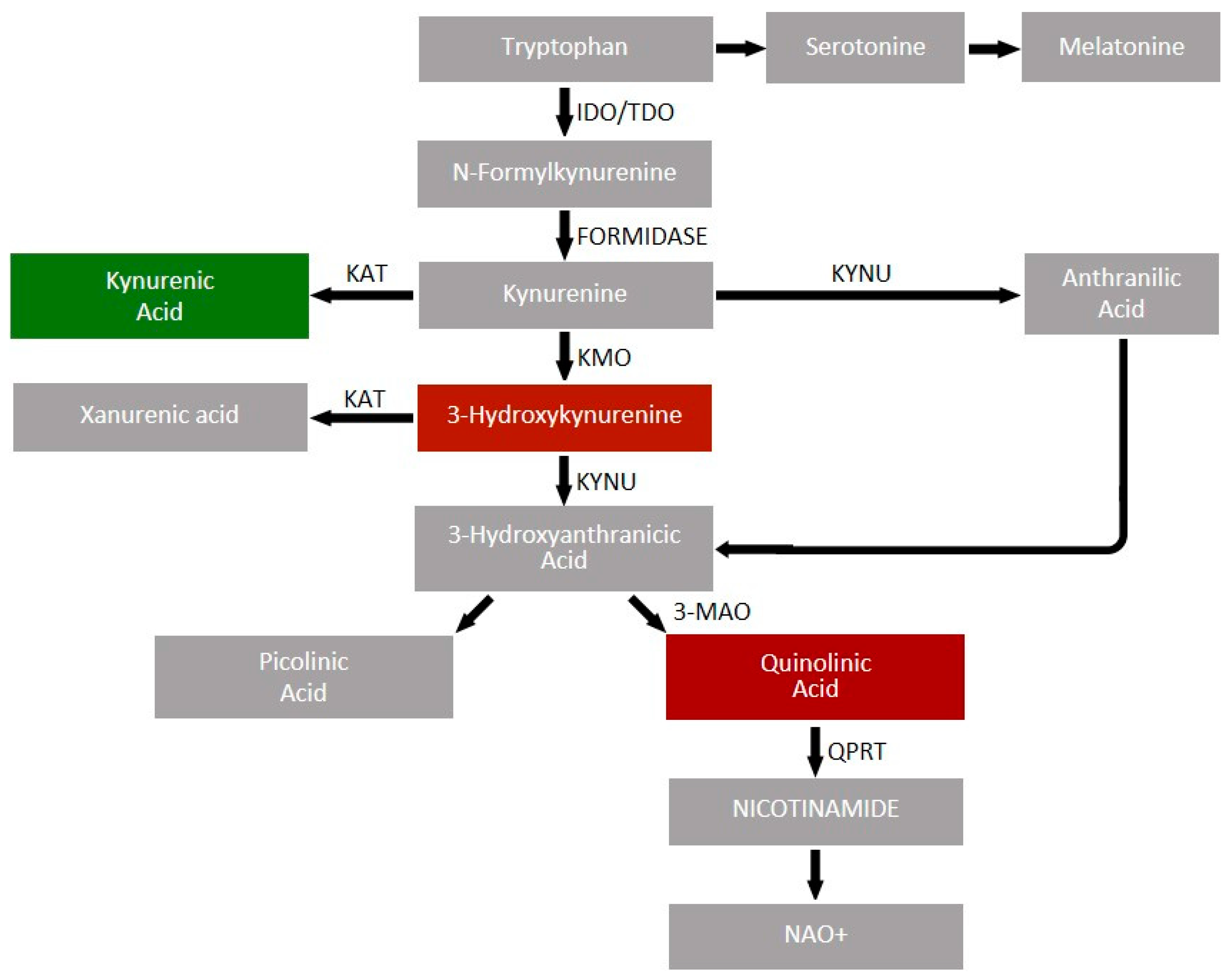
4.3. Kynurenine Pathway
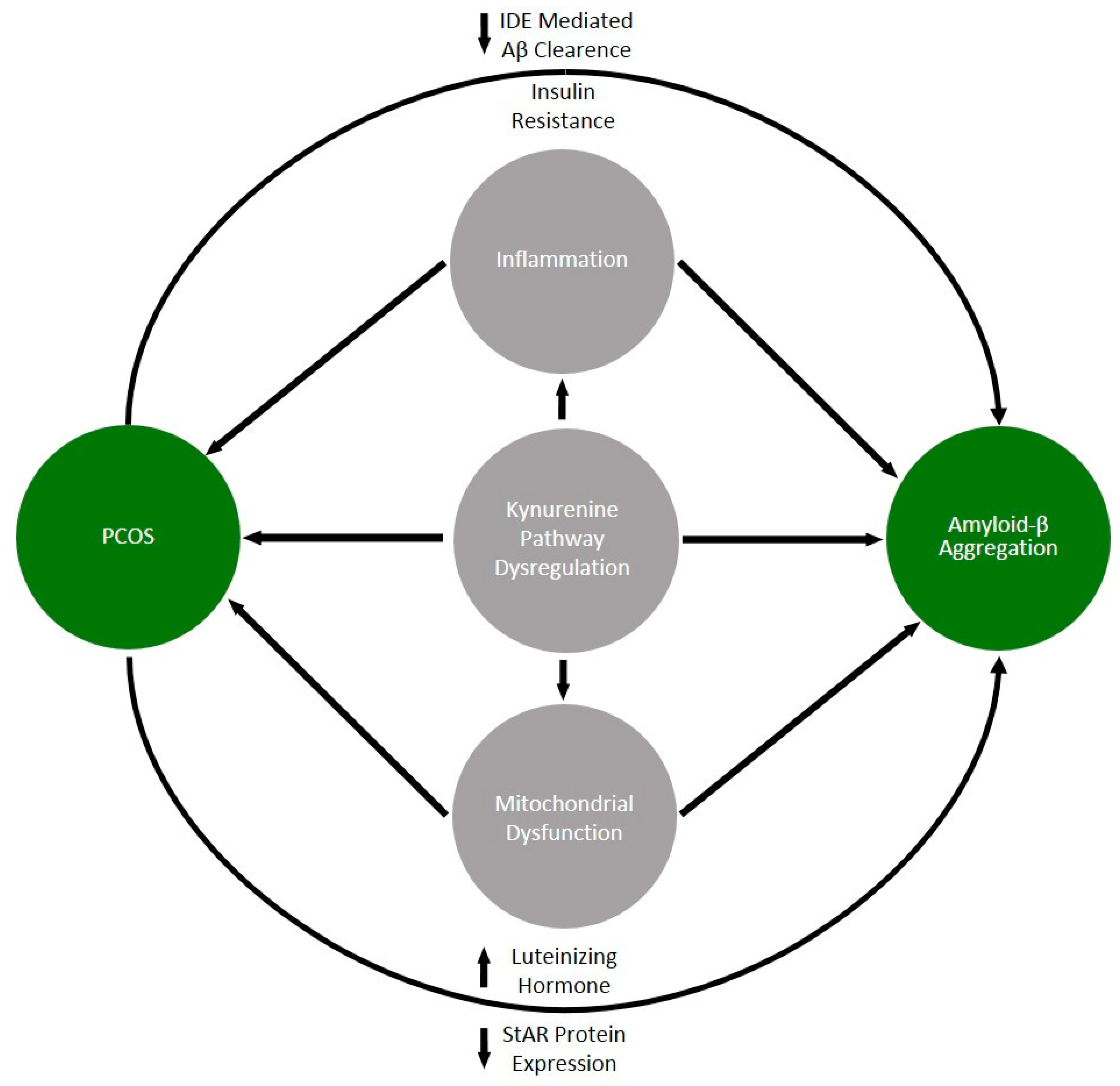
5. Common Denominators of PCOS and Alzheimer Disease
5.1. Insulin Resistance and Metabolic Disorders
5.2. The Role of Luteinizing Hormone and Anti-Mullerian Hormone (AMH)
5.3. Dysfunction of the Kynurenine Pathway
5.4. Other Biomarkers
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer’s Disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-X.; Tian, Y.; Wang, Z.-T.; Ma, Y.-H.; Tan, L.; Yu, J.-T. The Epidemiology of Alzheimer’s Disease Modifiable Risk Factors and Prevention. J. Prev. Alzheimer’s Dis. 2021, 8, 313–321. [Google Scholar] [CrossRef]
- 2023 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [CrossRef] [PubMed]
- Mendez, M.F. Early-Onset Alzheimer’s Disease. Neurol. Clin. 2017, 35, 263–281. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Ferrucci, L.; Kapogiannis, D. Effects of Monoclonal Antibodies against Amyloid-β on Clinical and Biomarker Outcomes and Adverse Event Risks: A Systematic Review and Meta-Analysis of Phase III RCTs in Alzheimer’s Disease. Ageing Res. Rev. 2021, 68, 101339. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Azziz, R.; Adashi, E.Y. Stein and Leventhal: 80 Years On. Am. J. Obstet. Gynecol. 2016, 214, 247.e1–247.e11. [Google Scholar] [CrossRef]
- Azziz, R.; Carmina, E.; Chen, Z.; Dunaif, A.; Laven, J.S.E.; Legro, R.S.; Lizneva, D.; Natterson-Horowtiz, B.; Teede, H.J.; Yildiz, B.O. Polycystic Ovary Syndrome. Nat. Rev. Dis. Primers 2016, 2, 16057. [Google Scholar] [CrossRef]
- Mirza, F.G.; Tahlak, M.A.; Rjeili, R.B.; Hazari, K.; Ennab, F.; Hodgman, C.; Khamis, A.H.; Atiomo, W. Polycystic Ovarian Syndrome (PCOS): Does the Challenge End at Conception? Int. J. Environ. Res. Public Health 2022, 19, 14914. [Google Scholar] [CrossRef]
- Weaver, D.F. Amyloid Beta Is an Early Responder Cytokine and Immunopeptide of the Innate Immune System. Alzheimer’s Dement. 2020, 6, e12100. [Google Scholar] [CrossRef]
- Butler, A.E.; Moin, A.S.M.; Sathyapalan, T.; Atkin, S.L. A Cross-Sectional Study of Alzheimer-Related Proteins in Women with Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2024, 25, 1158. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.K.; Kamboj, P.; Goswami, K. Pathophysiology and management of Alzheimer’s disease: An overview. J. Anal. Pharm. Res. 2018, 9, 226–235. [Google Scholar] [CrossRef]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Kasten, T.; Sigurdson, W.; Bateman, R.J. Amyloid-Beta Isoform Metabolism Quantitation by Stable Isotope Labeled Kinetics. Anal. Biochem. 2013, 440, 56–62. [Google Scholar] [CrossRef]
- Begcevic, I.; Brinc, D.; Brown, M.; Martinez-Morillo, E.; Goldhardt, O.; Grimmer, T.; Magdolen, V.; Batruch, I.; Diamandis, E.P. Brain-Related Proteins as Potential CSF Biomarkers of Alzheimer’s Disease: A Targeted Mass Spectrometry Approach. J. Proteom. 2018, 182, 12–20. [Google Scholar] [CrossRef]
- Jack, C.R.; Lowe, V.J.; Weigand, S.D.; Wiste, H.J.; Senjem, M.L.; Knopman, D.S.; Shiung, M.M.; Gunter, J.L.; Boeve, B.F.; Kemp, B.J.; et al. Serial PIB and MRI in Normal, Mild Cognitive Impairment and Alzheimer’s Disease: Implications for Sequence of Pathological Events in Alzheimer’s Disease. Brain 2009, 132, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Matsuyama, S.; Iso, H.; Umeda, T.; Takuma, H.; Ohnishi, K.; Ishibashi, K.; Teraoka, R.; Sakama, N.; Yamashita, T.; et al. A Mouse Model of Amyloid β Oligomers: Their Contribution to Synaptic Alteration, Abnormal Tau Phosphorylation, Glial Activation, and Neuronal Loss In Vivo. J. Neurosci. 2010, 30, 4845–4856. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Kim, K.; Lim, H.J.; Kim, H.Y.; Shin, J.; Park, I.; Cho, I.; Kim, H.Y.; Kim, S.; McLean, C.; et al. Early Onset Diagnosis in Alzheimer’s Disease Patients via Amyloid-β Oligomers-Sensing Probe in Cerebrospinal Fluid. Nat. Commun. 2024, 15, 1004. [Google Scholar] [CrossRef]
- Savage, M.J.; Kalinina, J.; Wolfe, A.; Tugusheva, K.; Korn, R.; Cash-Mason, T.; Maxwell, J.W.; Hatcher, N.G.; Haugabook, S.J.; Wu, G.; et al. A Sensitive Aβ Oligomer Assay Discriminates Alzheimer’s and Aged Control Cerebrospinal Fluid. J. Neurosci. 2014, 34, 2884–2897. [Google Scholar] [CrossRef]
- La Joie, R.; Visani, A.V.; Baker, S.L.; Brown, J.A.; Bourakova, V.; Cha, J.; Chaudhary, K.; Edwards, L.; Iaccarino, L.; Janabi, M.; et al. Prospective Longitudinal Atrophy in Alzheimer’s Disease Correlates with the Intensity and Topography of Baseline Tau-PET. Sci. Transl. Med. 2020, 12, eaau5732. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes with Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-R.; Huang, J.-B.; Yang, S.-L.; Hong, F.-F. Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules 2022, 27, 1816. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, P.J.; Price, D.L.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer Disease: Evidence for Selective Loss of Cholinergic Neurons in the Nucleus Basalis. Ann. Neurol. 1981, 10, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Rodriguez, J.J.; Pacheco-Herrero, M.; Thyssen, D.; Murillo-Carretero, M.I.; Berrocoso, E.; Spires-Jones, T.L.; Bacskai, B.J.; Garcia-Alloza, M. Rapid β-Amyloid Deposition and Cognitive Impairment after Cholinergic Denervation in APP/PS1 Mice. J. Neuropathol. Exp. Neurol. 2013, 72, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Kuo, Y.M.; Spiegel, K.; Emmerling, M.R.; Sue, L.I.; Kokjohn, K.; Roher, A.E. The Cholinergic Deficit Coincides with Abeta Deposition at the Earliest Histopathologic Stages of Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2000, 59, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Potter, P.E.; Rauschkolb, P.K.; Pandya, Y.; Sue, L.I.; Sabbagh, M.N.; Walker, D.G.; Beach, T.G. Pre- and Post-Synaptic Cortical Cholinergic Deficits Are Proportional to Amyloid Plaque Presence and Density at Preclinical Stages of Alzheimer’s Disease. Acta Neuropathol. 