
Endothelial Cell Dysfunction Due to Molecules Secreted by Macrophages in Sepsis
 ,
,
Abstract
:1. Introduction
2. Basic Functions of Macrophages and Endothelial Cells
3. Macrophages Secrete a Variety of Substances to Wake Up Endothelial Cells
3.1. Macrophages Increase Endothelial Cell Adhesion and Ability to Recruit Leukocytes
3.2. Macrophages Increase Endothelial Cell Permeability in Various Ways
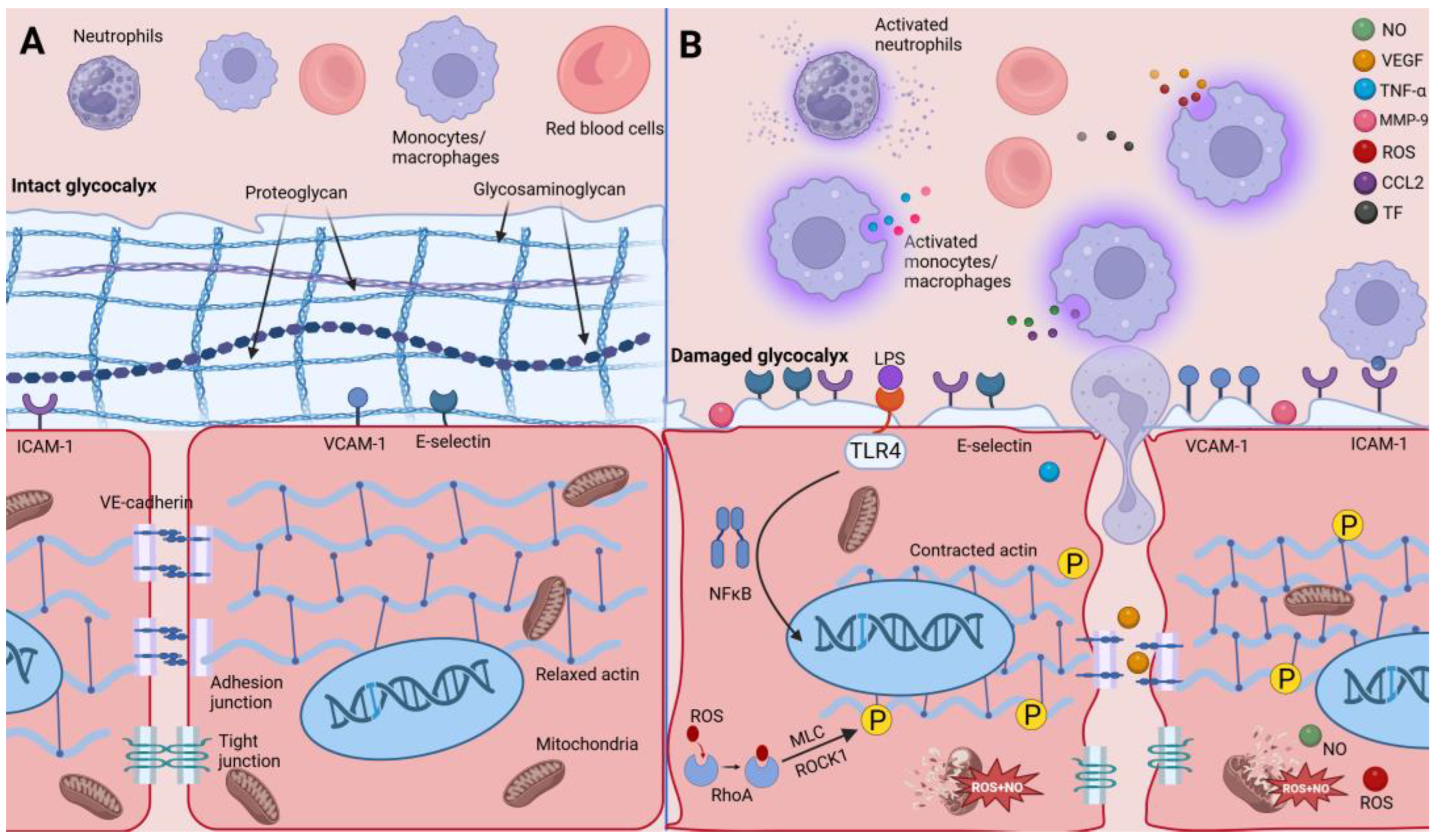
3.2.1. Injured Gatekeepers—Glycocalyx of Endothelial Cells
3.2.2. Impaired Endothelial Cell Junctions
3.2.3. Impaired Endothelial Cytoskeleton
3.2.4. Enhanced Endothelial Cell Apoptosis
3.3. Macrophages Enhance Endothelial Cell Procoagulant Capacity

{kind=link}
| Molecules Secreted by Macrophages | Affected Functions | Possible Mechanisms | References |
|---|---|---|---|
| CCL2 | adhesion | Positive feedback effect with endothelial cells | [21,23,26] |
| TNF-α | adhesion | Activates ZIPK/ NF-κB | [31] |
| permeability | Activates heparanase; upregulates MMP-9 | [50,51] | |
| coagulability | Downregulates eNOS expression | [103,104] | |
| ROS | permeability | Upregulates Nox2; activates RhoA/ROCK1/MLC | [59,60,82,85] |
| VEGF | permeability | Promotes FA kinase phosphorylation of VE-cadherin | [74] |
| MMP-9 | permeability | Regulates macrophage polarization | [54] |
| NO | permeability | Impairs mitochondrial energy production | [96] |
| TF | coagulability | Initiate exogenous coagulation pathway | [100] |
4. Potential Therapeutic Target—Modulation of Macrophage Polarization
4.1. Regulation of Macrophage Polarization by miRNA
4.2. Regulation of Macrophage Polarization by Medicines
| Substance of Intervention | M1-Type Macrophage | M2-Type Macrophage | Potential Signaling Pathwayor Metabolic Pathway | Reference | Year |
|---|---|---|---|---|---|
| miR-164a | ↓ | TLR4-NF-κB | [119] | 2015 | |
| miR-21 | ↑ | CSF1-R | [122] | 2015 | |
| ↑ | STAT3 | [123] | 2015 | ||
| miR-23a-3p | ↑ | PLK1-STAT1/STAT3 | [124] | 2022 | |
| Luteolin | ↓ | p-STAT3 | [125] | 2020 | |
| ↑ | p-STAT6 | ||||
| TPX | ↓ | ↑ | p65 and NF-κB | [126] | 2018 |
| Canagliflozin | ↓ | ↑ | glucose metabolism | [127] | 2019 |
| Liraglutide | [129] | 2022 |
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.-L.; Sakr, Y.; Sprung, C.L.; Ranieri, V.M.; Reinhart, K.; Gerlach, H.; Moreno, R.; Carlet, J.; Le Gall, J.-R.; Payen, D.; et al. Sepsis in European Intensive Care Units: Results of the SOAP Study. Crit. Care Med. 2006, 34, 344–353. [Google Scholar] [CrossRef]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, Regional, and National Sepsis Incidence and Mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Joffre, J.; Hellman, J.; Ince, C.; Ait-Oufella, H. Endothelial Responses in Sepsis. Am. J. Resp. Crit. Care 2020, 202, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and Functions of Tissue Macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef]
- Sica, A.; Erreni, M.; Allavena, P.; Porta, C. Macrophage Polarization in Pathology. Cell. Mol. Life Sci. 2015, 72, 4111–4126. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Haemmig, S.; Chen, J.; McCoy, M.; Cheng, H.S.; Zhou, H.; Pérez-Cremades, D.; Cheng, X.; Sun, X.; Haneo-Mejia, J.; et al. Endothelial Cell-Specific Deletion of a microRNA Accelerates Atherosclerosis. Atherosclerosis 2022, 350, 9–18. [Google Scholar] [CrossRef]
