


Mechanisms of Resistance to Anti-PD-1 Immunotherapy in Melanoma and Strategies to Overcome It

 , ,
, ,  and
and 
Abstract
1. Introduction
2. PD-1 and PD-L1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Resistance Phenotype | Drug Exposure Requirement | Best Response | Confirmatory Scan for PD Requirement | Confirmatory Scan Time Frame | Neoadjuvant Therapy Resistance | Adjuvant Therapy Resistance |
|---|---|---|---|---|---|---|
| Primary resistance | ≥6 weeks | PD; SD for <6 months * | Yes * | ≥4 weeks after initial disease progression ** | major pathological response before the surgery + requirements for primary resistance | Timing of last dose prior to PD <12 weeks + confirmatory biopsy *** |
| Secondary Resistance | ≥6 months | CR, PR, SD for >6 months * | Yes * | ≥4 weeks after disease progression ** | no major pathological response before the surgery + requirements for secondary resistance | Timing of last dose prior to PD ≥12 weeks + confirmatory biopsy *** |
3. Primary Resistance Mechanisms
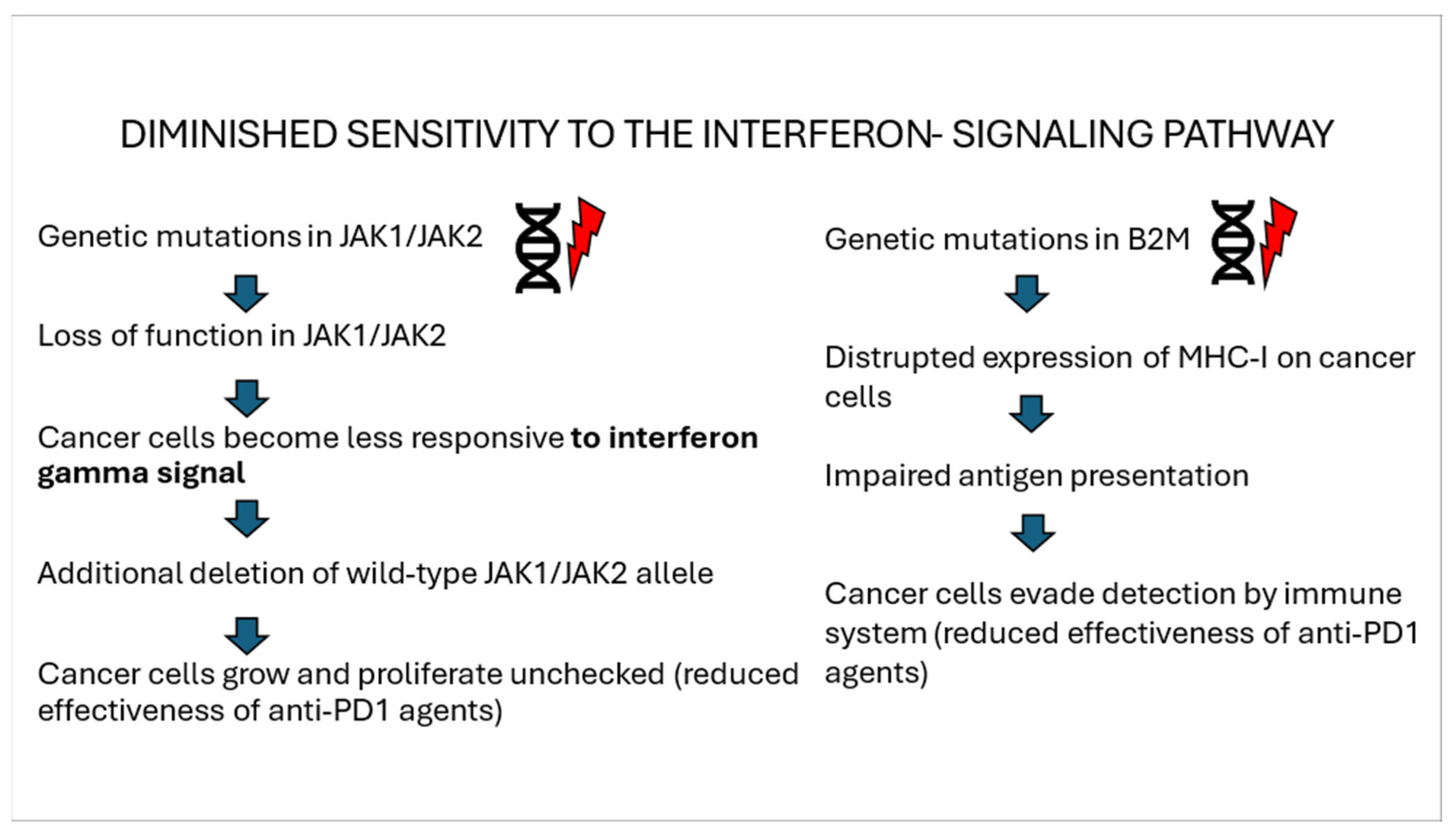
3.1. Interferon Gamma Signaling Pathway
3.2. Insufficient Antigen Presentation and/or Recognition
3.3. Immunosuppressive Tumor Microenvironment
3.4. Intrinsic Oncogenic Pathway Altering TME
4. Innate Resistance Mechanisms
5. Secondary Resistance Mechanisms
5.1. Tumor Intrinsic Mechanisms
5.1.1. Mutations Within the Interferon Signaling Pathway
5.1.2. Mutations in Antigen Presentation Pathway Genes
5.1.3. Loss of Tumor Suppressor Genes
5.2. Tumor-Extrinsic Mechanisms
5.2.1. T-Cell Exhaustion and Memory T Cells
5.2.2. Immunosuppressive Microenvironment
5.2.3. Metabolic Competition
6. Management of Patients with Melanoma Resistant to Anti-PD1 Therapy
7. Emerging Insights and Potential Therapeutic Strategies
| Gene | Immune Influence | Study |
|---|---|---|
| EZH2 | Affects antigen presentation and T-cell infiltration. | [149] |
| HDAC6 | Increases IL-10 and PD-L1, suppressing the immune response. | [150] |
| RASSF5 & ITGB2 | Impairs immunogenicity generation. | [150] |
| KDM5B | Recruits SETDB1 and modifies immune gene expression. | [151] |
| SETDB1 | Regulates immune-related gene clusters and antigen presentation. | [147,152,153] |
| FTO | Elevates PD-1 expression via autophagy. | [154] |
| LAG3 | Negatively regulates T cells by binding to MHC II. | [155,156,157] |
| STING | Modulates metabolism, MHC I, and PD-L1 expression. | [158] |
| NLRP3 | Influences inflammatory signals and macrophage activity. | [159,160,161] |
| PAI-1 | Affects macrophage polarisation, autophagy cycle | [162,163,164] |
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Karasarides, M.; Cogdill, A.P.; Robbins, P.B.; Bowden, M.; Burton, E.M.; Butterfield, L.H.; Cesano, A.; Hammer, C.; Haymaker, C.L.; Horak, C.E.; et al. Hallmarks of Resistance to Immune-Checkpoint Inhibitors. Cancer Immunol. Res. 2022, 10, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Gravbrot, N.; Gilbert-Gard, K.; Mehta, P.; Ghotmi, Y.; Banerjee, M.; Mazis, C.; Sundararajan, S. Therapeutic Monoclonal Antibodies Targeting Immune Checkpoints for the Treatment of Solid Tumors. Antibodies 2019, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Seto, T.; Sam, D.; Pan, M. Mechanisms of Primary and Secondary Resistance to Immune Checkpoint Inhibitors in Cancer. Med. Sci. 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fuereder, T. Resistance to immune checkpoint inhibitors. Next steps and combinational approaches. Mag. Eur. Med. Oncol. 2019, 12, 123–127. [Google Scholar] [CrossRef]
- Veldman, J.; Visser, L.; Berg, A.V.D.; Diepstra, A. Primary and acquired resistance mechanisms to immune checkpoint inhibition in Hodgkin lymphoma. Cancer Treat. Rev. 2020, 82, 101931. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.J.; Weber, J.S.; et al. Association of Pembrolizumab with Tumor Response and Survival Among Patients with Advanced Melanoma. JAMA 2016, 315, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Handel, E.E.; McKeown, J.; Wei, J.; Kankaria, R.A.; Burnette, H.; Johnson, D.B.; Lawless, A.; Czapla, J.; Sullivan, R.J.; Albrecht, L.J.; et al. Outcomes following long-term disease control with immune checkpoint inhibitors in patients with advanced melanoma. Eur. J. Cancer 2025, 215, 115171. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Long, G.V.; Carlino, M.S.; McNeil, C.; Ribas, A.; Gaudy-Marqueste, C.; Schachter, J.; Nyakas, M.; Kee, D.; Petrella, T.M.; Blaustein, A.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: 10-year follow-up of the phase III KEYNOTE-006 study. Ann. Oncol. 