2011, 122, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C. Genetic Diagnosis and Prognosis of Alzheimer’s Disease: Challenges and Opportunities. Expert Rev. Mol. Diagn. 2015, 15, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer Disease: Risk, Mechanisms, and Therapy. Nat. Rev. Neurol. 2013, 9, 106. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of Age, Sex, and Ethnicity on the Association between Apolipoprotein E Genotype and Alzheimer Disease. A Meta-Analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Exceptionally Low Likelihood of Alzheimer’s Dementia in APOE2 Homozygotes from a 5,000-Person Neuropathological Study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A Mutation in APP Protects against Alzheimer’s Disease and Age-Related Cognitive Decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Viveros Salazar, S.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-Human TREM2 Induces Microglia Proliferation and Reduces Pathology in an Alzheimer’s Disease Model. J. Exp. Med. 2020, 217, e20200785. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [PubMed]
- Mantzavinos, V.; Alexiou, A. Biomarkers for Alzheimer’s Disease Diagnosis. Curr. Alzheimer Res. 2017, 14, 1149–1154. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, J.; Cheng, X.; Nie, X.; He, B. Insulin Resistance in Polycystic Ovary Syndrome across Various Tissues: An Updated Review of Pathogenesis, Evaluation, and Treatment. J. Ovarian Res. 2023, 16, 9. [Google Scholar] [CrossRef]
- Legro, R.S.; Castracane, V.D.; Kauffman, R.P. Detecting Insulin Resistance in Polycystic Ovary Syndrome: Purposes and Pitfalls. Obstet. Gynecol. Surv. 2004, 59, 141–154. [Google Scholar] [CrossRef]
- DeUgarte, C.M.; Bartolucci, A.A.; Azziz, R. Prevalence of Insulin Resistance in the Polycystic Ovary Syndrome Using the Homeostasis Model Assessment. Fertil. Steril. 2005, 83, 1454–1460. [Google Scholar] [CrossRef]
- Dunaif, A.; Segal, K.R.; Futterweit, W.; Dobrjansky, A. Profound Peripheral Insulin Resistance, Independent of Obesity, in Polycystic Ovary Syndrome. Diabetes 1989, 38, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Teede, H.J.; Tay, C.T.; Laven, J.J.E.; Dokras, A.; Moran, L.J.; Piltonen, T.T.; Costello, M.F.; Boivin, J.; Redman, L.M.; Boyle, J.A.; et al. Recommendations from the 2023 International Evidence-Based Guideline for the Assessment and Management of Polycystic Ovary Syndrome. Eur. J. Endocrinol. 2023, 189, G43–G64. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, A.; Kudła, M.; Spaczyński, R.Z.; Dębski, R.; Męczekalski, B.; Wielgoś, M.; Ruchała, M.; Małecka-Tendera, E.; Kos-Kudła, B.; Jędrzejuk, D.; et al. The Polycystic Ovary Syndrome: A Position Statement from the Polish Society of Endocrinology, the Polish Society of Gynaecologists and Obstetricians, and the Polish Society of Gynaecological Endocrinology. Endokrynol. Pol. 2018, 69, 328–344. [Google Scholar] [CrossRef]
- Szeliga, A.; Rudnicka, E.; Maciejewska-Jeske, M.; Kucharski, M.; Kostrzak, A.; Hajbos, M.; Niwczyk, O.; Smolarczyk, R.; Meczekalski, B. Neuroendocrine Determinants of Polycystic Ovary Syndrome. Int. J. Environ. Res. Public Health 2022, 19, 3089. [Google Scholar] [CrossRef] [PubMed]
- Malini, N.A.; Roy, G.K. Influence of Insulin on LH, Testosterone and SHBG in Various PCOS Categories Based on the Mode of Secretion of LH in Relation to FSH Levels. Acta Endocrinol. 2021, 17, 313–318. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Nikiforov, N.G.; Eid, A.H.; Nedosugova, L.V.; Starodubova, A.V.; Popkova, T.V.; Bezsonov, E.E.; Orekhov, A.N. Mitochondrial Dysfunction and Chronic Inflammation in Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2021, 22, 3923. [Google Scholar] [CrossRef]
- Qi, X.; Yun, C.; Pang, Y.; Qiao, J. The Impact of the Gut Microbiota on the Reproductive and Metabolic Endocrine System. Gut Microbes 2021, 13, 1–21. [Google Scholar] [CrossRef]
- He, F.; Li, Y. Role of Gut Microbiota in the Development of Insulin Resistance and the Mechanism Underlying Polycystic Ovary Syndrome: A Review. J. Ovarian Res. 2020, 13, 73. [Google Scholar] [CrossRef]
- Wild, R.A.; Rizzo, M.; Clifton, S.; Carmina, E. Lipid Levels in Polycystic Ovary Syndrome: Systematic Review and Meta-Analysis. Fertil. Steril. 2011, 95, 1073–1079.e11. [Google Scholar] [CrossRef]
- Rudnicka, E.; Suchta, K.; Grymowicz, M.; Calik-Ksepka, A.; Smolarczyk, K.; Duszewska, A.M.; Smolarczyk, R.; Meczekalski, B. Chronic Low Grade Inflammation in Pathogenesis of PCOS. Int. J. Mol. Sci. 2021, 22, 3789. [Google Scholar] [CrossRef]
- Lim, S.S.; Kakoly, N.S.; Tan, J.W.J.; Fitzgerald, G.; Bahri Khomami, M.; Joham, A.E.; Cooray, S.D.; Misso, M.L.; Norman, R.J.; Harrison, C.L.; et al. Metabolic Syndrome in Polycystic Ovary Syndrome: A Systematic Review, Meta-Analysis and Meta-Regression. Obes. Rev. 2019, 20, 339–352. [Google Scholar] [CrossRef]
- Legro, R.S.; Kunselman, A.R.; Dunaif, A. Prevalence and Predictors of Dyslipidemia in Women with Polycystic Ovary Syndrome. Am. J. Med. 2001, 111, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Burnatowska, E.; Wikarek, A.; Oboza, P.; Ogarek, N.; Glinianowicz, M.; Kocelak, P.; Olszanecka-Glinianowicz, M. Emotional Eating and Binge Eating Disorders and Night Eating Syndrome in Polycystic Ovary Syndrome—A Vicious Circle of Disease: A Systematic Review. Nutrients 2023, 15, 295. [Google Scholar] [CrossRef] [PubMed]
- Zehravi, M.; Maqbool, M.; Ara, I. Depression and Anxiety in Women with Polycystic Ovarian Syndrome: A Literature Survey. Int. J. Adolesc. Med. Health 2021, 33, 367–373. [Google Scholar] [CrossRef]
- Kolhe, J.V.; Chhipa, A.S.; Butani, S.; Chavda, V.; Patel, S.S. PCOS and Depression: Common Links and Potential Targets. Reprod. Sci. 2022, 29, 3106–3123. [Google Scholar] [CrossRef]
- Zeng, X.; Xie, Y.-J.; Liu, Y.-T.; Long, S.-L.; Mo, Z.-C. Polycystic Ovarian Syndrome: Correlation between Hyperandrogenism, Insulin Resistance and Obesity. Clin. Chim. Acta 2020, 502, 214–221. [Google Scholar] [CrossRef]