- Theret, M.; Mounier, R.; Rossi, F. The Origins and Non-Canonical Functions of Macrophages in Development and Regeneration. Development 2019, 146, dev156000. [Google Scholar] [CrossRef]
- Stein, M.; Keshav, S.; Harris, N.; Gordon, S. Interleukin 4 Potently Enhances Murine Macrophage Mannose Receptor Activity: A Marker of Alternative Immunologic Macrophage Activation. J. Exp. Med. 1992, 176, 287–292. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1-M2 Polarization Balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M.; Van Der Poll, T. Endothelial Barrier Dysfunction in Septic Shock. J. Intern. Med. 2015, 277, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute Respiratory Distress Syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, K.; Zieger, B. Endothelial Cells and Coagulation. Cell Tissue Res. 2022, 387, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Zoulikha, M.; Xiao, Q.; Boafo, G.F.; Sallam, M.A.; Chen, Z.; He, W. Pulmonary Delivery of siRNA against Acute Lung Injury/Acute Respiratory Distress Syndrome. Acta Pharm. Sin. B 2022, 12, 600–620. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Yin, J.; Zheng, Z.; Li, L.; Feng, X. Endothelial Cell Dynamics in Sepsis-Induced Acute Lung Injury and Acute Respiratory Distress Syndrome: Pathogenesis and Therapeutic Implications. Cell Commun. Signal. 2024, 22, 241. [Google Scholar] [CrossRef]
- Goldenberg, N.M.; Steinberg, B.E.; Slutsky, A.S.; Lee, W.L. Broken Barriers: A New Take on Sepsis Pathogenesis. Sci. Transl. Med. 2011, 3, 88ps25. [Google Scholar] [CrossRef] [PubMed]
- Hasan, D.; Chalouhi, N.; Jabbour, P.; Hashimoto, T. Macrophage Imbalance (M1 vs. M2) and Upregulation of Mast Cells in Wall of Ruptured Human Cerebral Aneurysms: Preliminary Results. J. Neuroinflamm. 2012, 9, 222. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving Functions of Endothelial Cells in Inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Hu, J.; Chen, R.; An, J.; Wang, Y.; Liang, M.; Huang, K. Dauricine Attenuates Vascular Endothelial Inflammation Through Inhibiting NF-κB Pathway. Front. Pharmacol. 2021, 12, 758962. [Google Scholar] [CrossRef]
- Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines 2017, 5, 34. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Singh, S.; Anshita, D.; Ravichandiran, V. MCP-1: Function, Regulation, and Involvement in Disease. Int. Immunopharmacol. 2021, 101, 107598. [Google Scholar] [CrossRef]
- Tacke, F.; Zimmermann, H.W. Macrophage Heterogeneity in Liver Injury and Fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver Macrophages in Tissue Homeostasis and Disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Dal-Secco, D.; Wang, J.; Zeng, Z.; Kolaczkowska, E.; Wong, C.H.Y.; Petri, B.; Ransohoff, R.M.; Charo, I.F.; Jenne, C.N.; Kubes, P. A Dynamic Spectrum of Monocytes Arising from the in Situ Reprogramming of CCR2+ Monocytes at a Site of Sterile Injury. J. Exp. Med. 2015, 212, 447–456. [Google Scholar] [CrossRef]
- Cappenberg, A.; Kardell, M.; Zarbock, A. Selectin-Mediated Signaling—Shedding Light on the Regulation of Integrin Activity in Neutrophils. Cells 2022, 11, 1310. [Google Scholar] [CrossRef]
