
Neuroinflammation—A Crucial Factor in the Pathophysiology of Depression—A Comprehensive Review

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Neuroinflammation and Depression: A Complex Relationship
2.1. Neuroinflammation: Cellular and Molecular Aspects
2.2. Peripheral and Central Immune System Interactions in Depression
2.3. Bidirectional Relationship: Can Depression Itself Trigger Neuroinflammation?
3. Biological Mechanisms Underlying Neuroinflammation in Depression
3.1. The HPA Axis and Stress
3.2. Cytokines as Mediators of Depression
3.3. Microglial and Astrocytic Activation in the Brain
3.4. Macrophages and Other Peripheral Immune Cells in CNS Inflammation
3.5. The Role of the BBB in Mood Disorders
3.6. Neurogenesis and Neuroplasticity in Depression
3.7. Oxidative Stress and Its Impact on Neurotransmitter Systems
4. The Gut–Brain Axis and Neuroinflammation in Depression
4.1. Gut Microbiota Composition in Depression
4.2. Gut Permeability and Its Effect on Neuroinflammation
4.3. Microbial-Derived Metabolites and Their Role in Neurotransmitter Production
5. Clinical Implications: From Mechanisms to Treatments
5.1. The Role of Anti-Inflammatory Drugs in Depression
5.2. Current and Emerging Biomarkers for Personalized Treatment Approaches
5.2.1. Interleukin-1 Family: IL-1β and IL-1RA
5.2.2. Chemokines: CCL2 and CXCL12
5.2.3. Interferon-Gamma (IFN-γ)
5.2.4. Transforming Growth Factor-Beta (TGF-β)
5.2.5. Soluble Tumor Necrosis Factor Receptors (sTNFRs)
5.2.6. Neopterin
5.2.7. Kynurenine Pathway Metabolites
5.3. How Depression Therapies Target Neuroinflammation
6. Conclusions
- ➢
- Development of more precise biomarkers for neuroinflammation in depression, which could help identify patients who may benefit from anti-inflammatory interventions;
- ➢
- Exploration of novel anti-inflammatory agents and immunomodulatory therapies as potential treatments for depression, particularly in cases where traditional antidepressants have proven ineffective;
- ➢
- Further elucidation of the mechanisms by which gut microbiota influence neuroinflammation and mood, potentially leading to probiotic or prebiotic interventions for depression;
- ➢
- Investigation of personalized treatment approaches that consider an individual inflammatory profile, genetic predisposition, and environmental factors;
- ➢
- Longitudinal studies to better understand the temporal relationship between neuroinflammation and depression, including potential preventive strategies targeting inflammatory processes.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Troubat, R.; Barone, P.; Leman, S.; Desmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; et al. Neuroinflammation and Depression: A Review. Eur. J. Neurosci. 2021, 53, 151–171. [Google Scholar] [CrossRef]
- Enache, D.; Pariante, C.M.; Mondelli, V. Markers of central inflammation in major depressive disorder: A systematic review and meta-analysis of studies examining cerebrospinal fluid, positron emission tomography and post-mortem brain tissue. Brain Behav. Immun. 2019, 81, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Frid, L.M.; Kessler, U.; Ousdal, O.T.; Hammar, Å.; Haavik, J.; Riemer, F.; Hirnstein, M.; Ersland, L.; Erchinger, V.J.; Ronold, E.H.; et al. Neurobiological Mechanisms of ECT and TMS Treatment in Depression: Study Protocol of a Multimodal Magnetic Resonance Investigation. BMC Psychiatry 2023, 23, 791. [Google Scholar]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar]
- He, Y.; Han, Y.; Liao, X.; Zou, M.; Wang, Y. Biology of cyclooxygenase-2: An application in depression therapeutics. Front. Psychiatry 2022, 13, 1037588. [Google Scholar] [CrossRef]
- Brites, D.; Fernandes, A. Neuroinflammation and Depression: Microglia Activation, Extracellular Microvesicles and microRNA Dysregulation. Front. Cell Neurosci. 2015, 9, 476. [Google Scholar]
- Wang, H.; He, Y.; Sun, Z.; Ren, S.; Liu, M.; Wang, G.; Yang, J. Microglia in depression: An overview of microglia in the pathogenesis and treatment of depression. J. Neuroinflamm. 2022, 19, 132. [Google Scholar] [CrossRef]
- Sălcudean, A.; Ősz, B.E.; Nan, A.G.; Sasu, A.B.; Cozma, M.M.; Tilinca, M.C.; Bocsak-Pop, A.R.; Strete, E.G. The efficacy of antidepresive treatment on COVID-19 pandemic-related depressive symptoms—A prospective, single-center, questionnaire-based study. Farmacia 2023, 71, 631–637. [Google Scholar]
- Remus, J.L.; Dantzer, R. Inflammation Models of Depression in Rodents: Relevance to Psychotropic Drug Discovery. Int. J. Neuropsychopharmacol. 2016, 19, pyw028. [Google Scholar] [CrossRef]
- Willette, A.A.; Pappas, C.; Hoth, N.; Wang, Q.; Klinedinst, B.; Willette, S.A.; Larsen, B.; Pollpeter, A.; Li, T.; Le, S.; et al. Inflammation, Negative Affect, and Amyloid Burden in Alzheimer’s Disease: Insights from the Kynurenine Pathway. Brain Behav. Immun. 2021, 95, 216–225. [Google Scholar] [CrossRef]
- Najjar, S.; Pearlman, D.M.; Alper, K.; Najjar, A.; Devinsky, O. Neuroinflammation and psychiatric illness. J. Neuroinflamm. 2013, 10, 816. [Google Scholar]
- Kim, Y.K.; Na, K.S.; Myint, A.M.; Leonard, B.E. The role of pro-inflammatory cytokines in neuroinflammation, neurogenesis, and the neuroendocrine system in major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 277–284. [Google Scholar] [PubMed]
- Decker Ramirez, E.B.; Arnold, M.E.; Schank, J.R. Vicarious defeat stress induces increased alcohol consumption in female mice: Role of neurokinin-1 receptor and interleukin-6. Addict. Biol. 2024, 29, e13357. [Google Scholar] [CrossRef]
- Peng, Z.; Peng, S.; Lin, K.; Zhao, B.; Wei, L.; Tuo, Q.; Liao, D.; Yuan, T.; Shi, Z. Chronic stress-induced depression requires the recruitment of peripheral Th17 cells into the brain. J. Neuroinflamm. 2022, 19, 186. [Google Scholar]
- Rodrigues, R.S.; Moreira, J.B.; Mateus, J.M.; Barateiro, A.; Paulo, S.L.; Vaz, S.H.; Lourenço, D.M.; Ribeiro, F.F.; Soares, R.; Loureiro-Campos, E.; et al. Cannabinoid Type 2 Receptor Inhibition Enhances the Antidepressant and Proneurogenic Effects of Physical Exercise after Chronic Stress. Transl Psychiatry 2024, 14, 170. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhai, Q.; Wang, S.; Li, Q.; Liu, J.; Meng, F.; Wang, W.; Zhang, J.; Wang, D.; Zhao, D.; et al. Cryptotanshinone Ameliorates CUS-Induced Depressive-like Behaviors in Mice. Transl Neurosci 2021, 12, 469–481. [Google Scholar] [CrossRef]
- Strauss, C.; Cavanagh, K.; Oliver, A.; Pettman, D. Mindfulness-based interventions for people diagnosed with a current episode of an anxiety or depressive disorder: A meta-analysis of randomized controlled trials. PLoS ONE 2014, 9, e96110. [Google Scholar]
- Balanzá-Martínez, V.; Cervera-Martínez, J. Lifestyle Prescription for Depression with a Focus on Nature Exposure and Screen Time: A Narrative Review. Int. J. Environ. Res. Public Health 2022, 19, 5094. [Google Scholar] [CrossRef]
- Sălcudean, A.; Popovici, R.A.; Pitic, D.E.; Sârbu, D.; Boroghina, A.; Jomaa, M.; Salehi, M.A.; Kher, A.A.M.; Lica, M.M.; Bodo, C.R.; et al. Unraveling the Complex Interplay Between Neuroinflammation and Depression: A Comprehensive Review. Int. J. Mol. Sci. 2025, 26, 1645. [Google Scholar] [CrossRef]