- Parker, J.; O’Brien, C.; Hawrelak, J.; Gersh, F.L. Polycystic Ovary Syndrome: An Evolutionary Adaptation to Lifestyle and the Environment. Int. J. Environ. Res. Public Health 2022, 19, 1336. [Google Scholar] [CrossRef] [PubMed]
- Paczkowska, K.; Rachoń, D.; Berg, A.; Rybka, J.; Kapczyńska, K.; Bolanowski, M.; Daroszewski, J. Specific Alteration of Branched-Chain Amino Acid Profile in Polycystic Ovary Syndrome. Biomedicines 2023, 11, 108. [Google Scholar] [CrossRef]
- Szmygin, H.; Lenart-Lipinska, M.; Szydelko, J.; Wozniak, S.; Matyjaszek-Matuszek, B. Branched-Chain Amino Acids as a Novel Biomarker of Metabolic Disturbances in Women with Polycystic Ovary Syndrome—Literature Review. Ginekol. Pol. 2022, 93, 665–669. [Google Scholar] [CrossRef]
- Di Guardo, F.; Ciotta, L.; Monteleone, M.; Palumbo, M. Male Equivalent Polycystic Ovarian Syndrome: Hormonal, Metabolic, and Clinical Aspects. Int. J. Fertil. Steril. 2020, 14, 79–83. [Google Scholar] [CrossRef]
- Cannarella, R.; Condorelli, R.A.; Mongioì, L.M.; La Vignera, S.; Calogero, A.E. Does a Male Polycystic Ovarian Syndrome Equivalent Exist? J. Endocrinol. Investig. 2018, 41, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.M.; Torchen, L.C.; Sung, Y.; Paparodis, R.; Legro, R.S.; Grebe, S.K.; Singh, R.J.; Taylor, R.L.; Dunaif, A. Evidence for Gonadotrophin Secretory and Steroidogenic Abnormalities in Brothers of Women with Polycystic Ovary Syndrome. Hum. Reprod. 2014, 29, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Paczkowska, K.; Sobczuk, J.; Zawadzka, K.; Jedrzejuk, D.; Zembska, A.; Konieczny, J.; Kaszubkiewicz-Wardęga, D.; Bolanowski, M.; Daroszewski, J. Circulating Levels of Irisin and Meteorin-like Protein in PCOS and Its Correlation with Metabolic Parameters. Endokrynol. Pol. 2024, 75, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Siomkajło, M.; Rybka, J.; Mierzchała-Pasierb, M.; Gamian, A.; Stankiewicz-Olczyk, J.; Bolanowski, M.; Daroszewski, J. Specific Plasma Amino Acid Disturbances Associated with Metabolic Syndrome. Endocrine 2017, 58, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Siomkajło, M.; Daroszewski, J. Branched Chain Amino Acids: Passive Biomarkers or the Key to the Pathogenesis of Cardiometabolic Diseases? Adv. Clin. Exp. Med. 2019, 28, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Pukajło, K.; Łaczmański, Ł.; Kolackov, K.; Kuliczkowska-Płaksej, J.; Bolanowski, M.; Milewicz, A.; Daroszewski, J. Irisin Plasma Concentration in PCOS and Healthy Subjects Is Related to Body Fat Content and Android Fat Distribution. Gynecol. Endocrinol. 2015, 31, 907–911. [Google Scholar] [CrossRef]
- Paczkowska, K.; Rachoń, D.; Berg, A.; Rybka, J.; Kapczyńska, K.; Bolanowski, M.; Daroszewski, J. Alteration of Branched-Chain and Aromatic Amino Acid Profile as a Novel Approach in Studying Polycystic Ovary Syndrome Pathogenesis. Nutrients 2023, 15, 4153. [Google Scholar] [CrossRef]
- Zuraikat, F.M.; Wood, R.A.; Barragán, R.; St-Onge, M.-P. Sleep and Diet: Mounting Evidence of a Cyclical Relationship. Annu. Rev. Nutr. 2021, 41, 309–332. [Google Scholar] [CrossRef]
- Lieberman, H.R.; Agarwal, S.; Fulgoni, V.L. Tryptophan Intake in the US Adult Population Is Not Related to Liver or Kidney Function but Is Associated with Depression and Sleep Outcomes. J. Nutr. 2016, 146, 2609S–2615S. [Google Scholar] [CrossRef]
- Lynch, J.H.; Dudareva, N. Aromatic Amino Acids: A Complex Network Ripe for Future Exploration. Trends Plant Sci. 2020, 25, 670–681. [Google Scholar] [CrossRef]
- Wu, G. Amino Acids: Metabolism, Functions, and Nutrition. Amino Acids 2009, 37, 1–17. [Google Scholar] [CrossRef]
- Krupa, A.; Kowalska, I. The Kynurenine Pathway—New Linkage between Innate and Adaptive Immunity in Autoimmune Endocrinopathies. Int. J. Mol. Sci. 2021, 22, 9879. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.A.; Fiorotto, M.L. Regulation of Muscle Growth in Neonates. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Palego, L.; Betti, L.; Rossi, A.; Giannaccini, G. Tryptophan Biochemistry: Structural, Nutritional, Metabolic, and Medical Aspects in Humans. J. Amino Acids 2016, 2016, 8952520. [Google Scholar] [CrossRef]
- Mittal, R.; Debs, L.H.; Patel, A.P.; Nguyen, D.; Patel, K.; O’Connor, G.; Grati, M.; Mittal, J.; Yan, D.; Eshraghi, A.A.; et al. Neurotransmitters: The Critical Modulators Regulating Gut-Brain Axis. J. Cell. Physiol. 2017, 232, 2359–2372. [Google Scholar] [CrossRef] [PubMed]
- McMenamy, R.H.; Oncley, J.L. The Specific Binding of L-Tryptophan to Serum Albumin. J. Biol. Chem. 1958, 233, 1436–1447. [Google Scholar] [CrossRef] [PubMed]
- Mintz, K.J.; Mercado, G.; Zhou, Y.; Ji, Y.; Hettiarachchi, S.D.; Liyanage, P.Y.; Pandey, R.R.; Chusuei, C.C.; Dallman, J.; Leblanc, R.M. Tryptophan Carbon Dots and Their Ability to Cross the Blood-Brain Barrier. Colloids Surf. B Biointerfaces 2019, 176, 488–493. [Google Scholar] [CrossRef]
- Ranhotra, H.S. Discrete Interplay of Gut Microbiota L-Tryptophan Metabolites in Host Biology and Disease. Mol. Cell. Biochem. 2023, 1–18. [Google Scholar] [CrossRef]
- Lee, J.-H.; Wood, T.K.; Lee, J. Roles of Indole as an Interspecies and Interkingdom Signaling Molecule. Trends Microbiol. 2015, 23, 707–718. [Google Scholar] [CrossRef]
- Zhang, L.S.; Davies, S.S. Microbial Metabolism of Dietary Components to Bioactive Metabolites: Opportunities for New Therapeutic Interventions. Genome Med. 2016, 8, 46. [Google Scholar] [CrossRef]
- Williams, B.B.; Van Benschoten, A.H.; Cimermancic, P.; Donia, M.S.; Zimmermann, M.; Taketani, M.; Ishihara, A.; Kashyap, P.C.; Fraser, J.S.; Fischbach, M.A. Discovery and Characterization of Gut Microbiota Decarboxylases That Can Produce the Neurotransmitter Tryptamine. Cell Host Microbe 2014, 16, 495–503. [Google Scholar] [CrossRef]
- Badawy, A.A.-B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef]