2024, 35, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Amaral, T.; Seeber, O.; Mersi, E.; Sanchez, S.; Thomas, I.; Meiwes, A.; Forschner, A.; Leiter, U.; Eigentler, T.; Keim, U.; et al. Primary Resistance to PD-1-Based Immunotherapy-A Study in 319 Patients with Stage IV Melanoma. Cancers 2020, 12, 1027. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hepner, A.; Versluis, J.M.; Wallace, R.; Allayous, C.; Brown, L.J.; Trojaniello, C.; Gerard, C.L.; Jansen, Y.J.L.; Bhave, P.; Neyns, B.; et al. The features and management of acquired resistance to PD1-based therapy in metastatic melanoma. Eur. J. Cancer 2024, 196, 113441. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Sobczuk, P.; Rogala, P.; Świtaj, T.; Placzke, J.; Kozak, K.; Mariuk-Jarema, A.; Spałek, M.; Dudzisz-Śledź, M.; Teterycz, P.; et al. Efficacy of immunotherapy beyond RECIST progression in advanced melanoma: A real-world evidence. Cancer Immunol. Immunother. 2022, 71, 1949–1958. [Google Scholar] [CrossRef]
- Ziogas, D.C.; Theocharopoulos, C.; Koutouratsas, T.; Haanen, J.; Gogas, H. Mechanisms of resistance to immune checkpoint inhibitors in melanoma: What we have to overcome? Cancer Treat. Rev. 2023, 113, 102499. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.Y.; Otsuji, M.; Gorski, K.; Huang, X.; Slansky, J.E.; Pai, S.I.; Shalabi, A.; Shin, T.; Pardoll, D.M.; Tsuchiya, H. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 2001, 193, 839–846. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Intlekofer, A.M.; Thompson, C.B. At the bench: Preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J. Leukoc. Biol. 2013, 94, 25–39. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007, 8, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ribas, A. Tumor immunotherapy directed at PD-1. N. Engl. J. Med. 2012, 366, 2517–2519. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.A.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 2006, 203, 883–895. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fife, B.T.; Pauken, K.E.; Eagar, T.N.; Obu, T.; Wu, J.; Tang, Q.; Azuma, M.; Krummel, M.F.; Bluestone, J.A. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol. 2009, 10, 1185–1192. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kluger, H.M.; Tawbi, H.A.; Ascierto, M.L.; Bowden, M.; Callahan, M.K.; Cha, E.; Chen, H.X.; Drake, C.G.; Feltquate, D.M.; Ferris, R.L.; et al. Defining tumor resistance to PD-1 pathway blockade: Recommendations from the first meeting of the SITC Immunotherapy Resistance Taskforce. J. Immunother. Cancer 2020, 8, e000398. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes with Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients with Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lipson, E.J.; Tawbi, H.A.-H.; Schadendorf, D.; Ascierto, P.A.; Matamala, L.; Gutiérrez, E.C.; Rutkowski, P.; Gogas, H.; Lao, C.D.; Menezes, J.J.d.; et al. Relatlimab (RELA) plus nivolumab (NIVO) versus NIVO in first-line advanced melanoma: Primary phase III results from RELATIVITY-047 (CA224-047). J. Clin. Oncol. 2021, 39, 9503. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- del Campo, A.B.; Kyte, J.A.; Carretero, J.; Zinchencko, S.; Méndez, R.; González-Aseguinolaza, G.; Ruiz-Cabello, F.; Aamdal, S.; Gaudernack, G.; Garrido, F.; et al. Immune escape of cancer cells with beta2-microglobulin loss over the course of metastatic melanoma. Int. J. Cancer 2014, 134, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Forschner, A.; Battke, F.; Hadaschik, D.; Schulze, M.; Weißgraeber, S.; Han, C.T.; Kopp, M.; Frick, M.; Klumpp, B.; Tietze, N.; et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma—Results of a prospective biomarker study. J. Immunother. Cancer 2019, 7, 180. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ladányi, A.; Kiss, J.; Somlai, B.; Gilde, K.; Fejos, Z.; Mohos, A.; Gaudi, I.; Tímár, J. Density of DC-LAMP(+) mature dendritic cells in combination with activated T lymphocytes infiltrating primary cutaneous melanoma is a strong independent prognostic factor. Cancer Immunol. Immunother. 2007, 56, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, W.; Wang, Y.; Li, W.; Yu, G.; Li, Z.; Fang, C.; Shen, Y.; Sun, Z.; Han, L.; et al. IL-37b suppresses T cell priming by modulating dendritic cell maturation and cytokine production via dampening ERK/NF-κB/S6K signalings. Acta Biochim. Biophys. Sin. 2015, 47, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Lindenberg, J.J.; van de Ven, R.; Lougheed, S.M.; Zomer, A.; Santegoets, S.J.; Griffioen, A.W.; Hooijberg, E.; van den Eertwegh, A.J.; Thijssen, V.L.; Scheper, R.J.; et al. Functional characterization of a STAT3-dependent dendritic cell-derived CD14(+) cell population arising upon IL-10-driven maturation. Oncoimmunology 2013, 2, e23837. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Strauss, L.; Bergmann, C.; Szczepanski, M.; Gooding, W.; Johnson, J.T.; Whiteside, T.L. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin. Cancer Res. 2007, 13, 4345–4354. [Google Scholar] [CrossRef] [PubMed]