- Xu, Y.; Zheng, Z.; Deng, H.; Wu, C.; Shi, C.; Xu, H. Correlation Between the Plasma Cortisol Level and the Characteristics of Magnetic Resonance Spectroscopy of the Prefrontal Cortex and Hippocampus in Depressed Patients. Int. J. Med. Imaging 2016, 4, 39–43. [Google Scholar]
- Schmaal, L.; Veltman, D.J.; van Erp, T.G.M.; Sämann, P.G.; Frodl, T.; Jahanshad, N.; Loehrer, E.; Tiemeier, H.; Hofman, A.; Niessen, W.J.; et al. Subcortical brain alterations in major depressive disorder: Findings from the ENIGMA Major Depressive Disorder working group. Mol. Psychiatry 2015, 21, 806–812. [Google Scholar] [CrossRef]
- Hasanova, L. The changes of serotonin-modulating anticonsolidation protein and dihydropyrimidinase-related protein 2 in the amygdala and blood of depressive rats. Azerbaijan J. Physiol. 2022, 2, 7–12. [Google Scholar] [CrossRef]
- Schmaal, L.; Hibar, D.; Sämann, P.; Hall, G.; Baune, B.; Jahanshad, N.; Veltman, D. Cortical abnormalities in adults and adolescents with major depression based on brain scans from 20 cohorts worldwide in the enigma major depressive disorder working group. Mol. Psychiatry 2016, 22, 900–909. [Google Scholar] [CrossRef]
- Whittle, S.; Rakesh, D.; Simmons, J.; Schwartz, O.; Allen, N.; Vijayakumar, N. Associations Between Brain Structural Development and Onset of Depressive Disorder During Adolescence and Emerging Adulthood: Results From a 15-Year Longitudinal Study. Biol. Psychiatry 2024, 95, S59. [Google Scholar] [CrossRef]
- Wu, B.; Li, X.; Zhou, J.; Zhang, M.; Long, Q. Altered Whole-Brain Functional Networks in Drug-Naïve, First-Episode Adolescents with Major Depression Disorder. J. Magn. Reson. Imaging 2020, 52, 1790–1798. [Google Scholar] [CrossRef] [PubMed]
- Joseph, C.; Wang, L.; Wu, R.; Manning, K.J.; Steffens, D.C. Structural brain changes and neuroticism in late-life depression: A neural basis for depression subtypes. Int. Psychogeriatr. 2021, 33, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Tripp, A.; Oh, H.; Guilloux, J.P.; Martinowich, K.; Lewis, D.A.; Sibille, E. Brain-derived neurotrophic factor signaling and subgenual anterior cingulate cortex dysfunction in major depressive disorder. Am. J. Psychiatry 2012, 169, 1194–1202. [Google Scholar] [CrossRef]
- Connolly, C.G.; Wu, J.; Ho, T.C.; Hoeft, F.; Wolkowitz, O.; Eisendrath, S.; Frank, G.; Hendren, R.; Max, J.E.; Paulus, M.P.; et al. Resting-state functional connectivity of subgenual anterior cingulate cortex in depressed adolescents. Biol. Psychiatry 2013, 74, 898–907. [Google Scholar] [CrossRef]
- Rolls, E.T.; Cheng, W.; Gong, W.; Qiu, J.; Zhou, C.; Zhang, J.; Lv, W.; Ruan, H.; Wei, D.; Cheng, K.; et al. Functional Connectivity of the Anterior Cingulate Cortex in Depression and in Health. Cereb. Cortex 2019, 29, 3617–3630. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, K.; Li, J.; Zhu, H.; Chen, B.; Liu, X. Disrupted topological organization of functional brain networks in Alzheimer’s disease patients with depressive symptoms. BMC Psychiatry 2022, 22, 810. [Google Scholar] [CrossRef]
- Ory, D.; Celen, S.; Verbruggen, A.; Bormans, G. PET radioligands for in vivo visualization of neuroinflammation. Curr. Pharm. Des. 2014, 20, 5897–5913. [Google Scholar] [CrossRef] [PubMed]
- Pišlar, A.; Tratnjek, L.; Glavan, G.; Zidar, N.; Živin, M.; Kos, J. Neuroinflammation-Induced Upregulation of Glial Cathepsin X Expression and Activity in vivo. Front. Mol. Neurosci. 2020, 13, 575453. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Herrmann, U.S.; Falsig, J.; Abakumova, I.; Nuvolone, M.; Schwarz, P.; Frauenknecht, K.; Rushing, E.J.; Aguzzi, A. A neuroprotective role for microglia in prion diseases. J. Exp. Med. 2016, 213, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.A.; Chesebro, B. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 2019, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhang, Y.; Yue, J.; Shi, Y.; Xiao, B.; Xiao, W.; Luo, Z. Non-coding RNAs: The Neuroinflammatory Regulators in Neurodegenerative Diseases. Front. Neurol. 2022, 13, 929290. [Google Scholar] [CrossRef]
- Wilcock, D.M. A changing perspective on the role of neuroinflammation in Alzheimer’s disease. Int. J. Alzheimers Dis. 2012, 2012, 495243. [Google Scholar] [CrossRef] [PubMed]
- Roboon, J.; Hattori, T.; Ishii, H.; Takarada-Iemata, M.; Nguyen, D.T.; Heer, C.D.; O’Meally, D.; Brenner, C.; Yamamoto, Y.; Okamoto, H.; et al. Inhibition of CD38 and supplementation of nicotinamide riboside ameliorate lipopolysaccharide-induced microglial and astrocytic neuroinflammation by increasing NAD. J. Neurochem. 2021, 158, 311–327. [Google Scholar] [CrossRef]
- Cerami, C.; Iaccarino, L.; Perani, D. Molecular Imaging of Neuroinflammation in Neurodegenerative Dementias: The Role of In Vivo PET Imaging. Int. J. Mol. Sci. 2017, 18, 993. [Google Scholar] [CrossRef]
- McCarrey, A.C.; Pacheco, J.; Carlson, O.D.; Egan, J.M.; Thambisetty, M.; An, Y.; Ferrucci, L.; Resnick, S.M. Interleukin-6 is linked to longitudinal rates of cortical thinning in aging. Transl. Neurosci. 2014, 5, 1–7. [Google Scholar] [CrossRef]
- Li, T.; Lu, L.; Pember, E.; Li, X.; Zhang, B.; Zhu, Z. New Insights into Neuroinflammation Involved in Pathogenic Mechanism of Alzheimer’s Disease and Its Potential for Therapeutic Intervention. Cells 2022, 11, 1925. [Google Scholar] [CrossRef]
- Li, Y.; Chen, X.; Zhou, M.; Feng, S.; Peng, X.; Wang, Y. Microglial TLR4/NLRP3 Inflammasome Signaling in Alzheimer’s Disease. J. Alzheimers Dis. 2024, 97, 75–88. [Google Scholar] [PubMed]
- Pacheco, R. T-cell based immunotherapies for Parkinson’s disease. Explor. Neuroprot Ther. 2021, 1, 72–85. [Google Scholar] [CrossRef]
- More, S.V.; Kumar, H.; Kim, I.S.; Song, S.Y.; Choi, D.K. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediat. Inflamm. 2013, 2013, 952375. [Google Scholar] [CrossRef] [PubMed]
- Tufekci, K.U.; Genc, S.; Genc, K. The endotoxin-induced neuroinflammation model of Parkinson’s disease. Park. Dis. 2011, 2011, 487450. [Google Scholar]
- González, H.; Pacheco, R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J. Neuroinflamm. 2014, 11, 201. [Google Scholar]
- Liu, L.; Zhao, Z.; Lu, L.; Liu, J.; Wu, X.; Sun, J.; Wei, Y.; Dong, J. The role of HMGB1 in neuroinflammation and tissue repair: A potential therapeutic target for depression? Tradit. Med. Mod. Med. 2018, 1, 85–93. [Google Scholar]
- Xu, X.; Piao, H.N.; Aosai, F.; Zeng, X.Y.; Cheng, J.H.; Cui, Y.X.; Li, J.; Ma, J.; Piao, H.R.; Jin, X.; et al. Arctigenin protects against depression by inhibiting microglial activation and neuroinflammation via HMGB1/TLR4/NF-κB and TNF-α/TNFR1/NF-κB pathways. Br. J. Pharmacol. 2020, 177, 5224–5245. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.X.; Xie, X.H.; Yao, L.; Wang, W.; Zhang, H.; Chen, M.M.; Sun, S.; Nie, Z.W.; Nagy, C.; Liu, Z. Human in vivo evidence of reduced astrocyte activation and neuroinflammation in patients with treatment-resistant depression following electroconvulsive therapy. Psychiatry Clin. Neurosci. 2023, 77, 653–664. [Google Scholar] [CrossRef]
- Melnikov, M.; Lopatina, A. Th17-cells in depression: Implication in multiple sclerosis. Front. Immunol. 2022, 13, 1010304. [Google Scholar] [CrossRef]
- Pasqualetti, G.; Brooks, D.J.; Edison, P. The role of neuroinflammation in dementias. Curr. Neurol. Neurosci. Rep. 2015, 15, 17. [Google Scholar]