- Anquetil, F.; Mondanelli, G.; Gonzalez, N.; Rodriguez Calvo, T.; Zapardiel Gonzalo, J.; Krogvold, L.; Dahl-Jørgensen, K.; Van den Eynde, B.; Orabona, C.; Grohmann, U.; et al. Loss of IDO1 Expression From Human Pancreatic β-Cells Precedes Their Destruction During the Development of Type 1 Diabetes. Diabetes 2018, 67, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Jamshed, L.; Debnath, A.; Jamshed, S.; Wish, J.V.; Raine, J.C.; Tomy, G.T.; Thomas, P.J.; Holloway, A.C. An Emerging Cross-Species Marker for Organismal Health: Tryptophan-Kynurenine Pathway. Int. J. Mol. Sci. 2022, 23, 6300. [Google Scholar] [CrossRef]
- Savitz, J. The Kynurenine Pathway: A Finger in Every Pie. Mol. Psychiatry 2020, 25, 131–147. [Google Scholar] [CrossRef]
- Castro-Portuguez, R.; Sutphin, G.L. Kynurenine Pathway, NAD+ Synthesis, and Mitochondrial Function: Targeting Tryptophan Metabolism to Promote Longevity and Healthspan. Exp. Gerontol. 2020, 132, 110841. [Google Scholar] [CrossRef]
- Kubacka, J.; Staniszewska, M.; Sadok, I.; Sypniewska, G.; Stefanska, A. The Kynurenine Pathway in Obese Middle-Aged Women with Normoglycemia and Type 2 Diabetes. Metabolites 2022, 12, 492. [Google Scholar] [CrossRef] [PubMed]
- Schwieler, L.; Trepci, A.; Krzyzanowski, S.; Hermansson, S.; Granqvist, M.; Piehl, F.; Venckunas, T.; Brazaitis, M.; Kamandulis, S.; Lindqvist, D.; et al. A Novel, Robust Method for Quantification of Multiple Kynurenine Pathway Metabolites in the Cerebrospinal Fluid. Bioanalysis 2020, 12, 379–392. [Google Scholar] [CrossRef]
- Arnone, D.; Saraykar, S.; Salem, H.; Teixeira, A.L.; Dantzer, R.; Selvaraj, S. Role of Kynurenine Pathway and Its Metabolites in Mood Disorders: A Systematic Review and Meta-Analysis of Clinical Studies. Neurosci. Biobehav. Rev. 2018, 92, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Wejksza, K.; Rzeski, W.; Turski, W.A. Kynurenic Acid Protects against the Homocysteine-Induced Impairment of Endothelial Cells. Pharmacol. Rep. 2009, 61, 751–756. [Google Scholar] [CrossRef]
- Walczak, K.; Wnorowski, A.; Turski, W.A.; Plech, T. Kynurenic Acid and Cancer: Facts and Controversies. Cell. Mol. Life Sci. 2020, 77, 1531–1550. [Google Scholar] [CrossRef] [PubMed]
- Kamel, R.; Baetz, D.; Gueguen, N.; Lebeau, L.; Barbelivien, A.; Guihot, A.-L.; Allawa, L.; Gallet, J.; Beaumont, J.; Ovize, M.; et al. Kynurenic Acid: A Novel Player in Cardioprotection against Myocardial Ischemia/Reperfusion Injuries. Pharmaceuticals 2023, 16, 1381. [Google Scholar] [CrossRef]
- Pawlowski, T.; Pawlak, D.; Inglot, M.; Zalewska, M.; Marciniak, D.; Bugajska, J.; Janocha-Litwin, J.; Malyszczak, K. The Role of Anthranilic Acid in the Increase of Depressive Symptoms and Major Depressive Disorder during Treatment for Hepatitis C with Pegylated Interferon-A2a and Oral Ribavirin. J. Psychiatry Neurosci. JPN 2021, 46, E166–E175. [Google Scholar] [CrossRef] [PubMed]
- Favennec, M.; Hennart, B.; Verbanck, M.; Pigeyre, M.; Caiazzo, R.; Raverdy, V.; Verkindt, H.; Leloire, A.; Guillemin, G.J.; Yengo, L.; et al. Post-Bariatric Surgery Changes in Quinolinic and Xanthurenic Acid Concentrations Are Associated with Glucose Homeostasis. PLoS ONE 2016, 11, e0158051. [Google Scholar] [CrossRef]
- Fazio, F.; Carrizzo, A.; Lionetto, L.; Damato, A.; Capocci, L.; Ambrosio, M.; Battaglia, G.; Bruno, V.; Madonna, M.; Simmaco, M.; et al. Vasorelaxing Action of the Kynurenine Metabolite, Xanthurenic Acid: The Missing Link in Endotoxin-Induced Hypotension? Front. Pharmacol. 2017, 8, 214. [Google Scholar] [CrossRef]
- Ruffmann, R.; Schlick, R.; Chirigos, M.A.; Budzynsky, W.; Varesio, L. Antiproliferative Activity of Picolinic Acid Due to Macrophage Activation. Drugs Exp. Clin. Res. 1987, 13, 607–614. [Google Scholar] [PubMed]
- Grant, R.S.; Coggan, S.E.; Smythe, G.A. The Physiological Action of Picolinic Acid in the Human Brain. Int. J. Tryptophan Res. 2009, 2, 71–79. [Google Scholar] [CrossRef]
- Farup, P.G.; Hamarsland, H.; Mølmen, K.S.; Ellefsen, S.; Hestad, K. The Kynurenine Pathway in Healthy Subjects and Subjects with Obesity, Depression and Chronic Obstructive Pulmonary Disease. Pharmaceuticals 2023, 16, 351. [Google Scholar] [CrossRef]
- Kiluk, M.; Lewkowicz, J.; Kowalska, I.; Pawlak, D.; Łagoda, K.; Tankiewicz-Kwedlo, A. Alterations of the Kynurenine Pathway in Patients with Type 1 Diabetes Are Associated with Metabolic Control of Diabetes. Pol. Arch. Intern. Med. 2023, 133, 16581. [Google Scholar] [CrossRef]
- Benitez, T.; VanDerWoude, E.; Han, Y.; Byun, J.; Konje, V.C.; Gillespie, B.W.; Saran, R.; Mathew, A.V. Kynurenine Pathway Metabolites Predict Subclinical Atherosclerotic Disease and New Cardiovascular Events in Chronic Kidney Disease. Clin. Kidney J. 2022, 15, 1952–1965. [Google Scholar] [CrossRef]
- Kiluk, M.; Lewkowicz, J.; Pawlak, D.; Tankiewicz-Kwedlo, A. Crosstalk between Tryptophan Metabolism via Kynurenine Pathway and Carbohydrate Metabolism in the Context of Cardio-Metabolic Risk—Review. J. Clin. Med. 2021, 10, 2484. [Google Scholar] [CrossRef] [PubMed]
- Ala, M.; Eftekhar, S.P. The Footprint of Kynurenine Pathway in Cardiovascular Diseases. Int. J. Tryptophan Res. 2022, 15, 11786469221096643. [Google Scholar] [CrossRef] [PubMed]
- Gouasmi, R.; Ferraro-Peyret, C.; Nancey, S.; Coste, I.; Renno, T.; Chaveroux, C.; Aznar, N.; Ansieau, S. The Kynurenine Pathway and Cancer: Why Keep It Simple When You Can Make It Complicated. Cancers 2022, 14, 2793. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; Darlington, L.G. The Kynurenine Pathway as a Therapeutic Target in Cognitive and Neurodegenerative Disorders. Br. J. Pharmacol. 2013, 169, 1211–1227. [Google Scholar] [CrossRef] [PubMed]
- Heidari, F.; Ramezani, A.; Erfani, N.; Razmkhah, M. Indoleamine 2, 3-Dioxygenase: A Professional Immunomodulator and Its Potential Functions in Immune Related Diseases. Int. Rev. Immunol. 2022, 41, 346–363. [Google Scholar] [CrossRef] [PubMed]