- Lammel, C.; Zwirchmayr, J.; Seigner, J.; Rollinger, J.M.; De Martin, R. Peucedanum Ostruthium Inhibits E-Selectin and VCAM-1 Expression in Endothelial Cells through Interference with NF-κB Signaling. Biomolecules 2020, 10, 1215. [Google Scholar] [CrossRef]
- Yao, L.; Setiadi, H.; Xia, L.; Laszik, Z.; Taylor, F.B.; McEver, R.P. Divergent Inducible Expression of P-Selectin and E-Selectin in Mice and Primates. Blood 1999, 94, 3820–3828. [Google Scholar] [CrossRef]
- Zhang, F.; Yu, W.; Hargrove, J.L.; Greenspan, P.; Dean, R.G.; Taylor, E.W.; Hartle, D.K. Inhibition of TNF-α Induced ICAM-1, VCAM-1 and E-Selectin Expression by Selenium. Atherosclerosis 2002, 161, 381–386. [Google Scholar] [CrossRef]
- Zeng, W.; Sun, Z.; Ma, T.; Song, X.; Li, S.; Zhang, Q.; Yuan, W.; Li, J.; Liu, L.; Zhu, M.; et al. Elevated ZIPK Is Required for TNF-α-Induced Cell Adhesion Molecule Expression and Leucocyte Adhesion in Endothelial Cells. Acta Biochim. Biophys. Sin. 2021, 53, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tang, Y.; Tang, J.; Liu, X.; Zi, S.; Li, S.; Chen, H.; Liu, A.; Huang, W.; Xie, J.; et al. Endothelial Cell-derived Extracellular Vesicles Expressing Surface VCAM1 Promote Sepsis-related Acute Lung Injury by Targeting and Reprogramming Monocytes. J. Extracell. Vesicles 2024, 13, e12423. [Google Scholar] [CrossRef] [PubMed]
- Sackstein, R. Glycosyltransferase-Programmed Stereosubstitution (GPS) to Create HCELL: Engineering a Roadmap for Cell Migration. Immunol. Rev. 2009, 230, 51–74. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the Site of Inflammation: The Leukocyte Adhesion Cascade Updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Novel Insights into Leukocyte Extravasation: Current Opinion in Hematology. Available online: https://journals.lww.com/co-hematology/Abstract/2012/05000/Novel_insights_into_leukocyte_extravasation.13.aspx (accessed on 20 May 2024).
- Jacobs, P.P.; Sackstein, R. CD44 and HCELL: Preventing Hematogenous Metastasis at Step 1. FEBS Lett. 2011, 585, 3148–3158. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Ding, Q.; Chen, S.; Lü, S.; Zhang, Y.; Long, M. E-Selectin Negatively Regulates Polymorphonuclear Neutrophil Transmigration through Altered Endothelial Junction Integrity. FASEB J. 2021, 35, e21521. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.-L.; Lefrant, J.-Y.; Kotfis, K.; Nanchal, R.; Martin-Loeches, I.; Wittebole, X.; Sakka, S.G.; Pickkers, P.; Moreno, R.; Sakr, Y.; et al. Comparison of European ICU Patients in 2012 (ICON) versus 2002 (SOAP). Intensive Care Med. 2018, 44, 337–344. [Google Scholar] [CrossRef] [PubMed]
- McNicholas, B.A.; Rooney, G.M.; Laffey, J.G. Lessons to Learn from Epidemiologic Studies in ARDS. Curr. Opin. Crit. Care 2018, 24, 41–48. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Chen, C.-S.; Yiang, G.-T.; Cheng, Y.-L.; Yong, S.-B.; Wu, M.-Y.; Li, C.-J. New Insights into the Immune Molecular Regulation of the Pathogenesis of Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2018, 19, 588. [Google Scholar] [CrossRef]
- Morphological Aspects of Extracellular Polysaccharides—H. Stanley Bennett. 1963. Available online: https://journals.sagepub.com/doi/10.1177/11.1.14 (accessed on 20 May 2024).