- Liu, S.; Gao, X.; Zhou, S. New Target for Prevention and Treatment of Neuroinflammation: Microglia Iron Accumulation and Ferroptosis. ASN Neuro 2022, 14, 17590914221133236. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Li, J.; Zhang, X.; Xiong, W. MicroRNA-124 modulates neuroinflammation in acute methanol poisoning rats via targeting Krüppel-like factor-6. Bioengineered 2022, 13, 13507–13519. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.; Li, X.; Fu, M.; Gao, X.; Li, P.; Guo, W. NLRP3-Dependent Pyroptosis: A Candidate Therapeutic target for Depression. Front. Cell. Neurosci. 2022, 16, 863426. [Google Scholar] [CrossRef]
- Zhang, W.; Yan, Z.F.; Gao, J.H.; Sun, L.; Huang, X.Y.; Liu, Z.; Yu, S.Y.; Cao, C.J.; Zuo, L.J.; Chen, Z.J.; et al. Role and mechanism of microglial activation in iron-induced selective and progressive dopaminergic neurodegeneration. Mol. Neurobiol. 2014, 49, 1153–1165. [Google Scholar] [CrossRef]
- Du, H.; Li, C.H.; Gao, R.B.; Tan, Y.; Wang, B.; Peng, Y.; Yang, N.; Ning, Y.L.; Li, P.; Zhao, Y.; et al. Inhibition of the interaction between microglial adenosine 2A receptor and NLRP3 inflammasome attenuates neuroinflammation posttraumatic brain injury. CNS Neurosci. Ther. 2024, 30, e14408. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, J.K.; Kulka, M. Decoding Mast Cell-Microglia Communication in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 1093. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.T.; Wang, S.Q.; Su, J.; Xu, L.X.; Ji, Z.Y.; Zhang, R.Y.; Zhao, Q.W.; Ma, Z.Q.; Deng, X.Y.; Ma, S.P. Baicalin ameliorates neuroinflammation-induced depressive-like behavior through inhibition of toll-like receptor 4 expression via the PI3K/AKT/FoxO1 pathway. J. Neuroinflamm. 2019, 16, 95. [Google Scholar] [CrossRef]
- Robson, M.J.; Quinlan, M.A.; Blakely, R.D. Immune System Activation and Depression: Roles of Serotonin in the Central Nervous System and Periphery. ACS Chem. Neurosci. 2017, 8, 932–942. [Google Scholar] [CrossRef]
- Singhal, G.; Baune, B.T. Microglia: An interface between the loss of neuroplasticity and depression. Frontiers in Cellular Neuroscience. Front. Cell. Neurosci. 2017, 11, 270. [Google Scholar] [CrossRef]
- Culmsee, C.; Michels, S.; Scheu, S.; Arolt, V.; Dannlowski, U.; Alferink, J. Mitochondria, Microglia, and the Immune System-How Are They Linked in Affective Disorders? Front. Psychiatry 2019, 9, 739. [Google Scholar] [CrossRef]
- Jiang, H.; Long, X.; Wang, Y.; Zhang, X.; Chen, L.; Yang, X.; Zhao, B.; Zhang, Y.; Chai, Y.; Bao, T. Acupuncture Ameliorates Depression-Like Behaviors Through Modulating the Neuroinflammation Mediated by TLR4 Signaling Pathway in Rats Exposed to Chronic Restraint Stress. Mol. Neurobiol. 2024, 61, 2606–2619. [Google Scholar] [PubMed]
- Jia, X.; Gao, Z.; Hu, H. Microglia in depression: Current perspectives. Sci. China Life Sci. 2021, 64, 911–925. [Google Scholar]
- Ogłodek, E.; Szota, A.; Just, M.; Moś, D.; Araszkiewicz, A. The role of the neuroendocrine and immune systems in the pathogenesis of depression. Pharmacol. Rep. 2014, 66, 776–781. [Google Scholar]
- Wang, J.; Lai, S.; Zhou, T.; Xia, Z.; Li, W.; Sha, W.; Liu, J.; Chen, Y. Progranulin from different gliocytes in the nucleus accumbens exert distinct roles in FTD- and neuroinflammation-induced depression-like behaviors. J. Neuroinflamm. 2022, 19, 318. [Google Scholar]
- Lin, J.R.; Fang, S.C.; Tang, S.S.; Hu, M.; Long, Y.; Ghosh, A.; Sun, H.B.; Kong, L.Y.; Hong, H. Hippocampal CysLT1R knockdown or blockade represses LPS-induced depressive behaviors and neuroinflammatory response in mice. Acta Pharmacol. Sin. 2017, 38, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Alshaya, D.S. Genetic and epigenetic factors associated with depression: An updated overview. Saudi J. Biol. Sci. 2022, 29, 103311. [Google Scholar]
- Guo, L.; Zhu, Z.; Wang, G.; Cui, S.; Shen, M.; Song, Z.; Wang, J.H. MicroRNA-15b contributes to depression-like behavior in mice by affecting synaptic protein levels and function in the nucleus accumbens. J. Biol. Chem. 2020, 295, 6831–6848. [Google Scholar]
- Akiyama, S.; Nagai, H.; Oike, S.; Horikawa, I.; Shinohara, M.; Lu, Y.; Futamura, T.; Shinohara, R.; Kitaoka, S.; Furuyashiki, T. Chronic Social Defeat Stress Increases the Amounts of 12-Lipoxygenase Lipid Metabolites in the Nucleus Accumbens of Stress-Resilient Mice. Sci. Rep. 2022, 12, 11385. [Google Scholar]
- Kushnareva, E.Y.; Krupina, N.A.; Khlebnikova, N.N.; Zolotov, N.N.; Kryzhanovskii, G.N. Activities of proline-specific peptidases in brain structures of rats with experimental anxiety-depressive state caused by administration of dipeptidyl peptidase IV inhibitors in the early postnatal period. Bull. Exp. Biol. Med. 2011, 151, 675–679. [Google Scholar] [CrossRef]
- Cheng, L.; Huang, C.; Chen, Z. Tauroursodeoxycholic Acid Ameliorates Lipopolysaccharide-Induced Depression Like Behavior in Mice via the Inhibition of Neuroinflammation and Oxido-Nitrosative Stress. Pharmacology 2019, 103, 93–100. [Google Scholar] [CrossRef]
- Zehtabian, K. The role of neurodegeneration in depression: Inflammatory responses and treatment options. Brain 2024, 9, 46–56. [Google Scholar] [CrossRef]
- Colasanti, A.; Guo, Q.; Giannetti, P.; Wall, M.B.; Newbould, R.D.; Bishop, C.; Onega, M.; Nicholas, R.; Ciccarelli, O.; Muraro, P.A.; et al. Hippocampal Neuroinflammation, Functional Connectivity, and Depressive Symptoms in Multiple Sclerosis. Biol. Psychiatry 2016, 80, 62–72. [Google Scholar] [CrossRef]
- Setiawan, E.; Wilson, A.A.; Mizrahi, R.; Rusjan, P.M.; Miler, L.; Rajkowska, G.; Suridjan, I.; Kennedy, J.L.; Rekkas, P.V.; Houle, S.; et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 2015, 72, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Adzic, M.; Brkic, Z.; Mitic, M.; Francija, E.; Jovicic, M.J.; Radulovic, J.; Maric, N.P. Therapeutic Strategies for Treatment of Inflammation-related Depression. Curr. Neuropharmacol. 2018, 16, 176–209. [Google Scholar] [CrossRef] [PubMed]
- Melón, L.; Hooper, A.; Yang, X.; Moss, S.; Maguire, J. Inability to suppress the stress-induced activation of the HPA axis during the peripartum period engenders deficits in postpartum behaviors in mice. Psychoneuroendocrinology 2018, 90, 182–193. [Google Scholar] [CrossRef]
- Serrats, J.; Schiltz, J.; García-Bueno, B.; Rooijen, N.; Reyes, T.; Sawchenko, P. Dual roles for perivascular macrophages in immune-to-brain signaling. Neuron 2010, 65, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Zajkowska, Z.; Gullett, N.; Walsh, A.; Zonca, V.; Pedersen, G.A.; Souza, L.; Kieling, C.; Fisher, H.L.; Kohrt, B.A.; Mondelli, V. Cortisol and Development of Depression in Adolescence and Young Adulthood—A Systematic Review and Meta-Analysis. Psychoneuroendocrinology 2022, 136, 105625. [Google Scholar] [CrossRef]
- Hennings, J.M.; Ising, M.; Uhr, M.; Holsboer, F.; Lucae, S. Recurrent suicide attempts affect normalization of HPA axis dysregulation after recovery from major depression. Front. Psychiatry 2022, 13, 937582. [Google Scholar] [CrossRef]
- Răchită, A.I.C.; Strete, G.E.; Sălcudean, A.; Ghiga, D.V.; Huțanu, A.; Muntean, L.M.; Suciu, L.M.; Mărginean, C. The Relationship between Psychological Sufferings, Value of Maternal Cortisol during Third Trimester of Pregnancy and Breastfeeding Initiation. Medicina 2023, 59, 339. [Google Scholar] [CrossRef]