- Mole, D.J.; Webster, S.P.; Uings, I.; Zheng, X.; Binnie, M.; Wilson, K.; Hutchinson, J.P.; Mirguet, O.; Walker, A.; Beaufils, B.; et al. Kynurenine-3-Monooxygenase Inhibition Prevents Multiple Organ Failure in Rodent Models of Acute Pancreatitis. Nat. Med. 2016, 22, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Rojewska, E.; Ciapała, K.; Piotrowska, A.; Makuch, W.; Mika, J. Pharmacological inhibition of indoleamine 2,3-dioxygenase-2 and kynurenine 3-monooxygenase, enzymes of the kynurenine pathway, significantly diminishes neuropathic pain in a rat model. Front. Pharmacol. 2018, 9, 724. [Google Scholar] [CrossRef]
- Filippini, P.; Papa, N.D.; Sambataro, D.; Bufalo, A.D.; Locatelli, F.; Rutella, S. Emerging Concepts on Inhibitors of Indoleamine 2,3-Dioxygenase in Rheumatic Diseases. Curr. Med. Chem. 2019, 19, 5381–5393. [Google Scholar] [CrossRef]
- Chen, H.; Huang, Y.; Lv, X.; Xu, X.; Ma, Y.; Wang, H.; Yuan, C. Prevalence of Dementia and the Attributable Contributions of Modifiable Risk Factors in China. Gen. Psych. 2023, 36, e101044. [Google Scholar] [CrossRef]
- Zhou, F.; Chen, S. Hyperhomocysteinemia and Risk of Incident Cognitive Outcomes: An Updated Dose-Response Meta-Analysis of Prospective Cohort Studies. Ageing Res. Rev. 2019, 51, 55–66. [Google Scholar] [CrossRef]
- Skoog, I.; Lernfelt, B.; Landahl, S.; Palmertz, B.; Andreasson, L.A.; Nilsson, L.; Persson, G.; Odén, A.; Svanborg, A. 15-Year Longitudinal Study of Blood Pressure and Dementia. Lancet 1996, 347, 1141–1145. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Tzourio, C.; Soumaré, A.; Kaffashian, S.; Dartigues, J.-F.; Ancelin, M.-L.; Samieri, C.; Dufouil, C.; Debette, S. Differential Associations of Plasma Lipids with Incident Dementia and Dementia Subtypes in the 3C Study: A Longitudinal, Population-Based Prospective Cohort Study. PLoS Med. 2017, 14, e1002265. [Google Scholar] [CrossRef] [PubMed]
- McGrath, E.R.; Beiser, A.S.; DeCarli, C.; Plourde, K.L.; Vasan, R.S.; Greenberg, S.M.; Seshadri, S. Blood Pressure from Mid- to Late Life and Risk of Incident Dementia. Neurology 2017, 89, 2447–2454. [Google Scholar] [CrossRef] [PubMed]
- Tolppanen, A.-M.; Ngandu, T.; Kåreholt, I.; Laatikainen, T.; Rusanen, M.; Soininen, H.; Kivipelto, M. Midlife and Late-Life Body Mass Index and Late-Life Dementia: Results from a Prospective Population-Based Cohort. J. Alzheimer’s Dis. 2014, 38, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Willette, A.A.; Johnson, S.C.; Birdsill, A.; Sager, M.A.; Christian, B.; Baker, L.D.; Craft, S.; Oh, J.; Statz, E.; Hermann, B.P.; et al. Insulin Resistance Predicts Brain Amyloid Deposition in Late Middle-Aged Adults. Alzheimer’s Dement. 2015, 11, 504–510.e1. [Google Scholar] [CrossRef] [PubMed]
- Ekblad, L.L.; Johansson, J.; Helin, S.; Viitanen, M.; Laine, H.; Puukka, P.; Jula, A.; Rinne, J.O. Midlife Insulin Resistance, APOE Genotype, and Late-Life Brain Amyloid Accumulation. Neurology 2018, 90, e1150–e1157. [Google Scholar] [CrossRef] [PubMed]
- Castellano, C.-A.; Baillargeon, J.-P.; Nugent, S.; Tremblay, S.; Fortier, M.; Imbeault, H.; Duval, J.; Cunnane, S.C. Regional Brain Glucose Hypometabolism in Young Women with Polycystic Ovary Syndrome: Possible Link to Mild Insulin Resistance. PLoS ONE 2015, 10, e0144116. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Kawasaki, K.; Ishiwata, K.; Ishii, K. Reduced Uptake of 18F-FDG and 15O-H2O in Alzheimer’s Disease-Related Regions after Glucose Loading. J. Cereb. Blood Flow Metab. 2015, 35, 1380–1385. [Google Scholar] [CrossRef]
- Wium-Andersen, I.K.; Osler, M.; Jørgensen, M.B.; Rungby, J.; Wium-Andersen, M.K. Antidiabetic Medication and Risk of Dementia in Patients with Type 2 Diabetes: A Nested Case–Control Study. Eur. J. Endocrinol. 2019, 181, 499–507. [Google Scholar] [CrossRef]
- Lee, J.H.; Byun, M.S.; Yi, D.; Choe, Y.M.; Choi, H.J.; Baek, H.; Sohn, B.K.; Lee, J.-Y.; Kim, H.J.; Kim, J.W.; et al. Sex-Specific Association of Sex Hormones and Gonadotropins, with Brain Amyloid and Hippocampal Neurodegeneration. Neurobiol. Aging 2017, 58, 34–40. [Google Scholar] [CrossRef]
- Berry, A.; Tomidokoro, Y.; Ghiso, J.; Thornton, J. Human Chorionic Gonadotropin (a Luteinizing Hormone Homologue) Decreases Spatial Memory and Increases Brain Amyloid-β Levels in Female Rats. Horm. Behav. 2008, 54, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Li, J.; Wang, Y.; Wang, W.; Zhu, Q.; He, Y.; Lu, Y.; Wu, H.; Li, X.; Zhu, Z.; et al. Novel Role of CXCL14 in Modulating STAR Expression in Luteinized Granulosa Cells: Implication for Progesterone Synthesis in PCOS Patients. Transl. Res. 2021, 230, 55–67. [Google Scholar] [CrossRef]
- Manna, P.R.; Kshirsagar, S.; Pradeepkiran, J.A.; Rawat, P.; Kumar, S.; Reddy, A.P.; Reddy, P.H. Protective Function of StAR in Amyloid-β Accumulated Hippocampal Neurotoxicity and Neurosteroidogenesis: Mechanistic Insights into Alzheimer’s Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2023, 1869, 166738. [Google Scholar] [CrossRef]
- Willette, A.A.; Pappas, C.; Hoth, N.; Wang, Q.; Klinedinst, B.; Willette, S.A.; Larsen, B.; Pollpeter, A.; Li, T.; Le, S.; et al. Inflammation, Negative Affect, and Amyloid Burden in Alzheimer’s Disease: Insights from the Kynurenine Pathway. Brain Behav. Immun. 2021, 95, 216–225. [Google Scholar] [CrossRef]
- van der Velpen, V.; Teav, T.; Gallart-Ayala, H.; Mehl, F.; Konz, I.; Clark, C.; Oikonomidi, A.; Peyratout, G.; Henry, H.; Delorenzi, M.; et al. Systemic and Central Nervous System Metabolic Alterations in Alzheimer’s Disease. Alzheimer’s Res. Ther. 2019, 11, 93. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mu, L.; Zhang, C.; Long, X.; Zhang, Y.; Li, R.; Zhao, Y.; Qiao, J. Abnormal Activation of Tryptophan-Kynurenine Pathway in Women with Polycystic Ovary Syndrome. Front. Endocrinol. 2022, 13, 877807. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Cai, X.; Xu, X.; Xu, Z.; Ye, S.; Wang, Y.; Hong, Y.; Shen, B.; Liao, Q.; Xie, Z.; et al. Urinary Metabolomics Identified Metabolic Disturbance Associated with Polycystic Ovary Syndrome. Anal. Biochem. 2022, 647, 114665. [Google Scholar] [CrossRef]