- Reitsma, S.; Slaaf, D.W.; Vink, H.; Van Zandvoort, M.A.M.J.; Oude Egbrink, M.G.A. The Endothelial Glycocalyx: Composition, Functions, and Visualization. Pflug. Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef]
- Betteridge, K.B.; Arkill, K.P.; Neal, C.R.; Harper, S.J.; Foster, R.R.; Satchell, S.C.; Bates, D.O.; Salmon, A.H.J. Sialic Acids Regulate Microvessel Permeability, Revealed by Novel in Vivo Studies of Endothelial Glycocalyx Structure and Function. J. Physiol. 2017, 595, 5015–5035. [Google Scholar] [CrossRef]
- Yuan, S.Y.; Rigor, R.R. Structure and Function of Exchange Microvessels. In Regulation of Endothelial Barrier Function; Morgan & Claypool Life Sciences: Willston, VT, USA, 2010. [Google Scholar]
- Potter, D.R.; Jiang, J.; Damiano, E.R. The Recovery Time Course of the Endothelial Cell Glycocalyx in Vivo and Its Implications in Vitro. Circ. Res. 2009, 104, 1318–1325. [Google Scholar] [CrossRef]
- Giantsos-Adams, K.M.; Koo, A.J.-A.; Song, S.; Sakai, J.; Sankaran, J.; Shin, J.H.; Garcia-Cardena, G.; Dewey, C.F. Heparan Sulfate Regrowth Profiles Under Laminar Shear Flow Following Enzymatic Degradation. Cell. Mol. Bioeng. 2013, 6, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Haeger, S.M.; Suflita, M.A.; Zhang, F.; Dailey, K.L.; Colbert, J.F.; Ford, J.A.; Picon, M.A.; Stearman, R.S.; Lin, L.; et al. Fibroblast Growth Factor Signaling Mediates Pulmonary Endothelial Glycocalyx Reconstitution. Am. J. Respir. Cell Mol. Biol. 2017, 56, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Xu, S.; Zhou, Z.; Wang, F.; Mao, L.; Li, H.; Wu, C.; Wang, J.; Huang, Y.; Li, D.; et al. Fibroblast Growth Factor-2 Alleviates the Capillary Leakage and Inflammation in Sepsis. Mol. Med. 2020, 26, 108. [Google Scholar] [CrossRef] [PubMed]
- Nishida, R.; Suzuki, D.; Akimoto, Y.; Matsubara, S.; Hayakawa, J.; Ushiyama, A.; Sasa, K.; Miyamoto, Y.; Iijima, T.; Kamijo, R. Exploring the Pathophysiological Mechanism of Interstitial Edema Focusing on the Role of Macrophages and Their Interaction with the Glycocalyx. J. Oral Biosci. 2023, 65, 111–118. [Google Scholar] [CrossRef]
- Schmidt, E.P.; Yang, Y.; Janssen, W.J.; Gandjeva, A.; Perez, M.J.; Barthel, L.; Zemans, R.L.; Bowman, J.C.; Koyanagi, D.E.; Yunt, Z.X.; et al. The Pulmonary Endothelial Glycocalyx Regulates Neutrophil Adhesion and Lung Injury during Experimental Sepsis. Nat. Med. 2012, 18, 1217–1223. [Google Scholar] [CrossRef]
- Ramnath, R.; Foster, R.R.; Qiu, Y.; Cope, G.; Butler, M.J.; Salmon, A.H.; Mathieson, P.W.; Coward, R.J.; Welsh, G.I.; Satchell, S.C. Matrix Metalloproteinase 9-Mediated Shedding of Syndecan 4 in Response to Tumor Necrosis Factor α: A Contributor to Endothelial Cell Glycocalyx Dysfunction. FASEB J. 2014, 28, 4686–4699. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, J.-T.; Pan, Y.; Liu, X.-F.; Xu, J.-W.; Cui, W.-J.; Qiao, X.-R.; Dong, L. Syndecan-1 Shedding by Matrix Metalloproteinase-9 Signaling Regulates Alveolar Epithelial Tight Junction in Lipopolysaccharide-Induced Early Acute Lung Injury. J. Inflamm. Res. 2021, 14, 5801–5816. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Lei, J.; Lin, D.; Yang, C.; Yan, L. Changes in Biological Behaviors of Rat Dermal Fibroblasts Induced by High Expression of MMP9. World J. Emerg. Med. 2014, 5, 139–143. [Google Scholar] [CrossRef]
- Tong, Y.; Yu, Z.; Chen, Z.; Zhang, R.; Ding, X.; Yang, X.; Niu, X.; Li, M.; Zhang, L.; Billiar, T.R.; et al. The HIV Protease Inhibitor Saquinavir Attenuates Sepsis-Induced Acute Lung Injury and Promotes M2 Macrophage Polarization via Targeting Matrix Metalloproteinase-9. Cell Death Dis. 2021, 12, 67. [Google Scholar] [CrossRef]
- Herb, M.; Schramm, M. Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef]
- Gluschko, A.; Herb, M.; Wiegmann, K.; Krut, O.; Neiss, W.F.; Utermöhlen, O.; Krönke, M.; Schramm, M. The Β2 Integrin Mac-1 Induces Protective LC3-Associated Phagocytosis of Listeria Monocytogenes. Cell Host Microbe 2018, 23, 324–337. [Google Scholar] [CrossRef]
- Yang, S.; Lian, G. ROS and Diseases: Role in Metabolism and Energy Supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef]
- Van Golen, R.F.; Reiniers, M.J.; Vrisekoop, N.; Zuurbier, C.J.; Olthof, P.B.; Van Rheenen, J.; Van Gulik, T.M.; Parsons, B.J.; Heger, M. The Mechanisms and Physiological Relevance of Glycocalyx Degradation in Hepatic Ischemia/Reperfusion Injury. Antioxid. Redox Sign 2014, 21, 1098–1118. [Google Scholar] [CrossRef]