- Santarsieri, M.; Kumar, R.G.; Kochanek, P.M.; Berga, S.; Wagner, A.K. Variable neuroendocrine-immune dysfunction in individuals with unfavorable outcome after severe traumatic brain injury. Brain Behav. Immun. 2015, 45, 15–27. [Google Scholar] [CrossRef]
- Benatti, C.; Alboni, S.; Blom, J.M.C.; Mendlewicz, J.; Tascedda, F.; Brunello, N. Molecular changes associated with escitalopram response in a stress-based model of depression. Psychoneuroendocrinology 2018, 87, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Kumar, R.G.; Davis, K.; McCullough, E.H.; Berga, S.L.; Wagner, A.K. Longitudinal sex and stress hormone profiles among reproductive age and post-menopausal women after severe TBI: A case series analysis. Brain Inj. 2016, 30, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ting, S.-M.; Sun, G.; Roy-O’Reilly, M.; Mobley, A.S.; Bautista Garrido, J.; Zheng, X.; Obertas, L.; Jung, J.E.; Kruzel, M.; et al. Beneficial Role of Neutrophils Through Function of Lactoferrin After Intracerebral Hemorrhage. Stroke 2018, 49, 1241–1247. [Google Scholar] [CrossRef]
- Oh, D.-R.; Yoo, J.-S.; Kim, Y.; Kang, H.; Lee, H.; Lm, S.J.; Choi, E.; Jung, M.-A.; Bae, D.; Oh, K.-N.; et al. Vaccinium Bracteatum Leaf Extract Reverses Chronic Restraint Stress-Induced Depression-Like Behavior in Mice: Regulation of Hypothalamic-Pituitary-Adrenal Axis, Serotonin Turnover Systems, and ERK/Akt Phosphorylation. Front. Pharmacol. 2018, 9, 604. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.; Li, Z.; Li, Z.; Wang, L.; Hu, Z.; Lü, L.; Wang, Z.; Liu, Y.; Yin, Y.; Li, Z.; et al. Chronic Glucocorticoid Exposure Induces Depression-Like Phenotype in Rhesus Macaque (Macaca Mulatta). Front. Neurosci. 2019, 13, 188. [Google Scholar] [CrossRef]
- Chaki, S. Vasopressin V1B Receptor Antagonists as Potential Antidepressants. Int. J. Neuropsychopharmacol. 2021, 24, 450–463. [Google Scholar] [CrossRef]
- Juruena, M.F.; Bocharova, M.; Agustini, B.; Young, A.H. Atypical depression and non-atypical depression: Is HPA axis function a biomarker? A systematic review. J. Affect. Disord. 2018, 233, 45–67. [Google Scholar] [CrossRef] [PubMed]
- Booij, S.H.; Bouma, E.M.; de Jonge, P.; Ormel, J.; Oldehinkel, A.J. Chronicity of depressive problems and the cortisol response to psychosocial stress in adolescents: The TRAILS study. Psychoneuroendocrinology 2013, 38, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Barton, D.J.; Kumar, R.G.; McCullough, E.H.; Galang, G.; Arenth, P.M.; Berga, S.L.; Wagner, A.K. Persistent Hypogonadotropic Hypogonadism in Men After Severe Traumatic Brain Injury: Temporal Hormone Profiles and Outcome Prediction. J. Head. Trauma. Rehabil. 2016, 31, 277–287. [Google Scholar] [CrossRef]
- Choudhary, K.; Prasad, S.R.; Lokhande, K.B.; Murti, K.; Singh, S.; Ravichandiran, V.; Kumar, N. 4-Methylesculetin ameliorates LPS-induced depression-like behavior through the inhibition of NLRP3 inflammasome. Front. Pharmacol. 2023, 14, 1120508. [Google Scholar] [CrossRef]
- Woldeamanuel, Y.W.; Sanjanwala, B.M.; Cowan, R.P. Endogenous glucocorticoids may serve as biomarkers for migraine chronification. Ther. Adv. Chronic Dis. 2020, 11, 2040622320939793. [Google Scholar] [CrossRef]
- Gültekin, M.; Baydemir, R.; Ismailogullari, S.; Tanriverdi, F.; Mirza, M. Evaluation of serum cortisol and dehydroepiandrosterone sulfate levels in Parkinson patients with and without postural instability. J. Clin. Exp. Investig. 2014, 5, 376–380. [Google Scholar] [CrossRef]
- Hou, L.; Yang, L.; Zhu, C.; Miao, J.; Zhou, W.; Tang, Y.; Meng, H.; Liu, S. Cuscutae Semen Alleviates CUS-Induced Depression-like Behaviors in Mice via the Gut Microbiota-Neuroinflammation Axis. Front. Pharmacol. 2023, 14, 1107781. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, G.J.; Vallender, E.J.; Garrett, M.R.; Challagundla, L.; Overholser, J.C.; Jurjus, G.; Dieter, L.; Syed, M.; Romero, D.G.; Benghuzzi, H.; et al. Altered Neuro-Inflammatory Gene Expression in Hippocampus in Major Depressive Disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 82, 177–186. [Google Scholar] [CrossRef]
- Jeon, S.W.; Kim, Y.K. Neuroinflammation and cytokine abnormality in major depression: Cause or consequence in that illness? World J. Psychiatry 2016, 6, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Barua, C.C.; Haloi, P.; Saikia, B.; Sulakhiya, K.; Pathak, D.C.; Tamuli, S.; Rizavi, H.; Ren, X. Zanthoxylum Alatum Abrogates Lipopolysaccharide-Induced Depression-like Behaviours in Mice by Modulating Neuroinflammation and Monoamine Neurotransmitters in the Hippocampus. Pharm. Biol. 2018, 56, 245–252. [Google Scholar]
- Castaño Barrios, L.; Da Silva Pinheiro, A.P.; Gibaldi, D.; Silva, A.A.; Machado Rodrigues E Silva, P.; Roffê, E.; Da Costa Santiago, H.; Tostes Gazzinelli, R.; Mineo, J.R.; Silva, N.M.; et al. Behavioral Alterations in Long-Term Toxoplasma Gondii Infection of C57BL/6 Mice Are Associated with Neuroinflammation and Disruption of the Blood Brain Barrier. PLoS ONE 2021, 16, e0258199. [Google Scholar] [CrossRef]
- Chen, H.; Dong, M.; He, H.; Piao, X.; Han, X.; Li, R.; Jiang, H.; Li, X.; Li, B.; Cui, R. Ginsenoside Re Prevents Depression-like Behaviors via Inhibition of Inflammation, Oxidative Stress, and Activating BDNF/TrkB/ERK/CREB Signaling: An In Vivo and In Vitro Study. J. Agric. Food Chem. 2024, 72, 19838–19851. [Google Scholar] [CrossRef]
- Tsamakis, K.; Mueller, C.; Tsirigotis, P.; Tsiptsios, D.; Tsamakis, C.; Charakopoulos, E.; Charalampous, C.; Spandidos, D.; Douzenis, A.; Papageorgiou, C.; et al. Depression Following Graft-versus-Host Disease in a Patient with Acute Lymphoblastic Leukaemia: A Case Report. Mol. Clin. Oncol. 2019, 12, 208–211. [Google Scholar] [CrossRef]
- Capuron, L.; Miller, A.H. Immune system to brain signaling: Neuropsychopharmacological implications. Pharmacol. Ther. 2011, 130, 226–238. [Google Scholar] [CrossRef]
- Liang, L.; Wang, H.; Hu, Y.; Bian, H.; Xiao, L.; Wang, G. Oridonin relieves depressive-like behaviors by inhibiting neuroinflammation and autophagy impairment in rats subjected to chronic unpredictable mild stress. Phytother. Res. 2022, 36, 3335–3351. [Google Scholar] [PubMed]
- Cheataini, F.; Ballout, N.; Al Sagheer, T. The effect of neuroinflammation on the cerebral metabolism at baseline and after neural stimulation in neurodegenerative diseases. J. Neurosci. Res. 2023, 101, 1360–1379. [Google Scholar] [CrossRef] [PubMed]
- Benatti, C.; Blom, J.M.C.; Rigillo, G.; Alboni, S.; Zizzi, F.; Torta, R.; Brunello, N.; Tascedda, F. Disease-Induced Neuroinflammation and Depression. CNSNDDT 2016, 15, 414–433. [Google Scholar] [CrossRef]
- Cai, Y.-J.; Wang, F.; Chen, Z.-X.; Li, L.; Fan, H.; Wu, Z.-B.; Ge, J.-F.; Hu, W.; Wang, Q.-N.; Zhu, D.-F. Hashimoto’s Thyroiditis Induces Neuroinflammation and Emotional Alterations in Euthyroid Mice. J. Neuroinflammation 2018, 15, 299. [Google Scholar] [CrossRef]
- Bugge, E.; Wynn, R.; Mollnes, T.E.; Reitan, S.K.; Grønli, O.K. Cytokine Profiles and Diagnoses in Elderly, Hospitalized Psychiatric Patients. BMC Psychiatry 2018, 18, 315. [Google Scholar]
- Dugan, A.M.; Parrott, J.M.; Redus, L.; Hensler, J.G.; O’Connor, J.C. Low-Level Stress Induces Production of Neuroprotective Factors in Wild-Type but Not BDNF+/- Mice: Interleukin-10 and Kynurenic Acid. Int. J. Neuropsychopharmacol. 2015, 19, pyv089. [Google Scholar] [CrossRef]