- Hou, E.; Zhao, Y.; Hang, J.; Qiao, J. Metabolomics and Correlation Network Analysis of Follicular Fluid Reveals Associations between L-Tryptophan, l-Tyrosine and Polycystic Ovary Syndrome. Biomed. Chromatogr. 2021, 35, e4993. [Google Scholar] [CrossRef]
- Gomez, G.; Beason-Held, L.L.; Bilgel, M.; An, Y.; Wong, D.F.; Studenski, S.; Ferrucci, L.; Resnick, S.M. Metabolic Syndrome and Amyloid Accumulation in the Aging Brain. J. Alzheimer’s Dis. JAD 2018, 65, 629. [Google Scholar] [CrossRef]
- Langbaum, J.B.S.; Chen, K.; Lee, W.; Reschke, C.; Bandy, D.; Fleisher, A.S.; Alexander, G.E.; Foster, N.L.; Weiner, M.W.; Koeppe, R.A.; et al. Categorical and Correlational Analyses of Baseline Fluorodeoxyglucose Positron Emission Tomography Images from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Neuroimage 2009, 45, 1107–1116. [Google Scholar] [CrossRef]
- Baker, L.D.; Cross, D.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin Resistance Is Associated with Alzheimer-like Reductions in Regional Cerebral Glucose Metabolism for Cognitively Normal Adults with Pre-diabetes or Early Type 2 Diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Abosharaf, H.A.; Elsonbaty, Y.; Tousson, E.; Mohamed, T.M. Alzheimer’s Disease-related Brain Insulin Resistance and the Prospective Therapeutic Impact of Metformin. J. Neuroendocrinol. 2024, 36, e13356. [Google Scholar] [CrossRef] [PubMed]
- Morelli, L.; Llovera, R.E.; Mathov, I.; Lue, L.-F.; Frangione, B.; Ghiso, J.; Castaño, E.M. Insulin-Degrading Enzyme in Brain Microvessels: Proteolysis of Amyloid β Vasculotropic Variants and Reduced Activity in Cerebral Amyloid Angiopathy. J. Biol. Chem. 2004, 279, 56004–56013. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Eisenhauer, P.B.; Conn, K.; Lynch, J.A.; Wells, J.M.; David Ullman, M.; McKee, A.; Thatte, H.S.; Fine, R.E. Insulin Degrading Enzyme Is Expressed in the Human Cerebrovascular Endothelium and in Cultured Human Cerebrovascular Endothelial Cells. Neurosci. Lett. 2004, 371, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Häring, H.-U. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef] [PubMed]
- Kulstad, J.J.; Green, P.S.; Cook, D.G.; Watson, G.S.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Rhoads, K.; Mehta, P.D.; et al. Differential Modulation of Plasma β-Amyloid by Insulin in Patients with Alzheimer Disease. Neurology 2006, 66, 1506–1510. [Google Scholar] [CrossRef] [PubMed]
- Leal, M.C.; Surace, E.I.; Holgado, M.P.; Ferrari, C.C.; Tarelli, R.; Pitossi, F.; Wisniewski, T.; Castaño, E.M.; Morelli, L. Notch Signaling Proteins HES-1 and Hey-1 Bind to Insulin Degrading Enzyme (IDE) Proximal Promoter and Repress Its Transcription and Activity: Implications for Cellular Aβ Metabolism. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.A.; Eltarzy, M.A.; Abdel Salam, R.M.; Ahmed, M.A.E. Liraglutide Mends Cognitive Impairment by Averting Notch Signaling Pathway Overexpression in a Rat Model of Polycystic Ovary Syndrome. Life Sci. 2021, 265, 118731. [Google Scholar] [CrossRef] [PubMed]
- van Gils, V.; Rizzo, M.; Côté, J.; Viechtbauer, W.; Fanelli, G.; Salas-Salvadó, J.; Wimberley, T.; Bulló, M.; Fernandez-Aranda, F.; Dalsgaard, S.; et al. The Association of Glucose Metabolism Measures and Diabetes Status with Alzheimer’s Disease Biomarkers of Amyloid and Tau: A Systematic Review and Meta-Analysis. Neurosci. Biobehav. Rev. 2024, 159, 105604. [Google Scholar] [CrossRef]
- Panda, S.P.; Kesharwani, A.; Singh, G.D.; Prasanth, D.; Vatchavai, B.R.; Kumari, P.V.K.; Panda, S.K.; Mallick, S.P. Impose of KNDy/GnRH Neural Circuit in PCOS, Ageing, Cancer and Alzheimer’s Disease: StAR Actions in Prevention of Neuroendocrine Dysfunction. Ageing Res. Rev. 2023, 92, 102086. [Google Scholar] [CrossRef]
- Lei, Z.M.; Rao, C.V.; Kornyei, J.L.; Licht, P.; Hiatt, E.S. Novel Expression of Human Chorionic Gonadotropin/Luteinizing Hormone Receptor Gene in Brain. Endocrinology 1993, 132, 2262–2270. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.S.B.; Campbell, R.E. Polycystic Ovary Syndrome and the Neuroendocrine Consequences of Androgen Excess. Compr. Physiol. 2022, 12, 3347–3369. [Google Scholar] [CrossRef] [PubMed]
- Moursi, M.O.; Salem, H.; Ibrahim, A.R.; Marzouk, S.; Al-Meraghi, S.; Al-Ajmi, M.; Al-Naimi, A.; Alansari, L. The Role of Anti-Mullerian Hormone and Other Correlates in Patients with Polycystic Ovary Syndrome. Gynecol. Endocrinol. 2023, 39, 2247098. [Google Scholar] [CrossRef] [PubMed]
- Piltonen, T.T.; Komsi, E.; Morin-Papunen, L.C.; Korhonen, E.; Franks, S.; Järvelin, M.-R.; Arffman, R.K.; Ollila, M.-M. AMH as Part of the Diagnostic PCOS Workup in Large Epidemiological Studies. Eur. J. Endocrinol. 2023, 188, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Barbotin, A.-L.; Mimouni, N.E.H.; Kuchcinski, G.; Lopes, R.; Viard, R.; Rasika, S.; Mazur, D.; Silva, M.S.B.; Simon, V.; Boursier, A.; et al. Hypothalamic Neuroglial Plasticity Is Regulated by Anti-Müllerian Hormone and Disrupted in Polycystic Ovary Syndrome. eBioMedicine 2023, 90, 104535. [Google Scholar] [CrossRef] [PubMed]
- Tata, B.; El Houda Mimouni, N.; Barbotin, A.-L.; Malone, S.A.; Loyens, A.; Pigny, P.; Dewailly, D.; Catteau-Jonard, S.; Sundström-Poromaa, I.; Piltonen, T.T.; et al. Elevated Prenatal Anti-Müllerian Hormone Reprograms the Fetus and Induces Polycystic Ovary Syndrome in Adulthood. Nat. Med. 2018, 24, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Short, R.A.; O’Brien, P.C.; Graff-Radford, N.R.; Bowen, R.L. Elevated Gonadotropin Levels in Patients with Alzheimer Disease. Mayo Clin. Proc. 2001, 76, 906–909. [Google Scholar] [CrossRef] [PubMed]
- Verdile, G.; Yeap, B.B.; Clarnette, R.M.; Dhaliwal, S.; Burkhardt, M.S.; Chubb, S.A.P.; De Ruyck, K.; Rodrigues, M.; Mehta, P.D.; Foster, J.K.; et al. Luteinizing Hormone Levels Are Positively Correlated with Plasma Amyloid-β Protein Levels in Elderly Men. J. Alzheimer’s Dis. 2008, 14, 201–208. [Google Scholar] [CrossRef]