- Viguier, M.; Lemaître, F.; Verola, O.; Cho, M.S.; Gorochov, G.; Dubertret, L.; Bachelez, H.; Kourilsky, P.; Ferradini, L. Foxp3 expressing CD4+CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J. Immunol. 2004, 173, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, Y.; Chen, W.; Xu, Z.P.; Gu, W. PD-L1 Distribution and Perspective for Cancer Immunotherapy-Blockade, Knockdown, or Inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e1512. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.E9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hopkins-Donaldson, S.; Ziegler, A.; Kurtz, S.; Bigosch, C.; Kandioler, D.; Ludwig, C.; Zangemeister-Wittke, U.; Stahel, R. Silencing of death receptor and caspase-8 expression in small cell lung carcinoma cell lines and tumors by DNA methylation. Cell Death Differ. 2003, 10, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Pallini, R.; Lotti, F.; Sette, G.; Patti, M.; Bartucci, M.; Ricci-Vitiani, L.; Signore, M.; Stassi, G.; Larocca, L.M.; et al. Inhibition of DNA methylation sensitizes glioblastoma for tumor necrosis factor-related apoptosis-inducing ligand-mediated destruction. Cancer Res. 2005, 65, 11469–11477. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H.; Becker, J.C.; Straten, P. Regulators of apoptosis: Suitable targets for immune therapy of cancer. Nat. Rev. Drug Discov. 2005, 4, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Talukder, A.; Savage, N.M.; Singh, N.; Liu, K. JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer. Oncoimmunology 2017, 6, e1291106. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Piper, M.; Kluger, H.; Ruppin, E.; Hu-Lieskovan, S. Immune Resistance Mechanisms and the Road to Personalized Immunotherapy. Am. Soc. Clin. Oncol. Educ. Book 2023, 43, e390290. [Google Scholar] [CrossRef] [PubMed]
- Hulpke, S.; Tampé, R. The MHC I loading complex: A multitasking machinery in adaptive immunity. Trends Biochem. Sci. 2013, 38, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Scanlan, M.J.; Gure, A.O.; Jungbluth, A.A.; Old, L.J.; Chen, Y.T. Cancer/testis antigens: An expanding family of targets for cancer immunotherapy. Immunol. Rev. 2002, 188, 22–32. [Google Scholar] [CrossRef] [PubMed]
- James, S.R.; Link, P.A.; Karpf, A.R. Epigenetic regulation of X-linked cancer/germline antigen genes by DNMT1 and DNMT3b. Oncogene 2006, 25, 6975–6985. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, H.; Gu, J.; Lin, S.; Li, J.; Lu, W.; Wang, Y.; Zhu, J. Methylation profiles of thirty four promoter-CpG islands and concordant methylation behaviours of sixteen genes that may contribute to carcinogenesis of astrocytoma. BMC Cancer 2004, 4, 65. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yu, J.; Ni, M.; Xu, J.; Zhang, H.; Gao, B.; Gu, J.; Chen, J.; Zhang, L.; Wu, M.; Zhen, S.; et al. Methylation profiling of twenty promoter-CpG islands of genes which may contribute to hepatocellular carcinogenesis. BMC Cancer 2002, 2, 29. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ning, B.; Liu, Y.; Wang, M.; Li, Y.; Xu, T.; Wei, Y. The Predictive Value of Tumor Mutation Burden on Clinical Efficacy of Immune Checkpoint Inhibitors in Melanoma: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2022, 13, 748674. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rao, S.V.; Moran, A.E.; Graff, J.N. Predictors of response and resistance to checkpoint inhibitors in solid tumors. Ann. Transl. Med. 2017, 5, 468. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pulluri, B.; Kumar, A.; Shaheen, M.; Jeter, J.; Sundararajan, S. Tumor microenvironment changes leading to resistance of immune checkpoint inhibitors in metastatic melanoma and strategies to overcome resistance. Pharmacol. Res. 2017, 123, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Falcone, I.; Conciatori, F.; Bazzichetto, C.; Ferretti, G.; Cognetti, F.; Ciuffreda, L.; Milella, M. Tumor Microenvironment: Implications in Melanoma Resistance to Targeted Therapy and Immunotherapy. Cancers 2020, 12, 2870. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Lv, Z.; Xu, D.; Cui, J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark. Res. 2020, 8, 34. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Powderly, J.D.; Koeppen, H.; Hodi, F.S.; Sosman, J.A.; Gettinger, S.N.; Desai, R.; Tabernero, J.; Soria, J.-C.; Hamid, O.; Fine, G.D.; et al. Biomarkers and associations with the clinical activity of PD-L1 blockade in a MPDL3280A study. J. Clin. Oncol. 2013, 31, 3001. [Google Scholar] [CrossRef]