- Leite, J.A.; Orellana, A.M.A.; Kinoshita, P.F.; de Mello, N.P.; Scavone, C.; Kawamoto, E.M. Neuroinflammation and Neurotransmission Mechanisms Involved in Neuropsychiatric Disorders. In Mechanisms of Neuroinflammation; Abreu, A., Ed.; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, J.; Liu, Y.; Li, Z.; Li, X. TLR4-NF-κB Signal Involved in Depressive-Like Behaviors and Cytokine Expression of Frontal Cortex and Hippocampus in Stressed C57BL/6 and ob/ob Mice. Neural Plast. 2018, 2018, 7254016. [Google Scholar] [CrossRef]
- Gałecka, M.; Bliźniewska-Kowalska, K.; Orzechowska, A.; Szemraj, J.; Maes, M.; Berk, M.; Su, K.-P.; Gałecki, P. Inflammatory versus Anti-Inflammatory Profiles in Major Depressive Disorders—The Role of IL-17, IL-21, IL-23, IL-35 and Foxp3. JPM 2021, 11, 66. [Google Scholar] [PubMed]
- Liu, R.; Liu, L.; Ren, S.; Wei, C.; Wang, Y.; Li, D.; Zhang, W. The role of IL-33 in depression: A systematic review and meta-analysis. Front. Psychiatry 2023, 14, 1242367. [Google Scholar] [CrossRef]
- Silva, Z.M.; Toledo, D.N.M.; Pio, S.; Machado, B.A.A.; Santos, P.V.D.; Hó, F.G.; Medina, Y.N.; Cordeiro, P.H.D.M.; Perucci, L.O.; Pinto, K.M.D.C.; et al. Neuroserpin, IL-33 and IL-17A as Potential Markers of Mild Symptoms of Depressive Syndrome in Toxoplasma Gondii-Infected Pregnant Women. Front. Immunol. 2024, 15, 1394456. [Google Scholar]
- Kudinova, A.Y.; Deak, T.; Hueston, C.M.; McGeary, J.E.; Knopik, V.S.; Palmer, R.H.; Gibb, B.E. Cross-species evidence for the role of interleukin-33 in depression risk. J. Abnorm. Psychol. 2016, 125, 482–494. [Google Scholar] [CrossRef]
- Kikuchi, M.; Takase, K.; Hayakawa, M.; Hayakawa, H.; Tominaga, S.I.; Ohmori, T. Altered behavior in mice overexpressing soluble ST2. Mol. Brain 2020, 13, 74. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.A.; Wertz, J.; Zhu, D.; Cattaneo, A.; Musaelyan, K.; Nikkheslat, N.; Thuret, S.; Pariante, C.M.; Zunszain, P.A. Antidepressant Compounds Can Be Both Pro- and Anti-Inflammatory in Human Hippocampal Cells. Int. J. Neuropsychopharmacol. 2014, 18, pyu076. [Google Scholar] [CrossRef]
- Ma, X.; Wang, J.; Quan, Q.; Zhang, H.; Tian, Y.; Wang, L.; Liu, L. Sestrin2 attenuates depressive-like behaviors and neuroinflammation in CUMS mice through inhibiting ferroptosis. Neuroreport 2024, 35, 143–151. [Google Scholar] [PubMed]
- Chan, C.K.; Tian, F.; Pimentel Maldonado, D.; Mowry, E.M.; Fitzgerald, K.C. Depression in multiple sclerosis across the adult lifespan. Mult. Scler. 2021, 27, 1771–1780. [Google Scholar] [PubMed]
- Park, H.; Han, K.-M.; Jeon, H.; Lee, J.-S.; Lee, H.; Jeon, S.G.; Park, J.-H.; Kim, Y.G.; Lin, Y.; Lee, Y.-H.; et al. The MAO Inhibitor Tranylcypromine Alters LPS- and Aβ-Mediated Neuroinflammatory Responses in Wild-Type Mice and a Mouse Model of AD. Cells 2020, 9, 1982. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Han, Q.-Q.; Gong, W.-Q.; Pan, D.-H.; Wang, L.-Z.; Hu, W.; Yang, M.; Li, B.; Yu, J.; Liu, Q. Microglial Activation Mediates Chronic Mild Stress-Induced Depressive- and Anxiety-like Behavior in Adult Rats. J. Neuroinflammation. 2018, 15, 21. [Google Scholar] [PubMed]
- Hirshman, N.A.; Hughes, F.M.; Jin, H.; Harrison, W.T.; White, S.W.; Doan, I.; Harper, S.N.; Leidig, P.D.; Purves, J.T. Cyclophosphamide-Induced Cystitis Results in NLRP3-Mediated Inflammation in the Hippocampus and Symptoms of Depression in Rats. Am. J. Physiol. Renal Physiol. 2020, 318, F354–F362. [Google Scholar] [CrossRef]
- Bai, H.; Chen, S.; Yuan, T.; Xu, D.; Cui, S.; Li, X. Paeoniflorin ameliorates neuropathic pain-induced depression-like behaviors in mice by inhibiting hippocampal neuroinflammation activated via TLR4/NF-κB pathway. Korean J. Physiol. Pharmacol. 2021, 25, 217–225. [Google Scholar] [CrossRef]
- Monje, F.J.; Cabatic, M.; Divisch, I.; Kim, E.J.; Herkner, K.R.; Binder, B.R.; Pollak, D.D. Constant darkness induces IL-6-dependent depression-like behavior through the NF-κB signaling pathway. J. Neurosci. 2011, 31, 9075–9083. [Google Scholar] [CrossRef]
- Covelo, A.; Araque, A. Neuronal activity determines distinct gliotransmitter release from a single astrocyte. Elife 2018, 7, e32237. [Google Scholar] [CrossRef] [PubMed]
- Kano, S.-I.; Choi, E.Y.; Dohi, E.; Agarwal, S.; Chang, D.J.; Wilson, A.M.; Lo, B.D.; Rose, I.V.L.; Gonzalez, S.; Imai, T.; et al. Glutathione S-Transferases Promote Proinflammatory Astrocyte-Microglia Communication during Brain Inflammation. Sci. Signal 2019, 12, eaar2124. [Google Scholar]
- Ellis, R.J.; Fan, Y.; Grelotti, D.; Tang, B.; Letendre, S.; He, J.J. Astrocyte Activation is A Potential Mechanism Underlying Depressed Mood and Apathy in People with HIV. J. Neurol. Psychol. 2022, 9, 05. [Google Scholar] [CrossRef] [PubMed]
- Pannell, M.; Szulzewsky, F.; Matyash, V.; Wolf, S.A.; Kettenmann, H. The subpopulation of microglia sensitive to neurotransmitters/neurohormones is modulated by stimulation with LPS, interferon-γ, and IL-4. Glia 2014, 62, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lai, S.; Wang, R.; Zhou, T.; Dong, N.; Zhu, L.; Chen, T.; Zhang, X.; Chen, Y. Dopamine D3 Receptor in the Nucleus Accumbens Alleviates Neuroinflammation in a Mouse Model of Depressive-like Behavior. Brain Behav. Immun. 2022, 101, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Ruan, L.; Qian, L.; Liu, X.; Le, Y. Norepinephrine promotes microglia to uptake and degrade amyloid beta peptide through upregulation of mouse formyl peptide receptor 2 and induction of insulin-degrading enzyme. J. Neurosci. 2010, 30, 11848–11857. [Google Scholar] [PubMed]
- Böttcher, C.; Fernández-Zapata, C.; Snijders, G.J.L.; Schlickeiser, S.; Sneeboer, M.A.M.; Kunkel, D.; De Witte, L.D.; Priller, J. Single-cell mass cytometry of microglia in major depressive disorder reveals a non-inflammatory phenotype with increased homeostatic marker expression. Transl. Psychiatry 2020, 10, 310. [Google Scholar] [CrossRef]
- Zhang, M.M.; Guo, M.X.; Zhang, Q.P.; Chen, X.Q.; Li, N.Z.; Liu, Q.; Cheng, J.; Wang, S.L.; Xu, G.H.; Li, C.F.; et al. IL-1R/C3aR signaling regulates synaptic pruning in the prefrontal cortex of depression. Cell Biosci. 2022, 12, 90. [Google Scholar] [CrossRef]
- Steiner, J.; Walter, M.; Gos, T.; Guillemin, G.J.; Bernstein, H.-G.; Sarnyai, Z.; Mawrin, C.; Brisch, R.; Bielau, H.; Meyer zu Schwabedissen, L.; et al. Severe Depression Is Associated with Increased Microglial Quinolinic Acid in Subregions of the Anterior Cingulate Gyrus: Evidence for an Immune-Modulated Glutamatergic Neurotransmission? J. Neuroinflammation 2011, 8, 94. [Google Scholar] [CrossRef]
- Villarreal, A.; Vidos, C.; Monteverde Busso, M.; Cieri, M.B.; Ramos, A.J. Pathological Neuroinflammatory Conversion of Reactive Astrocytes Is Induced by Microglia and Involves Chromatin Remodeling. Front. Pharmacol. 2021, 12, 689346. [Google Scholar] [CrossRef]
- Puñal, V.M.; Paisley, C.E.; Brecha, F.S.; Lee, M.A.; Perelli, R.M.; Wang, J.; O’Koren, E.G.; Ackley, C.R.; Saban, D.R.; Reese, B.E.; et al. Large-scale death of retinal astrocytes during normal development is non-apoptotic and implemented by microglia. PLoS Biol. 2019, 17, e3000492. [Google Scholar]