- Perović, M.; Wugalter, K.; Einstein, G. Review of the Effects of Polycystic Ovary Syndrome on Cognition: Looking beyond the Androgen Hypothesis. Front. Neuroendocrinol. 2022, 67, 101038. [Google Scholar] [CrossRef]
- Lai, W.; Li, X.; Zhu, H.; Zhu, X.; Tan, H.; Feng, P.; Chen, L.; Luo, C. Plasma Luteinizing Hormone Level Affects the Brain Activity of Patients with Polycystic Ovary Syndrome. Psychoneuroendocrinology 2020, 112, 104535. [Google Scholar] [CrossRef]
- Bowen, R.L.; Perry, G.; Xiong, C.; Smith, M.A.; Atwood, C.S. A Clinical Study of Lupron Depot in the Treatment of Women with Alzheimer’s Disease: Preservation of Cognitive Function in Patients Taking an Acetylcholinesterase Inhibitor and Treated with High Dose Lupron over 48 Weeks. J. Alzheimer’s Dis. 2015, 44, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Casadesus, G.; Webber, K.M.; Atwood, C.S.; Pappolla, M.A.; Perry, G.; Bowen, R.L.; Smith, M.A. Luteinizing Hormone Modulates Cognition and Amyloid-Beta Deposition in Alzheimer APP Transgenic Mice. Biochim. Biophys. Acta 2006, 1762, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.A.; Udiawar, M.; Berlot, R.; Jones, D.K.; O’Sullivan, M.J. White Matter Microstructure and Cognitive Function in Young Women with Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2016, 101, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.R.; Stetson, C.L.; Slominski, A.T.; Pruitt, K. Role of the Steroidogenic Acute Regulatory Protein in Health and Disease. Endocrine 2016, 51, 7–21. [Google Scholar] [CrossRef]
- Manna, P.R.; Reddy, A.P.; Pradeepkiran, J.A.; Kshirsagar, S.; Reddy, P.H. Regulation of Retinoid Mediated StAR Transcription and Steroidogenesis in Hippocampal Neuronal Cells: Implications for StAR in Protecting Alzheimer’s Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2023, 1869, 166596. [Google Scholar] [CrossRef]
- Zhou, J.; Singh, N.; Galske, J.; Hudobenko, J.; Hu, X.; Yan, R. BACE1 Regulates Expression of Clusterin in Astrocytes for Enhancing Clearance of β-Amyloid Peptides. Mol. Neurodegener. 2023, 18, 31. [Google Scholar] [CrossRef]
- Meethal, S.V.; Smith, M.A.; Bowen, R.L.; Atwood, C.S. The Gonadotropin Connection in Alzheimer’s Disease. Endocrine 2005, 26, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Ding, X.; Zhu, J. Kisspeptin and Polycystic Ovary Syndrome. Front. Endocrinol. 2019, 10, 298. [Google Scholar] [CrossRef]
- Milton, N.G.N.; Chilumuri, A.; Rocha-Ferreira, E.; Nercessian, A.N.; Ashioti, M. Kisspeptin Prevention of Amyloid-β Peptide Neurotoxicity in Vitro. ACS Chem. Neurosci. 2012, 3, 706–719. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Brew, B.J.; Noonan, C.E.; Takikawa, O.; Cullen, K.M. Indoleamine 2,3 Dioxygenase and Quinolinic Acid Immunoreactivity in Alzheimer’s Disease Hippocampus. Neuropathol. Appl. Neurobiol. 2005, 31, 395–404. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Brew, B.J. Implications of the Kynurenine Pathway and Quinolinic Acid in Alzheimer’s Disease. Redox Rep. 2002, 7, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Ting, K.; Cullen, K.M.; Braidy, N.; Brew, B.J.; Guillemin, G.J. The Excitotoxin Quinolinic Acid Induces Tau Phosphorylation in Human Neurons. PLoS ONE 2009, 4, e6344. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Goozee, K.; Lim, C.K.; James, I.; Shen, K.; Jacobs, K.R.; Sohrabi, H.R.; Shah, T.; Asih, P.R.; Dave, P.; et al. Alterations in Serum Kynurenine Pathway Metabolites in Individuals with High Neocortical Amyloid-β Load: A Pilot Study. Sci. Rep. 2018, 8, 8008. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Zetterberg, H.; Goozee, K.; Lim, C.K.; Jacobs, K.R.; Ashton, N.J.; Hye, A.; Pedrini, S.; Sohrabi, H.R.; Shah, T.; et al. Plasma Neurofilament Light Chain and Amyloid-β Are Associated with the Kynurenine Pathway Metabolites in Preclinical Alzheimer’s Disease. J. Neuroinflam. 2019, 16, 186. [Google Scholar] [CrossRef] [PubMed]
- Meier-Stephenson, F.S.; Meier-Stephenson, V.C.; Carter, M.D.; Meek, A.R.; Wang, Y.; Pan, L.; Chen, Q.; Jacobo, S.; Wu, F.; Lu, E.; et al. Alzheimer’s Disease as an Autoimmune Disorder of Innate Immunity Endogenously Modulated by Tryptophan Metabolites. Alzheimer’s Dement. 2022, 8, e12283. [Google Scholar] [CrossRef] [PubMed]
- Stover, K.R.; Stafford, P.M.; Damian, A.C.; Pasangulapati, J.P.; Goodwin-Tindall, J.; López Vásquez, L.M.; Lee, S.; Yang, S.-P.; Reed, M.A.; Barden, C.J.; et al. Development and Optimization of a Target Engagement Model of Brain IDO Inhibition for Alzheimer’s Disease. Curr. Alzheimer Res. 2023, 20, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Shi, L.; He, Z.N.T.; Kuang, C.; Han, T.; Yang, Q. The Protective Effect of IDO1 Inhibition in Aβ-Treated Neurons and APP/PS1 Mice. Am. J. Alzheimer’s Dis. Other Demen. 2023, 38, 15333175231214861. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, B.S.; Inam, M.E.; Enduru, N.; Quevedo, J.; Zhao, Z. The Kynurenine Pathway in Alzheimer’s Disease: A Meta-Analysis of Central and Peripheral Levels. Braz. J. Psychiatry 2023, 45, 286–297. [Google Scholar] [CrossRef]
- Jovanovic, F.; Sudhakar, A.; Knezevic, N.N. The kynurenine pathway and polycystic ovary syndrome: Inflammation as a common denominator. Int. J. Tryptophan Res. 2022, 15, 11786469221099214. [Google Scholar] [CrossRef]
- García-Sáenz, M.R.; Ferreira-Hermosillo, A.; Lobaton-Ginsberg, M. Proinflammatory cytokines in polycystic ovarian syndrome. Rev. Med. Inst. Mex. Seguro Soc. 2022, 60, 569–576. [Google Scholar]
- Moreno-Asso, A.; Altıntaş, A.; McIlvenna, L.C.; Patten, R.K.; Botella, J.; McAinch, A.J.; Rodgers, R.J.; Barrès, R.; Stepto, N.K. Non-Cell Autonomous Mechanisms Control Mitochondrial Gene Dysregulation in Polycystic Ovary Syndrome. J. Mol. Endocrinol. 2021, 68, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bao, Y.; Zhou, X.; Zheng, L. Polycystic Ovary Syndrome and Mitochondrial Dysfunction. Reprod. Biol. Endocrinol. 2019, 17, 67. [Google Scholar] [CrossRef]