- Chen, W.; Ten Dijke, P. Immunoregulation by members of the TGFβ superfamily. Nat. Rev. Immunol. 2016, 16, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Vilain, R.E.; Menzies, A.M.; Wilmott, J.S.; Kakavand, H.; Madore, J.; Guminski, A.; Liniker, E.; Kong, B.Y.; Cooper, A.J.; Howle, J.R.; et al. Dynamic Changes in PD-L1 Expression and Immune Infiltrates Early During Treatment Predict Response to PD-1 Blockade in Melanoma. Clin. Cancer Res. 2017, 23, 5024–5033. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ngiow, S.F.; Young, A.; Jacquelot, N.; Yamazaki, T.; Enot, D.; Zitvogel, L.; Smyth, M.J. A Threshold Level of Intratumor CD8+ T-cell PD1 Expression Dictates Therapeutic Response to Anti-PD1. Cancer Res. 2015, 75, 3800–3811. [Google Scholar] [CrossRef] [PubMed]
- Mognol, G.P.; Spreafico, R.; Wong, V.; Scott-Browne, J.P.; Togher, S.; Hoffmann, A.; Hogan, P.G.; Rao, A.; Trifari, S. Exhaustion-associated regulatory regions in CD8(+) tumor-infiltrating T cells. Proc. Natl. Acad. Sci. USA 2017, 114, e2776–e2785. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sakaguchi, S.; Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T. Regulatory T cells: How do they suppress immune responses? Int. Immunol. 2009, 21, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Manzotti, C.N.; Liu, M.; Burke, F.; Mead, K.I.; Sansom, D.M. CD86 and CD80 differentially modulate the suppressive function of human regulatory T cells. J. Immunol. 2004, 172, 2778–2784. [Google Scholar] [CrossRef] [PubMed]
- Tekguc, M.; Wing, J.B.; Osaki, M.; Long, J.; Sakaguchi, S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2023739118. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hofer, F.; Di Sario, G.; Musiu, C.; Sartoris, S.; De Sanctis, F.; Ugel, S. A Complex Metabolic Network Confers Immunosuppressive Functions to Myeloid-Derived Suppressor Cells (MDSCs) within the Tumour Microenvironment. Cells 2021, 10, 2700. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra67. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Espinosa, E.; Marquez-Rodas, I.; Soria, A.; Berrocal, A.; Manzano, J.L.; Gonzalez-Cao, M.; Martin-Algarra, S.; Spanish Melanoma, G. Predictive factors of response to immunotherapy-a review from the Spanish Melanoma Group (GEM). Ann. Transl. Med. 2017, 5, 389. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bourhis, M.; Palle, J.; Galy-Fauroux, I.; Terme, M. Direct and Indirect Modulation of T Cells by VEGF-A Counteracted by Anti-Angiogenic Treatment. Front. Immunol. 2021, 12, 616837. [Google Scholar] [CrossRef] [PubMed]
- Olbryt, M.; Rajczykowski, M.; Widlak, W. Biological Factors behind Melanoma Response to Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2020, 21, 4071. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Prendergast, G.C.; Smith, C.; Thomas, S.; Mandik-Nayak, L.; Laury-Kleintop, L.; Metz, R.; Muller, A.J. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunother. 2014, 63, 721–735. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Trojaniello, C.; Luke, J.J.; Ascierto, P.A. Therapeutic Advancements Across Clinical Stages in Melanoma, with a Focus on Targeted Immunotherapy. Front. Oncol. 2021, 11, 670726. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M. Filling the Tank: Keeping Antitumor T Cells Metabolically Fit for the Long Haul. Cancer Immunol. Res. 2016, 4, 1001–1006. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Mittal, D.; Stagg, J.; Smyth, M.J. Targeting cancer-derived adenosine: New therapeutic approaches. Cancer Discov. 2014, 4, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. The “cancer immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Weide, B.; Martens, A.; Hassel, J.C.; Berking, C.; Postow, M.A.; Bisschop, K.; Simeone, E.; Mangana, J.; Schilling, B.; Di Giacomo, A.M.; et al. Baseline Biomarkers for Outcome of Melanoma Patients Treated with Pembrolizumab. Clin. Cancer Res. 2016, 22, 5487–5496. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Diem, S.; Kasenda, B.; Spain, L.; Martin-Liberal, J.; Marconcini, R.; Gore, M.; Larkin, J. Serum lactate dehydrogenase as an early marker for outcome in patients treated with anti-PD-1 therapy in metastatic melanoma. Br. J. Cancer 2016, 114, 256–261. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zimmer, L.; Apuri, S.; Eroglu, Z.; Kottschade, L.A.; Forschner, A.; Gutzmer, R.; Schlaak, M.; Heinzerling, L.; Krackhardt, A.M.; Loquai, C.; et al. Ipilimumab alone or in combination with nivolumab after progression on anti-PD-1 therapy in advanced melanoma. Eur. J. Cancer 2017, 75, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Su, L.J.; Pinckney, J.; Critton, D.; Boyer, E.; Krishnakumar, A.; Corbett, M.; Rankin, A.L.; Dibella, R.; Campbell, L.; et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature 2019, 574, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mitsuhashi, K.; Nosho, K.; Sukawa, Y.; Matsunaga, Y.; Ito, M.; Kurihara, H.; Kanno, S.; Igarashi, H.; Naito, T.; Adachi, Y.; et al. Association of Fusobacterium species in pancreatic cancer tissues with molecular features and prognosis. Oncotarget 2015, 6, 7209–7220. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Round, J.L.; Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008, 453, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 2015, 42, 344–355. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host. Microbe. 2013, 14, 207–215. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Atefi, M.; Avramis, E.; Lassen, A.; Wong, D.J.; Robert, L.; Foulad, D.; Cerniglia, M.; Titz, B.; Chodon, T.; Graeber, T.G.; et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin. Cancer Res. 2014, 20, 3446–3457. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Falletta, P.; Sanchez-Del-Campo, L.; Chauhan, J.; Effern, M.; Kenyon, A.; Kershaw, C.J.; Siddaway, R.; Lisle, R.; Freter, R.; Daniels, M.J.; et al. Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes. Dev. 2017, 31, 18–33. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Verdegaal, E.M.; de Miranda, N.F.; Visser, M.; Harryvan, T.; van Buuren, M.M.; Andersen, R.S.; Hadrup, S.R.; van der Minne, C.E.; Schotte, R.; Spits, H.; et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature 2016, 536, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Nakayama, M.; Hayakawa, Y.; Kojima, Y.; Ikeda, H.; Imai, N.; Ogasawara, K.; Okumura, K.; Thomas, D.M.; Smyth, M.J. IFN-γ is required for cytotoxic T cell-dependent cancer genome immunoediting. Nat Commun 2017, 8, 14607. [Google Scholar] [CrossRef]
- Wang, K.; Coutifaris, P.; Brocks, D.; Wang, G.; Azar, T.; Solis, S.; Nandi, A.; Anderson, S.; Han, N.; Manne, S.; et al. Combination anti-PD-1 and anti-CTLA-4 therapy generates waves of clonal responses that include progenitor-exhausted CD8(+) T cells. Cancer Cell 2024, 42, 1582–1597. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xiao, Z.; Yan, R.; Liu, H.; Huang, X.; Liang, Z.; An, G.; Ge, Y. Preventive Treatment with PD-1 Antibody Increases Tissue-resident Memory T Cells Infiltration and Delays Esophageal Carcinogenesis. Cancer Prev. Res. 2023, 16, 669–679. [Google Scholar] [CrossRef]
- VanderWalde, A.; Bellasea, S.L.; Kendra, K.L.; Khushalani, N.I.; Campbell, K.M.; Scumpia, P.O.; Kuklinski, L.F.; Collichio, F.; Sosman, J.A.; Ikeguchi, A.; et al. Ipilimumab with or without nivolumab in PD-1 or PD-L1 blockade refractory metastatic melanoma: A randomized phase 2 trial. Nat. Med. 2023, 29, 2278–2285. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pires da Silva, I.; Ahmed, T.; Reijers, I.L.M.; Weppler, A.M.; Betof Warner, A.; Patrinely, J.R.; Serra-Bellver, P.; Allayous, C.; Mangana, J.; Nguyen, K.; et al. Ipilimumab alone or ipilimumab plus anti-PD-1 therapy in patients with metastatic melanoma resistant to anti-PD-(L)1 monotherapy: A multicentre, retrospective, cohort study. Lancet Oncol. 2021, 22, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Lewis, K.D.; Weise, A.; McKean, M.; Papadopoulos, K.P.; Crown, J.; Kim, T.M.; Lee, D.H.; Thomas, S.S.; Mehnert, J.; et al. Phase I Study of Fianlimab, a Human Lymphocyte Activation Gene-3 (LAG-3) Monoclonal Antibody, in Combination with Cemiplimab in Advanced Melanoma. J. Clin. Oncol. 2024, 42, 2928–2938. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Patel, S.P.; Dimou, A.; Victor, A.I.; Mooradian, M.; Buchbinder, E.I.; Hernandez-Aya, L.F.; Prieto, P.A.; Cohen, J.V.; Flaherty, K.; Kahn, S.; et al. Safety and efficacy of first-in-class CXCR1/2 inhibitor SX-682 in combination with pembrolizumab (pem) in patients (pts) with metastatic melanoma (mMEL) with disease progression on anti–PD-1 therapy. J. Clin. Oncol. 2024, 42, 9508. [Google Scholar] [CrossRef]