- Du, R.H.; Wu, F.F.; Lu, M.; Shu, X.D.; Ding, J.H.; Wu, G.; Hu, G. Uncoupling protein 2 modulation of the NLRP3 inflammasome in astrocytes and its implications in depression. Redox Biol. 2016, 9, 178–187. [Google Scholar] [PubMed]
- Yu, X.; Wang, S.; Wu, W.; Chang, H.; Shan, P.; Yang, L.; Zhang, W.; Wang, X. Exploring New Mechanism of Depression from the Effects of Virus on Nerve Cells. Cells 2023, 12, 1767. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Peng, Y.L.; Liu, Y.Z.; Wu, T.Y.; Shen, X.L.; Zhou, J.R.; Sun, D.Y.; Huang, A.J.; Wang, X.; et al. Involvement of inflammasome activation in lipopolysaccharide-induced mice depressive-like behaviors. CNS Neurosci. Ther. 2014, 20, 119–124. [Google Scholar]
- Jiang, P.; Guo, Y.; Dang, R.; Yang, M.; Liao, D.; Li, H.; Sun, Z.; Feng, Q.; Xu, P. Salvianolic acid B protects against lipopolysaccharide-induced behavioral deficits and neuroinflammatory response: Involvement of autophagy and NLRP3 inflammasome. J. Neuroinflamm. 2017, 14, 239. [Google Scholar]
- Sun, D.; Gao, G.; Zhong, B.; Zhang, H.; Ding, S.; Sun, Z.; Zhang, Y.; Li, W. NLRP1 inflammasome involves in learning and memory impairments and neuronal damages during aging process in mice. Behav. Brain Funct. 2021, 17, 11. [Google Scholar]
- Mi, L.; Min, X.; Chai, Y.; Zhang, J.; Chen, X. NLRP1 Inflammasomes: A Potential Target for the Treatment of Several Types of Brain Injury. Front. Immunol. 2022, 13, 863774. [Google Scholar]
- Li, X.; Zhang, H.; Yang, L.; Dong, X.; Han, Y.; Su, Y.; Li, W.; Li, W. Inhibition of NLRP1 inflammasome improves autophagy dysfunction and Aβ disposition in APP/PS1 mice. Behav. Brain Funct. 2023, 19, 7. [Google Scholar]
- Alizadehmoghaddam, S.; Pourabdolhossein, F.; Najafzadehvarzi, H.; Sarbishegi, M.; Saleki, K.; Nouri, H.R. Crocin attenuates the lipopolysaccharide-induced neuroinflammation via expression of AIM2 and NLRP1 inflammasome in an experimental model of Parkinson’s disease. Heliyon 2024, 10, e25523. [Google Scholar] [CrossRef]
- Hu, B.; Zhang, J.; Huang, J.; Luo, B.; Zeng, X.; Jia, J. NLRP3/1-mediated pyroptosis: Beneficial clues for the development of novel therapies for Alzheimer’s disease. Neural Regen. Res. 2024, 19, 2400–2410. [Google Scholar]
- Shen, Y.; Qian, L.; Luo, H.; Li, X.; Ruan, Y.; Fan, R.; Si, Z.; Chen, Y.; Li, L.; Liu, Y. The Significance of NLRP Inflammasome in Neuropsychiatric Disorders. Brain Sci. 2022, 12, 1057. [Google Scholar] [CrossRef]
- Lian, H.; Litvinchuk, A.; Chiang, A.C.; Aithmitti, N.; Jankowsky, J.L.; Zheng, H. Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J. Neurosci. 2016, 36, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Leng, L.; Zhuang, K.; Liu, Z.; Huang, C.; Gao, Y.; Chen, G.; Lin, H.; Hu, Y.; Wu, D.; Shi, M.; et al. Menin Deficiency Leads to Depressive-like Behaviors in Mice by Modulating Astrocyte-Mediated Neuroinflammation. Neuron 2018, 100, 551–563.e7. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Shen, Y.; Liu, P.; Shen, Y.; Hu, Y.; Li, P.; Zhang, Y.; Miao, F.; Zhang, J. NLRC5 Deficiency Reduces LPS-Induced Microglial Activation via Inhibition of NF-κB Signaling and Ameliorates Mice’s Depressive-like Behavior. Int. J. Mol. Sci. 2023, 24, 13265. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, Y.; Duan, Q.; Ni, K.; Jiao, Y.; Zhu, J.; Sun, J.; Zhang, W.; Ma, Z. Dehydrocorydaline alleviates sleep deprivation-induced persistent postoperative pain in adolescent mice through inhibiting microglial P2Y12 receptor expression in the spinal cord. Mol. Pain 2023, 19, 17448069231216234. [Google Scholar] [CrossRef]
- Zhao, Q.; Wu, X.; Yan, S.; Xie, X.; Fan, Y.; Zhang, J.; Peng, C.; You, Z. The antidepressant-like effects of pioglitazone in a chronic mild stress mouse model are associated with PPARγ-mediated alteration of microglial activation phenotypes. J. Neuroinflamm. 2016, 13, 259. [Google Scholar] [CrossRef]
- He, L.; Zheng, Y.; Huang, L.; Ye, J.; Ye, Y.; Luo, H.; Chen, X.; Yao, W.; Chen, J.; Zhang, J.C. Nrf2 regulates the arginase 1+ microglia phenotype through the initiation of TREM2 transcription, ameliorating depression-like behavior in mice. Transl. Psychiatry 2022, 12, 459. [Google Scholar] [CrossRef]
- Fang, Y.; Guo, H.; Wang, Q.; Liu, C.; Ge, S.; Yan, B. The role and mechanism of NLRP3 inflammasome-mediated astrocyte activation in dehydrocorydaline against CUMS-induced depression. Front. Pharmacol. 2022, 13, 1008249. [Google Scholar] [CrossRef]
- Cui, Y.; Yang, Y.; Ni, Z.; Dong, Y.; Cai, G.; Foncelle, A.; Ma, S.; Sang, K.; Tang, S.; Li, Y.; et al. Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature 2018, 554, 323–327. [Google Scholar] [CrossRef]
- Da Mesquita, S.; Herz, J.; Wall, M.; Dykstra, T.; de Lima, K.A.; Norris, G.T.; Dabhi, N.; Kennedy, T.; Baker, W.; Kipnis, J. Aging-associated deficit in CCR7 is linked to worsened glymphatic function, cognition, neuroinflammation, and β-amyloid pathology. Sci. Adv. 2021, 7, eabe4601. [Google Scholar] [CrossRef]
- Carmona-Mora, P.; Ander, B.P.; Jickling, G.C.; Dykstra-Aiello, C.; Zhan, X.; Ferino, E.; Hamade, F.; Amini, H.; Hull, H.; Sharp, F.R.; et al. Distinct peripheral blood monocyte and neutrophil transcriptional programs following intracerebral hemorrhage and different etiologies of ischemic stroke. J. Cereb. Blood Flow. Metab. 2021, 41, 1398–1416. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, S.A.; Wen, J.; Shum, J.; Doerner, J.; Herlitz, L.; Putterman, C. CSF-1R inhibition attenuates renal and neuropsychiatric disease in murine lupus. Clin. Immunol. 2017, 185, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef]
- Shelton, R.C.; Miller, A.H. Inflammation in depression: Is adiposity a cause? Dialogues Clin. Neurosci. 2011, 13, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Du, B.; Zhang, H.; Lu, X.; Chen, C.; Fan, C.; Bi, X. NLR Is Associated with Geriatric Depression in Chinese Women: A Community-Based Cross-Sectional Study in Eastern China. Front. Psychol. 2020, 10, 2941. [Google Scholar] [CrossRef]
- Benton, T.; Lynch, K.; Dubé, B.; Gettes, D.R.; Tustin, N.B.; Ping Lai, J.; Metzger, D.S.; Blume, J.; Douglas, S.D.; Evans, D.L.; et al. Selective serotonin reuptake inhibitor suppression of HIV infectivity and replication. Psychosom. Med. 2010, 72, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Luo, F.; Shi, G.; Chen, R.; Zhou, P. Depression and macrophages: A bibliometric and visual analysis from 2000 to 2022. Medicine 2023, 102, e34174. [Google Scholar] [CrossRef]
- Jiang, A.; Zhang, S.; Li, Z.; Liang, R.; Ren, S.; Li, J.; Pu, Y.; Yang, J. MiR-615-3p promotes the phagocytic capacity of splenic macrophages by targeting ligand-dependent nuclear receptor corepressors in cirrhosis-related portal hypertension. Exp. Biol. Med. 2011, 236, 672–680. [Google Scholar] [CrossRef]
- Greeson, J.M.; Gettes, D.R.; Spitsin, S.; Dubé, B.; Benton, T.D.; Lynch, K.G.; Douglas, S.D.; Evans, D.L. The Selective Serotonin Reuptake Inhibitor Citalopram Decreases Human Immunodeficiency Virus Receptor and Coreceptor Expression in Immune Cells. Biol. Psychiatry 2016, 80, 33–39. [Google Scholar] [CrossRef]
- Dey, A.; Hankey Giblin, P.A. Insights into Macrophage Heterogeneity and Cytokine-Induced Neuroinflammation in Major Depressive Disorder. Pharmaceuticals 2018, 11, 64. [Google Scholar] [CrossRef]