- Melhem, N.; Murata, S.; Marengo, L.; Goodfriend, E.; Zhong, Y.; Deal, M.; Brummit, B.; Krancevich, K.; Kaufman, B.; Douaihy, A.; et al. Activation of the Kynurenine Pathway and Mitochondrial Dysfunction in Suicidal Youth. Biol. Psychiatry 2021, 89, S81–S82. [Google Scholar] [CrossRef]
- Castellano-Gonzalez, G.; Jacobs, K.R.; Don, E.; Cole, N.J.; Adams, S.; Lim, C.K.; Lovejoy, D.B.; Guillemin, G.J. Kynurenine 3-Monooxygenase Activity in Human Primary Neurons and Effect on Cellular Bioenergetics Identifies New Neurotoxic Mechanisms. Neurotox. Res. 2019, 35, 530–541. [Google Scholar] [CrossRef]
- Schwarz, M.J.; Guillemin, G.J.; Teipel, S.J.; Buerger, K.; Hampel, H. Increased 3-Hydroxykynurenine Serum Concentrations Differentiate Alzheimer’s Disease Patients from Controls. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 345–352. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Sehgal, A.; Singh, S.; Bhatia, S.; Al-Harrasi, A.; Zengin, G.; Bumbu, A.G.; Andronie-Cioara, F.L.; Nechifor, A.C.; et al. The Footprint of Kynurenine Pathway in Neurodegeneration: Janus-Faced Role in Parkinson’s Disorder and Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 6737. [Google Scholar] [CrossRef]
- Sas, K.; Robotka, H.; Toldi, J.; Vécsei, L. Mitochondria, Metabolic Disturbances, Oxidative Stress and the Kynurenine System, with Focus on Neurodegenerative Disorders. J. Neurol. Sci. 2007, 257, 221–239. [Google Scholar] [CrossRef] [PubMed]
- Sabuncu, T.; Vural, H.; Harma, M.; Harma, M. Oxidative Stress in Polycystic Ovary Syndrome and Its Contribution to the Risk of Cardiovascular Disease☆. Clin. Biochem. 2001, 34, 407–413. [Google Scholar] [CrossRef]
- Ye, M.; Hu, B.; Shi, W.; Guo, F.; Xu, C.; Li, S. Mitochondrial DNA 4977 Bp Deletion in Peripheral Blood Is Associated with Polycystic Ovary Syndrome. Front. Endocrinol. 2021, 12, 675581. [Google Scholar] [CrossRef]
- Jang, S.; Park, S.-H. Antidiabetic Drug Metformin Protects Neuronal Cells against Quinolinic Acid-Induced Excitotoxicity by Decreasing Intracellular Calcium. Chonnam Med. J. 2018, 54, 24–30. [Google Scholar] [CrossRef]
- Kickstein, E.; Krauss, S.; Thornhill, P.; Rutschow, D.; Zeller, R.; Sharkey, J.; Williamson, R.; Fuchs, M.; Köhler, A.; Glossmann, H.; et al. Biguanide Metformin Acts on Tau Phosphorylation via mTOR/Protein Phosphatase 2A (PP2A) Signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 21830–21835. [Google Scholar] [CrossRef]
- Dangla-Valls, A.; Molinuevo, J.L.; Altirriba, J.; Sánchez-Valle, R.; Alcolea, D.; Fortea, J.; Rami, L.; Balasa, M.; Muñoz-García, C.; Ezquerra, M.; et al. CSF microRNA Profiling in Alzheimer’s Disease: A Screening and Validation Study. Mol. Neurobiol. 2017, 54, 6647–6654. [Google Scholar] [CrossRef]
- Walker, A.K.; Wing, E.E.; Banks, W.A.; Dantzer, R. Leucine Competes with Kynurenine for Blood-to-Brain Transport and Prevents Lipopolysaccharide-Induced Depression-like Behavior in Mice. Mol. Psychiatry 2019, 24, 1523–1532. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Method | Finding | Model | Putative AD-PCOS Link | Reference |
|---|---|---|---|---|
| PiB-PET | HOMA positively correlates with Aβ accumulation in AD-related brain areas | AD patients | IR present in PCOS might promote Aβ aggregation | [115,116] |
| 18FDG-PET | Glucose hypometabolism in AD-related brain areas when compared to healthy subjects | PCOS patients | PCOS is a risk factor for cellular energy efficiency in AD-related areas | [117] |
| 18FDG-PET | Glucose hypometabolism in AD-related brain areas positively correlates with insulin and glucose concentration | Healthy subjects | Hyperinsulinemia worsens cellular energy efficiency in AD-related areas. | [118] |
| Laboratory | Dose-dependent negative correlation between usage of antidiabetic medication and odds of dementia | Diabetic patients | IR and metabolic syndrome increase the risk of dementia | [119] |
| PiB-PET | LH and FSH positively correlates with amyloid burden | Older-age subjects | Excessive LH secretion present in PCOS might promote Aβ aggregation | [120] |
| Laboratory | Treatment with bHCG promotes Aβ aggregation | Ovariectomized rats | LH might have amyloidogenic properties | [121] |
| Laboratory | StAR protein lowered expression in AD | AD patients | Lowered StAR expression leads to increased Aβ aggregation; expression of this protein is lowered also in PCOS | [122,123] |
| Laboratory | Kynurenine pathway overactivation present in both PCOS and AD | PCOS and AD patients | [124,125,126,127,128] | |
| Laboratory | 3-HK levels are positively correlated with HOMA IR | Diabetic patients | Kynurenine dysregulation is linked to IR | [87] |
| Laboratory | Amyloid precursor protein concentration is increased in PCOS group compared to healthy subjects | PCOS patients | In PCOS Aβ aggregation might be more likely | [11] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobczuk, J.; Paczkowska, K.; Andrusiów, S.; Bolanowski, M.; Daroszewski, J. Are Women with Polycystic Ovary Syndrome at Increased Risk of Alzheimer Disease? Lessons from Insulin Resistance, Tryptophan and Gonadotropin Disturbances and Their Link with Amyloid-Beta Aggregation. Biomolecules 2024, 14, 918. https://doi.org/10.3390/biom14080918
Sobczuk J, Paczkowska K, Andrusiów S, Bolanowski M, Daroszewski J. Are Women with Polycystic Ovary Syndrome at Increased Risk of Alzheimer Disease? Lessons from Insulin Resistance, Tryptophan and Gonadotropin Disturbances and Their Link with Amyloid-Beta Aggregation. Biomolecules. 2024; 14(8):918. https://doi.org/10.3390/biom14080918
Chicago/Turabian StyleSobczuk, Joachim, Katarzyna Paczkowska, Szymon Andrusiów, Marek Bolanowski, and Jacek Daroszewski. 2024. "Are Women with Polycystic Ovary Syndrome at Increased Risk of Alzheimer Disease? Lessons from Insulin Resistance, Tryptophan and Gonadotropin Disturbances and Their Link with Amyloid-Beta Aggregation" Biomolecules 14, no. 8: 918. https://doi.org/10.3390/biom14080918