- Weiss, S.A.; Sznol, M.; Shaheen, M.; Berciano-Guerrero, M.; Couselo, E.M.; Rodríguez-Abreu, D.; Boni, V.; Schuchter, L.M.; Gonzalez-Cao, M.; Arance, A.; et al. A Phase II Trial of the CD40 Agonistic Antibody Sotigalimab (APX005M) in Combination with Nivolumab in Subjects with Metastatic Melanoma with Confirmed Disease Progression on Anti-PD-1 Therapy. Clin. Cancer Res. 2024, 30, 74–81. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.J.; Eroglu, Z.; Brockstein, B.; Poklepovic, A.S.; Bajaj, M.; Babu, S.; Hallmeyer, S.; Velasco, M.; Lutzky, J.; Higgs, E.; et al. Pembrolizumab Plus Ipilimumab Following Anti-PD-1/L1 Failure in Melanoma. J. Clin. Oncol. 2021, 39, 2647–2655. [Google Scholar] [CrossRef] [PubMed]
- Vanderwalde, A.M.; Moon, J.; Kendra, K.; Khushalani, N.I.; Collichio, F.; Sosman, J.A.; Ikeguchi, A.; Victor, A.I.; Truong, T.-G.; Chmielowski, B.; et al. Abstract CT013: S1616: Ipilimumab plus nivolumab versus ipilimumab alone in patients with metastatic or unresectable melanoma that did not respond to anti-PD-1 therapy. Cancer Res. 2022, 82 (Suppl. S12), CT013-CT013. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Melero, I.; Bhatia, S.; Bono, P.; Sanborn, R.E.; Lipson, E.J.; Callahan, M.K.; Gajewski, T.; Gomez-Roca, C.A.; Hodi, F.S.; et al. Initial efficacy of anti-lymphocyte activation gene-3 (anti–LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti–PD-1/PD-L1 therapy. J. Clin. Oncol. 2017, 35, 9520. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Lipson, E.J.; Dummer, R.; Larkin, J.; Long, G.V.; Sanborn, R.E.; Chiarion-Sileni, V.; Dréno, B.; Dalle, S.; Schadendorf, D.; et al. Nivolumab and Relatlimab in Patients with Advanced Melanoma That Had Progressed on Anti–Programmed Death-1/Programmed Death Ligand 1 Therapy: Results from the Phase I/IIa RELATIVITY-020 Trial. J. Clin. Oncol. 2023, 41, 2724–2735. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Lee, S.J.; Chmielowski, B.; Tarhini, A.A.; Cohen, G.I.; Truong, T.-G.; Moon, H.H.; Davar, D.; O’Rourke, M.; Stephenson, J.J.; et al. Combination dabrafenib and trametinib versus combination nivolumab and ipilimumab for patients with advanced BRAF-mutant melanoma: The DREAMseq trial—ECOG-ACRIN EA6134. J. Clin. Oncol. 2023, 41, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Rohaan, M.W.; Borch, T.H.; Van Den Berg, J.H.; Met, Ö.; Kessels, R.; Geukes Foppen, M.H.; Stoltenborg Granhøj, J.; Nuijen, B.; Nijenhuis, C.; Jedema, I.; et al. Tumor-Infiltrating Lymphocyte Therapy or Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2022, 387, 2113–2125. [Google Scholar] [CrossRef]
- Chesney, J.; Lewis, K.D.; Kluger, H.; Hamid, O.; Whitman, E.; Thomas, S.; Wermke, M.; Cusnir, M.; Domingo-Musibay, E.; Phan, G.Q.; et al. Efficacy and safety of lifileucel, a one-time autologous tumor-infiltrating lymphocyte (TIL) cell therapy, in patients with advanced melanoma after progression on immune checkpoint inhibitors and targeted therapies: Pooled analysis of consecutive cohorts of the C-144-01 study. J. Immunother. Cancer 2022, 10, e005755. [Google Scholar] [CrossRef]
- Gaughan, E.M.; Horton, B.J. Outcomes from Cytotoxic Chemotherapy Following Progression on Immunotherapy in Metastatic Melanoma: An Institutional Case-Series. Front. Oncol. 2022, 12, 855782. [Google Scholar] [CrossRef] [PubMed]
- Mastroianni, J.; Stickel, N.; Andrlova, H.; Hanke, K.; Melchinger, W.; Duquesne, S.; Schmidt, D.; Falk, M.; Andrieux, G.; Pfeifer, D.; et al. miR-146a Controls Immune Response in the Melanoma Microenvironment. Cancer Res. 2019, 79, 183–195. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zakharia, Y.; McWilliams, R.R.; Rixe, O.; Drabick, J.; Shaheen, M.F.; Grossmann, K.F.; Kolhe, R.; Pacholczyk, R.; Sadek, R.; Tennant, L.L.; et al. Phase II trial of the IDO pathway inhibitor indoximod plus pembrolizumab for the treatment of patients with advanced melanoma. J. Immunother. Cancer 2021, 9, e002057. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Kato, S.; Nesline, M.K.; Conroy, J.M.; DePietro, P.; Pabla, S.; Kurzrock, R. Indoleamine 2,3-dioxygenase (IDO) inhibitors and cancer immunotherapy. Cancer Treat. Rev. 2022, 110, 102461. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lazaro-Camp, V.J.; Salari, K.; Meng, X.; Yang, S. SETDB1 in cancer: Overexpression and its therapeutic implications. Am. J. Cancer Res. 2021, 11, 1803–1827. [Google Scholar] [PubMed] [PubMed Central]