- Beumer, W.; Gibney, S.M.; Drexhage, R.C.; Pont-Lezica, L.; Doorduin, J.; Klein, H.C.; Steiner, J.; Connor, T.J.; Harkin, A.; Versnel, M.A.; et al. The immune theory of psychiatric diseases: A key role for activated microglia and circulating monocytes. J. Leukoc. Biol. 2012, 92, 959–975. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Fan, C.; Wang, P.; Li, Y.; Yang, M.; Yu, S.Y. Hippocampal CA1 βCaMKII mediates neuroinflammatory responses via COX-2/PGE2 signaling pathways in depression. J. Neuroinflamm. 2018, 15, 338. [Google Scholar]
- Sharma, A. Systems Genomics Support for Immune and Inflammation Hypothesis of Depression. Curr. Neuropharmacol. 2016, 14, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Miller, A.H. Is depression an inflammatory disorder? Curr. Psychiatry Rep. 2011, 13, 467–475. [Google Scholar] [CrossRef]
- Alvarez-Mon, M.A.; Gómez, A.M.; Orozco, A.; Lahera, G.; Sosa, M.D.; Diaz, D.; Auba, E.; Albillos, A.; Monserrat, J.; Alvarez-Mon, M. Abnormal Distribution and Function of Circulating Monocytes and Enhanced Bacterial Translocation in Major Depressive Disorder. Front. Psychiatry 2019, 10, 812. [Google Scholar]
- Gao, X.; Duan, S.; Cao, Y.; Zhang, Y. Change of monocytes/macrophages in ulcerative colitis patients with symptoms of anxiety and depression. BMC Gastroenterol. 2023, 23, 67. [Google Scholar] [CrossRef]
- Shechter, R.; Miller, O.; Yovel, G.; Rosenzweig, N.; London, A.; Ruckh, J.; Kim, K.W.; Klein, E.; Kalchenko, V.; Bendel, P.; et al. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity 2013, 38, 555–569. [Google Scholar]
- Yan, P.; Kim, K.W.; Xiao, Q.; Ma, X.; Czerniewski, L.R.; Liu, H.; Rawnsley, D.R.; Yan, Y.; Randolph, G.J.; Epelman, S.; et al. Peripheral monocyte-derived cells counter amyloid plaque pathogenesis in a mouse model of Alzheimer’s disease. J. Clin. Investig. 2022, 132, e152565. [Google Scholar]
- Ma, Y.; Li, Y.; Jiang, L.; Wang, L.; Jiang, Z.; Wang, Y.; Zhang, Z.; Yang, G.Y. Macrophage depletion reduced brain injury following middle cerebral artery occlusion in mice. J. Neuroinflamm. 2016, 13, 38. [Google Scholar]
- Richter, N.; Wendt, S.; Georgieva, P.B.; Hambardzumyan, D.; Nolte, C.; Kettenmann, H. Glioma-associated microglia and macrophages/monocytes display distinct electrophysiological properties and do not communicate via gap junctions. Neurosci. Lett. 2014, 583, 130–135. [Google Scholar] [CrossRef]
- Lica, M.M.; Papai, A.; Salcudean, A.; Crainic, M.; Covaciu, C.G.; Mihai, A. Assessment of Psychopathology in Adolescents with Insulin-Dependent Diabetes (IDD) and the Impact on Treatment Management. Children 2021, 8, 414. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Lee, H.; Lee, G.; Oh, S.J.; Shin, M.K.; Shim, I.; Bae, H. CD4+CD25+ regulatory T cell depletion modulates anxiety and depression-like behaviors in mice. PLoS ONE 2012, 7, e42054. [Google Scholar] [CrossRef]
- Brachman, R.A.; Lehmann, M.L.; Maric, D.; Herkenham, M. Lymphocytes from chronically stressed mice confer antidepressant-like effects to naive mice. J. Neurosci. 2015, 35, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Lawson, M.A.; Kelley, K.W. Inflammation-associated depression: From serotonin to kynurenine. Psychoneuroendocrinology 2011, 36, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.; Dikshit, R.; Karia, S.; Sonavane, S.; Shah, N.; De Sousa, A. Neutrophil-lymphocyte Ratio and C-reactive Protein Level in Patients with Major Depressive Disorder Before and After Pharmacotherapy. East. Asian Arch. Psychiatry 2018, 28, 53–58. [Google Scholar] [PubMed]
- Daray, F.M.; Grendas, L.N.; Arena, Á.R.; Tifner, V.; Álvarez Casiani, R.I.; Olaviaga, A.; Chiapella, L.C.; Vázquez, G.; Penna, M.B.; Hunter, F.; et al. Decoding the inflammatory signature of the major depressive episode: Insights from peripheral immunophenotyping in active and remitted condition, a case-control study. Transl. Psychiatry 2024, 14, 254. [Google Scholar] [CrossRef]
- Beurel, E.; Harrington, L.E.; Jope, R.S. Inflammatory T helper 17 cells promote depression-like behavior in mice. Biol. Psychiatry 2013, 73, 622–630. [Google Scholar] [CrossRef]
- Austin, P.J.; Berglund, A.M.; Siu, S.; Fiore, N.T.; Gerke-Duncan, M.B.; Ollerenshaw, S.L.; Leigh, S.J.; Kunjan, P.A.; Kang, J.W.; Keay, K.A. Evidence for a distinct neuro-immune signature in rats that develop behavioural disability after nerve injury. J. Neuroinflamm. 2015, 12, 96. [Google Scholar] [CrossRef]
- Demir, S.; Atli, A.; Bulut, M.; İbiloğlu, A.O.; Güneş, M.; Kaya, M.C.; Demirpençe, Ö.; Sır, A. Neutrophil-lymphocyte ratio in patients with major depressive disorder undergoing no pharmacological therapy. Neuropsychiatr. Dis. Treat. 2015, 11, 2253–2258. [Google Scholar]
- Hagenberg, J.; BeCOME study group; OPTIMA study group; Brückl, T.M.; Erhart, M.; Kopf-Beck, J.; Ködel, M.; Rehawi, G.; Röh-Karamihalev, S.; Sauer, S.; et al. Dissecting depression symptoms: Multi-omics clustering uncovers immune-related subgroups and cell-type specific dysregulation. Brain Behav. Immun. 2025, 123, 353–369. [Google Scholar] [CrossRef]
- Casanova-Acebes, M.; Nicolás-Ávila, J.A.; Li, J.L.; García-Silva, S.; Balachander, A.; Rubio-Ponce, A.; Weiss, L.A.; Adrover, J.M.; Burrows, K.; A-González, N.; et al. Neutrophils instruct homeostatic and pathological states in naive tissues. J. Exp. Med. 2018, 215, 2778–2795. [Google Scholar] [PubMed]
- Tlili, A.; Pintard, C.; Hurtado-Nedelec, M.; Liu, D.; Marzaioli, V.; Thieblemont, N.; Dang, P.M.; El-Benna, J. ROCK2 interacts with p22phox to phos-phorylate p47phox and to control NADPH oxidase activation in human monocytes. Proc. Natl. Acad. Sci. USA 2023, 120, e2209184120. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.H.; Park, C.J.; Jeong, H.J.; Sunwoo, M.K.; Ahn, S.S.; Lee, S.K.; Lee, P.H.; Kim, Y.J.; Sohn, Y.H.; Chung, S.J. Association of choroid plexus volume with motor symptoms and dopaminergic degeneration in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2023, 94, 1047–1055. [Google Scholar] [CrossRef]
- Yu, P.; Venkat, P.; Chopp, M.; Zacharek, A.; Shen, Y.; Liang, L.; Landschoot-Ward, J.; Liu, Z.; Jiang, R.; Chen, J. Deficiency of tPA Exacerbates White Matter Damage, Neuroinflammation, Glymphatic Dysfunction and Cognitive Dysfunction in Aging Mice. Aging Dis. 2019, 10, 770–783. [Google Scholar]
- Yao, D.; Li, R.; Hao, J.; Huang, H.; Wang, X.; Ran, L.; Fang, Y.; He, Y.; Wang, W.; Liu, X.; et al. Melatonin alleviates depression-like behaviors and cognitive dysfunction in mice by regulating the circadian rhythm of AQP4 polarization. Transl. Psychiatry 2023, 13, 310. [Google Scholar] [PubMed]
- Yang, T.; Tang, Y.; Liu, X.; Gong, S.; Yao, E. Microglia synchronizes with the circadian rhythm of the glymphatic system and modulates glymphatic system function. IUBMB Life 2024, 76, 1209–1222. [Google Scholar] [CrossRef]
- Chi, L.; Cheng, X.; Lin, L.; Yang, T.; Sun, J.; Feng, Y.; Liang, F.; Pei, Z.; Teng, W. Porphyromonas gingivalis-Induced Cognitive Impairment Is Associated With Gut Dysbiosis, Neuroinflammation, and Glymphatic Dysfunction. Front. Cell Infect. Microbiol. 2021, 11, 755925. [Google Scholar]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Devinney, M.J.; Wong, M.K.; Wright, M.C.; Marcantonio, E.R.; Terrando, N.; Browndyke, J.N.; Whitson, H.E.; Cohen, H.J.; Nackley, A.G.; Klein, M.E.; et al. Role of Blood-Brain Barrier Dysfunction in Delirium following Non-cardiac Surgery in Older Adults. Ann. Neurol. 2023, 94, 1024–1035. [Google Scholar]