- Yu, H.; Kalogeris, T.; Korthuis, R.J. Reactive Species-Induced Microvascular Dysfunction in Ischemia/Reperfusion. Free Radic. Biol. Med. 2019, 135, 182–197. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion Injury and Reactive Oxygen Species: The Evolution of a Concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Petry, A.; Djordjevic, T.; Weitnauer, M.; Kietzmann, T.; Hess, J.; Görlach, A. NOX2 and NOX4 Mediate Proliferative Response in Endothelial Cells. Antioxid. Redox Sign 2006, 8, 1473–1484. [Google Scholar] [CrossRef]
- Wang, G.; Anrather, J.; Glass, M.J.; Tarsitano, M.J.; Zhou, P.; Frys, K.A.; Pickel, V.M.; Iadecola, C. Nox2, Ca2+, and Protein Kinase C Play a Role in Angiotensin II-Induced Free Radical Production in Nucleus Tractus Solitarius. Hypertension 2006, 48, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, L.A.; Brunn, G.J.; Platt, J.L. Heparanase, a Potential Regulator of Cell-Matrix Interactions. Trends Biochem. Sci. 2000, 25, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Komarova, Y.; Malik, A.B. Regulation of Endothelial Permeability via Paracellular and Transcellular Transport Pathways. Annu. Rev. Physiol. 2010, 72, 463–493. [Google Scholar] [CrossRef] [PubMed]
- Simionescu, M.; Simionescu, N.; Palade, G.E. Segmental Differentiations of Cell Junctions in the Vascular Endothelium. The Microvasculature. J. Cell Biol. 1975, 67, 863–885. [Google Scholar] [CrossRef] [PubMed]
- Gotsch, U.; Borges, E.; Bosse, R.; Böggemeyer, E.; Simon, M.; Mossmann, H.; Vestweber, D. VE-Cadherin Antibody Accelerates Neutrophil Recruitment in Vivo. J. Cell Sci. 1997, 110, 583–588. [Google Scholar] [CrossRef]
- Xiong, M.; Elson, G.; Legarda, D.; Leibovich, S.J. Production of Vascular Endothelial Growth Factor by Murine Macrophages: Regulation by Hypoxia, Lactate, and the Inducible Nitric Oxide Synthase Pathway. Am. J. Pathol. 1998, 153, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Sugishita, Y.; Shimizu, T.; Yao, A.; Kinugawa, K.-I.; Nojiri, T.; Harada, K.; Matsui, H.; Nagai, R.; Takahashi, T. Lipopolysaccharide Augments Expression and Secretion of Vascular Endothelial Growth Factor in Rat Ventricular Myocytes. Biochem. Biophys. Res. Commun. 2000, 268, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Nolan, A.; Weiden, M.D.; Thurston, G.; Gold, J.A. Vascular Endothelial Growth Factor Blockade Reduces Plasma Cytokines in a Murine Model of Polymicrobial Sepsis. Inflammation 2004, 28, 271–278. [Google Scholar] [CrossRef]
- Gavard, J.; Gutkind, J.S. VEGF Controls Endothelial-Cell Permeability by Promoting the Beta-Arrestin-Dependent Endocytosis of VE-Cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Garner, J.; Buckley, K.M.; Vincent, P.A.; Chiasson, C.M.; Dejana, E.; Faundez, V.; Kowalczyk, A.P. P120-Catenin Regulates Clathrin-Dependent Endocytosis of VE-Cadherin. Mol. Biol. Cell 2005, 16, 5141–5151. [Google Scholar] [CrossRef]
- Chen, X.L.; Nam, J.-O.; Jean, C.; Lawson, C.; Walsh, C.T.; Goka, E.; Lim, S.-T.; Tomar, A.; Tancioni, I.; Uryu, S.; et al. VEGF-Induced Vascular Permeability Is Mediated by FAK. Dev. Cell 2012, 22, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Jean, C.; Chen, X.L.; Nam, J.-O.; Tancioni, I.; Uryu, S.; Lawson, C.; Ward, K.K.; Walsh, C.T.; Miller, N.L.G.; Ghassemian, M.; et al. Inhibition of Endothelial FAK Activity Prevents Tumor Metastasis by Enhancing Barrier Function. J. Cell Biol. 2014, 204, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Birukov, K.G. Rho and Reactive Oxygen Species at Crossroads of Endothelial Permeability and Inflammation. Antioxid. Redox Sign 2019, 31, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Wojciak-Stothard, B.; Ridley, A.J. Rho GTPases and the Regulation of Endothelial Permeability. Vasc. Pharmacol. 2002, 39, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Spindler, V.; Schlegel, N.; Waschke, J. Role of GTPases in Control of Microvascular Permeability. Cardiovasc. Res. 2010, 87, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Hall, A. The Small GTP-Binding Protein Rho Regulates the Assembly of Focal Adhesions and Actin Stress Fibers in Response to Growth Factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- You, L.-J.; Li, P.-W.; Zhang, W.-W.; Feng, M.-F.; Zhao, W.-P.; Hou, H.-M.; Piao, X.-M.; Wang, L.-B.; Zhang, Y. Schisandrin A Ameliorates Increased Pulmonary Capillary Endothelial Permeability Accompanied with Sepsis through Inhibition of RhoA/ROCK1/MLC Pathways. Int. Immunopharmacol. 2023, 118, 110124. [Google Scholar] [CrossRef]