- Melero, I.; de Miguel Luken, M.; de Velasco, G.; Garralda, E.; Martín-Liberal, J.; Joerger, M.; Alonso, G.; Goebeler, M.-E.; Schuler, M.; König, D.; et al. Neutralizing GDF-15 can overcome anti-PD-1 and anti-PD-L1 resistance in solid tumours. Nature 2024, 637, 1218–1227. [Google Scholar] [CrossRef]
- Xu, T.; Dai, J.; Tang, L.; Yang, L.; Si, L.; Sheng, X.; Cui, C.; Chi, Z.; Kong, Y.; Guo, J. EZH2 Inhibitor Enhances the STING Agonist—Induced Antitumor Immunity in Melanoma. J. Investig. Dermatol. 2022, 142, 1158–1170. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Hernandez, M.; Munoz, Z.; Villagra, A. Enhancing Therapeutic Approaches for Melanoma Patients Targeting Epigenetic Modifiers. Cancers 2021, 13, 6180. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, S.M.; Cai, W.L.; Liu, X.; Thakral, D.; Luo, J.; Chan, L.H.; McGeary, M.K.; Song, E.; Blenman, K.R.M.; Micevic, G.; et al. KDM5B promotes immune evasion by recruiting SETDB1 to silence retroelements. Nature 2021, 598, 682–687. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Griffin, G.K.; Wu, J.; Iracheta-Vellve, A.; Patti, J.C.; Hsu, J.; Davis, T.; Dele-Oni, D.; Du, P.P.; Halawi, A.G.; Ishizuka, J.J.; et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021, 595, 309–314. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Strepkos, D.; Markouli, M.; Klonou, A.; Papavassiliou, A.G.; Piperi, C. Histone Methyltransferase SETDB1: A Common Denominator of Tumorigenesis with Therapeutic Potential. Cancer Res. 2021, 81, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wei, J.; Cui, Y.-H.; Park, G.; Shah, P.; Deng, Y.; Aplin, A.E.; Lu, Z.; Hwang, S.; He, C.; et al. m6A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat. Commun. 2019, 10, 2782. [Google Scholar] [CrossRef]
- Atkinson, V.; Khattak, A.; Haydon, A.; Eastgate, M.; Roy, A.; Prithviraj, P.; Mueller, C.; Brignone, C.; Triebel, F. Eftilagimod alpha, a soluble lymphocyte activation gene-3 (LAG-3) protein plus pembrolizumab in patients with metastatic melanoma. J. Immunother. Cancer 2020, 8, e001681. [Google Scholar] [CrossRef]
- Shen, R.; Postow, M.A.; Adamow, M.; Arora, A.; Hannum, M.; Maher, C.; Wong, P.; Curran, M.A.; Hollmann, T.J.; Jia, L.; et al. LAG-3 expression on peripheral blood cells identifies patients with poorer outcomes after immune checkpoint blockade. Sci. Transl. Med. 2021, 13, p.eabf5107. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sidaway, P. LAG3 inhibition improves outcomes. Nat. Rev. Clin. Oncol. 2022, 19, 149. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Barber, G.N. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016, 76, 6747–6759. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhao, W. NLRP3 Inflammasome-A Key Player in Antiviral Responses. Front. Immunol. 2020, 11, 211. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhuang, T.; Li, S.; Yi, X.; Guo, S.; Wang, Y.; Chen, J.; Liu, L.; Jian, Z.; Gao, T.; Kang, P.; et al. Tranilast Directly Targets NLRP3 to Protect Melanocytes from Keratinocyte-Derived IL-1β Under Oxidative Stress. Front. Cell Dev. Biol. 2020, 8, 588. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lee, H.E.; Lee, J.Y.; Yang, G.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Inhibition of NLRP3 inflammasome in tumor microenvironment leads to suppression of metastatic potential of cancer cells. Sci. Rep. 2019, 9, 12277. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tseng, Y.J.; Lee, C.H.; Chen, W.Y.; Yang, J.L.; Tzeng, H.T. Inhibition of PAI-1 Blocks PD-L1 Endocytosis and Improves the Response of Melanoma Cells to Immune Checkpoint Blockade. J. Investig. Dermatol. 2021, 141, 2690–2698. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wei, X.; He, J.; Tian, X.; Yuan, S.; Sun, L. Plasminogen activator inhibitor-1 in cancer research. Biomed. Pharmacother. 2018, 105, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Kubala, M.H.; Punj, V.; Placencio-Hickok, V.R.; Fang, H.; Fernandez, G.E.; Sposto, R.; DeClerck, Y.A. Plasminogen Activator Inhibitor-1 Promotes the Recruitment and Polarization of Macrophages in Cancer. Cell Rep. 2018, 25, 2177–2191. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]



| Primary Resistance—Main Mechanisms | Alterations Leading to Resistance |
|---|---|
| Resistance of IFN-γ-signaling pathway | - mutations in JAK1/2 - loss of PTEN expression |
| Insufficient antigen presentation and/or antigen recognition | - genetic mutation in HLA complex (beta-2 microglobulin) - post-transcriptional silencing by non-coding microRNAs - hypermethylation and deacetylation of the HLA promoter area - low tumor mutation burden - lack of neoantigens |
| Immunosuppressive tumor microenvironment | - increased presence of immunosuppressive cells (Tregs, MDSCs) - elevated levels of TAMs - elevated IDO - presence of Fusobacterium nucleatum |
| Intrinsic oncogenic pathway | - mutation in the WNT/β-catenin pathway - loss of PTEN |
| Secondary Resistance—Main Mechanisms | Alterations Leading to Resistance |
| Mutations within IFN-γ-signaling pathway | - acquired loss of function JAK1/2 mutation |
| Mutations in antigen presentation pathway | - loss of β2-microglobulin - loss of mutation-associated neoantigens - upregulation of NGFR, downregulation of MART-1 |
| Loss of tumor suppressor genes | - loss of PTEN |
| T-cell exhaustion and memory T cells | - upregulation of multiple co-inhibitory receptors (PD-1, TIM-3, TIGIT, LAG-3) - lack of TMEM |
| Immunosuppressive microenvironment | - increased presence of immunosuppressive cells (Tregs, MDSCs) - upregulating inhibitory pathways (EZH2, LAG-3, TIM-3, TIGIT, VISTA) |
| Metabolic component | - expression of IDO, IL-10, arginase, TGF-β |
| Cell | Observed Alteration Leading to Resistance |
|---|---|
| Tumor cell | Mutations in IFN-γ signaling pathway: - Loss of PTEN - Loss of function mutations in JAK1/2 |
| Tumor cell | Mutations in HLA antigen presentation complex: - beta-2-microglobulin complex - posttranscriptional by non-coding microRNA - hypermethylation/deacetylation of the HLA promotor area |
| Tumor cell | Low mutation burden, lack of neoantigens |
| Tregs | - secretion of IL-10, TGF-β, extracellular adenosine - absorption of IL-2 - reduction of CD80/86 presence (through CTLA-4 mediated process) |
| MDSCs | Promotion of angiogenesis and tumor invasion: - secretion of TGF-β and VEGF - secretion of ROS, NO, IL-10 Suppression of T-cell response: - arginine, tryptophan - secretion of ROS, NO, IL-10 |
| TAMs | Prevention of recruitment of CD8+ T cells |
| Tumor cell | Mutation in WNT/β-catenin |
| Tumor cell | Expression of IPRES (EMT): - upregulation of genes such as AXL, TWIST2, WNT5a, LOXL2, ROR2, TAGLN, and FAP |
| CD8+ T-cell | Exhaustion (TEX): - upregulation of coinhibitory receptors: PD-1, TIM-3, TIGIT, LAG-3 |
| Management of Anti-Pd1 Therapy Resistance | |||
|---|---|---|---|
| Nonclinical Trial Options (Approved By FDA) | Clinical Trials Options | ||
| Treatment | Response Rate | ||
| Ipilimumab 1 mg/kg + pembrolizumab [134] Ipilimumab 3 mg/kg + nivolumab [135] | 29% 29% | BNT111 + cemiplimab Phase II | (NCT04526899) |
| Nivolumab + relatlimab [136,137] | 11% | Avelumab (anti-PDL1) Phase I | NCT01772004)-completed |
| BRAF/MEK inhibitors for BRAF-mutated melanoma [138] | 48% | CXCR1/2 inhibitor SX-682 + pembrolizumab Phase I | (NCT03161431) |
| Tumor-infiltrating lymphocytes [139,140] | 49% | Nivolumab + PD-L1/IDO peptide vaccine Phase I/II | (NCT03047928) |
| Chemotherapy [141] | 22% | Sotigalimab + nivolumab Phase II | (NCT03123783) |
| BNT111 Phase I | (NCT02410733) | ||
| ONCOS-102 +/− balstilimab Phase II | (NCT0556149) | ||
| TransCon IL-2 β/γ monotherapy/ in combination Phase I/II | (NCT05081609) | ||
| PF-07329640 monotherapy/ in combination bevacizumab or sasanlimab Phase I | (NCT06448364) | ||
| Fianlimab + cemiplimab Phase I | (NCT03005782) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zielińska, M.K.; Ciążyńska, M.; Sulejczak, D.; Rutkowski, P.; Czarnecka, A.M. Mechanisms of Resistance to Anti-PD-1 Immunotherapy in Melanoma and Strategies to Overcome It. Biomolecules 2025, 15, 269. https://doi.org/10.3390/biom15020269
Zielińska MK, Ciążyńska M, Sulejczak D, Rutkowski P, Czarnecka AM. Mechanisms of Resistance to Anti-PD-1 Immunotherapy in Melanoma and Strategies to Overcome It. Biomolecules. 2025; 15(2):269. https://doi.org/10.3390/biom15020269
Chicago/Turabian StyleZielińska, Magdalena K., Magdalena Ciążyńska, Dorota Sulejczak, Piotr Rutkowski, and Anna M. Czarnecka. 2025. "Mechanisms of Resistance to Anti-PD-1 Immunotherapy in Melanoma and Strategies to Overcome It" Biomolecules 15, no. 2: 269. https://doi.org/10.3390/biom15020269
APA StyleZielińska, M. K., Ciążyńska, M., Sulejczak, D., Rutkowski, P., & Czarnecka, A. M. (2025). Mechanisms of Resistance to Anti-PD-1 Immunotherapy in Melanoma and Strategies to Overcome It. Biomolecules, 15(2), 269. https://doi.org/10.3390/biom15020269