- Petrache, I.; Crow, M.T.; Neuss, M.; Garcia, J.G.N. Central Involvement of Rho Family GTPases in TNF-Alpha-Mediated Bovine Pulmonary Endothelial Cell Apoptosis. Biochem. Biophys. Res. Commun. 2003, 306, 244–249. [Google Scholar] [CrossRef]
- Hinshaw, D.B.; Burger, J.M.; Armstrong, B.C.; Hyslop, P.A. Mechanism of Endothelial Cell Shape Change in Oxidant Injury. J. Surg. Res. 1989, 46, 339–349. [Google Scholar] [CrossRef]
- Zhao, Y.; Davis, H.W. Hydrogen Peroxide-Induced Cytoskeletal Rearrangement in Cultured Pulmonary Endothelial Cells. J. Cell Physiol. 1998, 174, 370–379. [Google Scholar] [CrossRef]
- Hastie, L.E.; Patton, W.F.; Hechtman, H.B.; Shepro, D. H2O2-Induced Filamin Redistribution in Endothelial Cells Is Modulated by the Cyclic AMP-Dependent Protein Kinase Pathway. J. Cell Physiol. 1997, 172, 373–381. [Google Scholar] [CrossRef]
- Holman, R.G.; Maier, R.V. Oxidant-Induced Endothelial Leak Correlates with Decreased Cellular Energy Levels. Am. Rev. Respir. Dis. 1990, 141, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.; Campbell, S.L. Mechanism of Redox-Mediated Guanine Nucleotide Exchange on Redox-Active Rho GTPases. J. Biol. Chem. 2005, 280, 31003–31010. [Google Scholar] [CrossRef] [PubMed]
- Kratzer, E.; Tian, Y.; Sarich, N.; Wu, T.; Meliton, A.; Leff, A.; Birukova, A.A. Oxidative Stress Contributes to Lung Injury and Barrier Dysfunction via Microtubule Destabilization. Am. J. Respir. Cell Mol. Biol. 2012, 47, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Ke, Y.; Tian, Y.; Ohmura, T.; Sitikov, A.; Sarich, N.; Montgomery, C.P.; Birukova, A.A. Staphylococcus Aureus-Induced Endothelial Permeability and Inflammation Are Mediated by Microtubule Destabilization. J. Biol. Chem. 2019, 294, 3369–3384. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, Y.; Gong, S.; Tang, J.; Zhang, J.; Li, F.; Yu, B.; Zhang, Y.; Kou, J. Ruscogenin Alleviates LPS-Induced Pulmonary Endothelial Cell Apoptosis by Suppressing TLR4 Signaling. Biomed. Pharmacother. 2020, 125, 109868. [Google Scholar] [CrossRef]
- Stefanec, T. Endothelial Apoptosis: Could It Have a Role in the Pathogenesis and Treatment of Disease? Chest 2000, 117, 841–854. [Google Scholar] [CrossRef] [PubMed]
- Polunovsky, V.A.; Wendt, C.H.; Ingbar, D.H.; Peterson, M.S.; Bitterman, P.B. Induction of Endothelial Cell Apoptosis by TNF Alpha: Modulation by Inhibitors of Protein Synthesis. Exp. Cell Res. 1994, 214, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Lindner, H.; Holler, E.; Ertl, B.; Multhoff, G.; Schreglmann, M.; Klauke, I.; Schultz-Hector, S.; Eissner, G. Peripheral Blood Mononuclear Cells Induce Programmed Cell Death in Human Endothelial Cells and May Prevent Repair: Role of Cytokines. Blood 1997, 89, 1931–1938. [Google Scholar] [CrossRef]
- Liang, Y.; Li, X.; Zhang, X.; Li, Z.; Wang, L.; Sun, Y.; Liu, Z.; Ma, X. Elevated Levels of Plasma TNF-α Are Associated with Microvascular Endothelial Dysfunction in Patients With Sepsis Through Activating the NF-κB and P38 Mitogen-Activated Protein Kinase in Endothelial Cells. Shock 2014, 41, 275–281. [Google Scholar] [CrossRef]
- Hosseinnejad, A.; Ludwig, N.; Mersmann, S.; Winnerbach, P.; Bleilevens, C.; Rossaint, R.; Rossaint, J.; Singh, S. Bioactive Nanogels Mimicking the Antithrombogenic Nitric Oxide-Release Function of the Endothelium. Small 2023, 19, 2205185. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.H.; Wang, X.L.; Wilcken, D.E. Nitric Oxide Induces and Inhibits Apoptosis through Different Pathways. FEBS Lett. 1998, 433, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.-L.; Fang, H.-X.; Liang, Y.; Zhao, Y.; Shi, C. MicroRNA-34a Promotes iNOS Secretion from Pulmonary Macrophages in Septic Suckling Rats through Activating STAT3 Pathway. Biomed. Pharmacother. 2018, 105, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Cassina, A.; Radi, R. Differential Inhibitory Action of Nitric Oxide and Peroxynitrite on Mitochondrial Electron Transport. Arch. Biochem. Biophys. 1996, 328, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Beltrán, B.; Mathur, A.; Duchen, M.R.; Erusalimsky, J.D.; Moncada, S. The Effect of Nitric Oxide on Cell Respiration: A Key to Understanding Its Role in Cell Survival or Death. Proc. Natl. Acad. Sci. USA 2000, 97, 14602–14607. [Google Scholar] [CrossRef]
- Jin, Y.-H.; Li, Z.-T.; Chen, H.; Jiang, X.-Q.; Zhang, Y.-Y.; Wu, F. Effect of Dexmedetomidine on Kidney Injury in Sepsis Rats through TLR4/MyD88/NF-κB/iNOS Signaling Pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 5020–5025. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, S.; Masereeuw, R.; Russel, F.G.M.; Pickkers, P. Selective iNOS Inhibition for the Treatment of Sepsis-Induced Acute Kidney Injury. Nat. Rev. Nephrol. 2009, 5, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Maneta, E.; Aivalioti, E.; Tual-Chalot, S.; Emini Veseli, B.; Gatsiou, A.; Stamatelopoulos, K.; Stellos, K. Endothelial Dysfunction and Immunothrombosis in Sepsis. Front. Immunol. 2023, 14, 1144229. [Google Scholar] [CrossRef]
- Hoppensteadt, D.; Tsuruta, K.; Cunanan, J.; Hirman, J.; Kaul, I.; Osawa, Y.; Fareed, J. Thrombin Generation Mediators and Markers in Sepsis-Associated Coagulopathy and Their Modulation by Recombinant Thrombomodulin. Clin. Appl. Thromb. Hemost. 2014, 20, 129–135. [Google Scholar] [CrossRef]
- Lv, S.; Han, M.; Yi, R.; Kwon, S.; Dai, C.; Wang, R. Anti-TNF-α Therapy for Patients with Sepsis: A Systematic Meta-Analysis. Int. J. Clin. Pract. 2014, 68, 520–528. [Google Scholar] [CrossRef]
- Yan, G.; You, B.; Chen, S.-P.; Liao, J.K.; Sun, J. Tumor Necrosis Factor-α Downregulates Endothelial Nitric Oxide Synthase mRNA Stability via Translation Elongation Factor 1-α 1. Circ. Res. 2008, 103, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, K.-S.; Kim, J.-H.; Lee, D.-K.; Park, M.; Choi, S.; Park, W.; Kim, S.; Choi, Y.K.; Hwang, J.Y.; et al. Aspirin Prevents TNF-α-Induced Endothelial Cell Dysfunction by Regulating the NF-κB-Dependent miR-155/eNOS Pathway: Role of a miR-155/eNOS Axis in Preeclampsia. Free Radic. Biol. Med. 2017, 104, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.T.; Beckmann, N.; Winer, L.K.; Kim, Y.; Goetzman, H.S.; Veile, R.E.; Gulbins, E.; Goodman, M.D.; Nomellini, V.; Caldwell, C.C. Amitriptyline Treatment Mitigates Sepsis-Induced Tumor Necrosis Factor Expression and Coagulopathy. Shock 2019, 51, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Xu, Z.; Zhang, C.; Xiong, J. Macrophage-Derived Exosomes Mediate Glomerular Endothelial Cell Dysfunction in Sepsis-Associated Acute Kidney Injury. Cell Biosci. 2023, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- Shakoory, B.; Carcillo, J.A.; Chatham, W.W.; Amdur, R.L.; Zhao, H.; Dinarello, C.A.; Cron, R.Q.; Opal, S.M. Interleukin-1 Receptor Blockade Is Associated with Reduced Mortality in Sepsis Patients With Features of Macrophage Activation Syndrome: Reanalysis of a Prior Phase III Trial*. Crit. Care Med. 2016, 44, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Kral-Pointner, J.B.; Schrottmaier, W.C.; Horvath, V.; Datler, H.; Hell, L.; Ay, C.; Niederreiter, B.; Jilma, B.; Schmid, J.A.; Assinger, A.; et al. Myeloid but Not Epithelial Tissue Factor Exerts Protective Anti-inflammatory Effects in Acid Aspiration-induced Acute Lung Injury. J. Thromb. Haemost. 2017, 15, 1625–1639. [Google Scholar] [CrossRef] [PubMed]
- Pawlinski, R.; Wang, J.-G.; Owens, A.P.; Williams, J.; Antoniak, S.; Tencati, M.; Luther, T.; Rowley, J.W.; Low, E.N.; Weyrich, A.S.; et al. Hematopoietic and Nonhematopoietic Cell Tissue Factor Activates the Coagulation Cascade in Endotoxemic Mice. Blood 2010, 116, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.A.J.; Preston, R.J.S.; O’Neill, L.A.J. Immunothrombosis and the Molecular Control of Tissue Factor by Pyroptosis: Prospects for New Anticoagulants. Biochem. J. 2022, 479, 731–750. [Google Scholar] [CrossRef]
- Chen, X.; Lao, Y.; Yi, J.; Yang, J.; He, S.; Chen, Y. SENP3 in Monocytes/Macrophages Up-regulates Tissue Factor and Mediates Lipopolysaccharide-induced Acute Lung Injury by Enhancing JNK Phosphorylation. J. Cell. Mol. Med. 2020, 24, 5454–5462. [Google Scholar] [CrossRef]
- Duan, H.; Li, L.; Shen, S.; Ma, Y.; Yin, X.; Liu, Z.; Yuan, C.; Wang, Y.; Zhang, J. Hydrogen Sulfide Reduces Cognitive Impairment in Rats After Subarachnoid Hemorrhage by Ameliorating Neuroinflammation Mediated by the TLR4/NF-κB Pathway in Microglia. Front. Cell. Neurosci. 2020, 14, 210. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Gao, Y.; Shou, S.; Chai, Y. The Roles of Macrophage Polarization in the Host Immune Response to Sepsis. Int. Immunopharmacol. 2021, 96, 107791. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Pentinmikko, N.; Ilmonen, M.; Salven, P. Dual Action of TGF-β Induces Vascular Growth in Vivo through Recruitment of Angiogenic VEGF-Producing Hematopoietic Effector Cells. Angiogenesis 2012, 15, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A Brief Review on the Mechanisms of miRNA Regulation. Genom. Proteom. Bioinform. 2009, 7, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Natoli, G. Transcriptional Regulation of Macrophage Polarization: Enabling Diversity with Identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, M.; Yu, G.; Bian, J.; Deng, X.; Wan, X.; Zhu, K. Serum miR-146a and miR-223 as Potential New Biomarkers for Sepsis. Biochem. Biophys. Res. Commun. 2010, 394, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Wang, X.; Zhang, X.; Ha, T.; Ma, H.; Liu, L.; Kalbfleisch, J.H.; Gao, X.; Kao, R.L.; Williams, D.L.; et al. Attenuation of Cardiac Dysfunction in Polymicrobial Sepsis by MicroRNA-146a Is Mediated via Targeting of IRAK1 and TRAF6 Expression. J. Immunol. 2015, 195, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Liu, Y.-T.; Fan, J. Exosomal Mediators in Sepsis and Inflammatory Organ Injury: Unraveling the Role of Exosomes in Intercellular Crosstalk and Organ Dysfunction. Mil. Med. Res. 2024, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Cui, B.; Zhang, W.; Ma, W.; Zhao, G.; Xing, L. Exosomal miR-21 Secreted by IL-1β-Primed-Mesenchymal Stem Cells Induces Macrophage M2 Polarization and Ameliorates Sepsis. Life Sci. 2021, 264, 118658. [Google Scholar] [CrossRef] [PubMed]
- Caescu, C.I.; Guo, X.; Tesfa, L.; Bhagat, T.D.; Verma, A.; Zheng, D.; Stanley, E.R. Colony Stimulating Factor-1 Receptor Signaling Networks Inhibit Mouse Macrophage Inflammatory Responses by Induction of microRNA-21. Blood 2015, 125, e1-13. [Google Scholar] [CrossRef]
- Wang, Z.; Brandt, S.; Medeiros, A.; Wang, S.; Wu, H.; Dent, A.; Serezani, C.H. MicroRNA 21 Is a Homeostatic Regulator of Macrophage Polarization and Prevents Prostaglandin E2-Mediated M2 Generation. PLoS ONE 2015, 10, e0115855. [Google Scholar] [CrossRef]
- Jiang, T.; Sun, L.; Zhu, J.; Li, N.; Gu, H.; Zhang, Y.; Li, M.; Xu, J. MicroRNA-23a-3p Promotes Macrophage M1 Polarization and Aggravates Lipopolysaccharide-Induced Acute Lung Injury by Regulating PLK1/STAT1/STAT3 Signalling. Int. J. Exp. Pathol. 2022, 103, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cao, M.; Xu, S.; Shi, J.; Mao, X.; Yao, X.; Liu, C. Luteolin Alters Macrophage Polarization to Inhibit Inflammation. Inflammation 2020, 43, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, Q.; Sun, W.; Chen, X.; Wang, Y.; Sun, Y.; Lin, L. 1,3,6,7-Tetrahydroxy-8-Prenylxanthone Ameliorates Inflammatory Responses Resulting from the Paracrine Interaction of Adipocytes and Macrophages. Br. J. Pharmacol. 2018, 175, 1590–1606. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin Reduces Inflammation and Fibrosis Biomarkers: A Potential Mechanism of Action for Beneficial Effects of SGLT2 Inhibitors in Diabetic Kidney Disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Song, C.; Zeng, Y.; Li, Y.; Li, H.; Liu, B.; Dai, M.; Pan, P. Canagliflozin Alleviates LPS-Induced Acute Lung Injury by Modulating Alveolar Macrophage Polarization. Int. Immunopharmacol. 2020, 88, 106969. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-L.; Wang, Z.-M.; Xu, C.; Che, F.-H.; Hu, X.-F.; Cao, R.; Xie, Y.-N.; Qiu, Y.; Shi, H.-B.; Liu, B.; et al. Liraglutide Attenuates Hepatic Ischemia-Reperfusion Injury by Modulating Macrophage Polarization. Front. Immunol. 2022, 13, 869050. [Google Scholar] [CrossRef] [PubMed]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.D.B.; De Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; De Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446. [Google Scholar] [CrossRef]
- Kim, Y.J.; Lee, S.; Jin, J.; Woo, H.; Choi, Y.-K.; Park, K.-G. Cassiaside C Inhibits M1 Polarization of Macrophages by Downregulating Glycolysis. Int. J. Mol. Sci. 2022, 23, 1696. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, H.; Zhang, W.; Jiang, L.; Tong, X.; Zheng, Y.; Xia, Z. Endothelial Cell Dysfunction Due to Molecules Secreted by Macrophages in Sepsis. Biomolecules 2024, 14, 980. https://doi.org/10.3390/biom14080980
He H, Zhang W, Jiang L, Tong X, Zheng Y, Xia Z. Endothelial Cell Dysfunction Due to Molecules Secreted by Macrophages in Sepsis. Biomolecules. 2024; 14(8):980. https://doi.org/10.3390/biom14080980
Chicago/Turabian StyleHe, Heng, Wei Zhang, Luofeng Jiang, Xirui Tong, Yongjun Zheng, and Zhaofan Xia. 2024. "Endothelial Cell Dysfunction Due to Molecules Secreted by Macrophages in Sepsis" Biomolecules 14, no. 8: 980. https://doi.org/10.3390/biom14080980