- Alluri, H.; Wilson, R.L.; Anasooya Shaji, C.; Wiggins-Dohlvik, K.; Patel, S.; Liu, Y.; Peng, X.; Beeram, M.R.; Davis, M.L.; Huang, J.H.; et al. Melatonin Preserves Blood-Brain Barrier Integrity and Permeability via Matrix Metalloproteinase-9 Inhibition. PLoS ONE 2016, 11, e0154427. [Google Scholar] [CrossRef]
- Xiong, J.; Gao, Y.; Li, X.; Li, K.; Li, Q.; Shen, J.; Han, Z.; Zhang, J. Losartan Treatment Could Improve the Outcome of TBI Mice. Front. Neurol. 2020, 11, 992. [Google Scholar] [CrossRef]
- Piers, T.M.; East, E.; Villegas-Llerena, C.; Sevastou, I.G.; Matarin, M.; Hardy, J.; Pocock, J.M. Soluble Fibrinogen Triggers Non-cell Autonomous ER Stress-Mediated Microglial-Induced Neurotoxicity. Front. Cell Neurosci. 2018, 12, 404. [Google Scholar] [CrossRef]
- Rodríguez, J.J.; Verkhratsky, A. Neurogenesis in Alzheimer’s disease. J. Anat. 2011, 219, 78–89. [Google Scholar] [CrossRef]
- Zeiss, C.J. Comparative Milestones in Rodent and Human Postnatal Central Nervous System Development. Toxicol. Pathol. 2021, 49, 1368–1373. [Google Scholar] [CrossRef] [PubMed]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Boström, E.; Westerlund, I.; Vial, C.; Buchholz, B.A.; et al. Dynamics of hippocampal neurogenesis in adult humans. Cell 2013, 153, 1219–1227. [Google Scholar] [PubMed]
- Braun, S.M.; Jessberger, S. Adult neurogenesis and its role in neuropsychiatric disease, brain repair and normal brain function. Neuropathol. Appl. Neurobiol. 2014, 40, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Dey, J.; Alam, M.T.; Chandra, S.; Gandhi, S.; Tripathi, P.P. Recalibrating the Existence of New Neurons in Adult Brain. ACS Chem. Neurosci. 2019, 10, 2091–2093. [Google Scholar]
- Eriksson, P.S.; Perfilieva, E.; Björk-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef]
- Sorrells, S.F.; Paredes, M.F.; Zhang, Z.; Kang, G.; Pastor-Alonso, O.; Biagiotti, S.; Page, C.E.; Sandoval, K.; Knox, A.; Connolly, A.; et al. Positive Controls in Adults and Children Support That Very Few, If Any, New Neurons Are Born in the Adult Human Hippocampus. J. Neurosci. 2021, 41, 2554–2565. [Google Scholar] [CrossRef]
- Dennis, C.V.; Suh, L.S.; Rodriguez, M.L.; Kril, J.J.; Sutherland, G.T. Human adult neurogenesis across the ages: An immunohistochemical study. Neuropathol. Appl. Neurobiol. 2016, 42, 621–638. [Google Scholar]
- Terstege, D.J.; Addo-Osafo, K.; Campbell Teskey, G.; Epp, J.R. New neurons in old brains: Implications of age in the analysis of neurogenesis in post-mortem tissue. Mol. Brain 2022, 15, 38. [Google Scholar] [PubMed]
- Mathews, K.J.; Allen, K.M.; Boerrigter, D.; Ball, H.; Shannon Weickert, C.; Double, K.L. Evidence for reduced neurogenesis in the aging human hippocampus despite stable stem cell markers. Aging Cell 2017, 16, 1195–1199. [Google Scholar] [PubMed]
- Braun, K.; Häberle, B.M.; Wittmann, M.T.; Lie, D.C. Enriched environment ameliorates adult hippocampal neurogenesis deficits in Tcf4 haploinsufficient mice. BMC Neurosci. 2020, 21, 50. [Google Scholar] [CrossRef] [PubMed]
- Lev-Vachnish, Y.; Cadury, S.; Rotter-Maskowitz, A.; Feldman, N.; Roichman, A.; Illouz, T.; Varvak, A.; Nicola, R.; Madar, R.; Okun, E. L-Lactate Promotes Adult Hippocampal Neurogenesis. Front. Neurosci. 2019, 13, 403. [Google Scholar]
- Lei, W.; Li, W.; Ge, L.; Chen, G. Non-engineered and Engineered Adult Neurogenesis in Mammalian Brains. Front. Neurosci. 2019, 13, 131. [Google Scholar]
- Petrik, D.; Encinas, J.M. Perspective: Of Mice and Men—How Widespread Is Adult Neurogenesis? Front. Neurosci. 2019, 13, 923. [Google Scholar]
- Liu, H.; Song, N. Molecular Mechanism of Adult Neurogenesis and its Association with Human Brain Diseases. J. Cent. Nerv. Syst. Dis. 2016, 8, 5–11. [Google Scholar] [CrossRef]
- Price, R.B.; Duman, R. Neuroplasticity in cognitive and psychological mechanisms of depression: An integrative model. Mol. Psychiatry 2020, 25, 530–543. [Google Scholar]
- Noda, Y.; Zomorrodi, R.; Vila-Rodriguez, F.; Downar, J.; Farzan, F.; Cash, R.F.H.; Rajji, T.K.; Daskalakis, Z.J.; Blumberger, D.M. Impaired neuroplasticity in the prefrontal cortex in depression indexed through paired associative stimulation. Depress. Anxiety 2018, 35, 448–456. [Google Scholar]
- Zhou, L.; Wu, Z.; Li, Y.; Xiao, L.; Wang, H.; Wang, G. Brief Maternal Separation Promotes Resilience to Anxiety-like and Depressive-like Behaviors in Female C57BL/6J Offspring with Imiquimod-Induced Psoriasis. Brain Sci. 2022, 12, 1250. [Google Scholar] [CrossRef]
- Dellarole, A.; Morton, P.; Brambilla, R.; Walters, W.; Summers, S.; Bernardes, D.; Grilli, M.; Bethea, J.R. Neuropathic pain-induced depressive-like behavior and hippocampal neurogenesis and plasticity are dependent on TNFR1 signaling. Brain Behav. Immun. 2014, 41, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Valente, M.M.; Bortolotto, V.; Cuccurazzu, B.; Ubezio, F.; Meneghini, V.; Francese, M.T.; Canonico, P.L.; Grilli, M. α2δ ligands act as positive modulators of adult hippocampal neurogenesis and prevent depression-like behavior induced by chronic restraint stress. Mol. Pharmacol. 2012, 82, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Borsini, A.; Giacobbe, J.; Mandal, G.; Boldrini, M. Acute and long-term effects of adolescence stress exposure on rodent adult hippocampal neurogenesis, cognition, and behaviour. Mol. Psychiatry 2023, 28, 4124–4137. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Xu, H.; Wang, L.; Zhang, Z. Comparison of Therapeutic Effects of TREK1 Blockers and Fluoxetine on Chronic Unpredicted Mild Stress Sensitive Rats. ACS Chem. Neurosci. 2018, 9, 2824–2831. [Google Scholar] [CrossRef]
- Mateus-Pinheiro, A.; Pinto, L.; Bessa, J.M.; Morais, M.; Alves, N.D.; Monteiro, S.; Patrício, P.; Almeida, O.F.; Sousa, N. Sustained remission from depressive-like behavior depends on hippocampal neurogenesis. Transl. Psychiatry 2013, 3, e210. [Google Scholar] [CrossRef] [PubMed]
- Déry, N.; Pilgrim, M.; Gibala, M.; Gillen, J.; Wojtowicz, J.M.; Macqueen, G.; Becker, S. Adult hippocampal neurogenesis reduces memory interference in humans: Opposing effects of aerobic exercise and depression. Front. Neurosci. 2013, 7, 66. [Google Scholar] [CrossRef]
- Liu, C.; Yan, L.; Qian, Y.; Song, P.; Wang, T.; Wei, M. The Extract of Acanthopanacis Cortex Relieves the Depression-Like Behavior and Modulates IL-17 Signaling in Chronic Mild Stress-Induced Depressive Mice. Dose Response 2023, 21, 15593258221148817. [Google Scholar] [CrossRef] [PubMed]
- Sanacora, G.; Treccani, G.; Popoli, M. Towards a glutamate hypothesis of depression: An emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 2012, 62, 63–77. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, W.; Liu, J.; Song, Y.; Liu, T.; Li, Z.; Wang, X.; Yang, N.; Li, Y.; Han, D.; et al. Metabolomic and Lipidomic Profiling of Preoperative CSF in Elderly Hip Fracture Patients with Postoperative Delirium. Front. Aging Neurosci. 2020, 12, 570210. [Google Scholar] [CrossRef]
- Xu, K.; Ren, Y.; Zhao, S.; Feng, J.; Wu, Q.; Gong, X.; Chen, J.; Xie, P. Oral D-ribose causes depressive-like behavior by altering glycerophospholipid metabolism via the gut-brain axis. Commun. Biol. 2024, 7, 69. [Google Scholar] [CrossRef]
- Kazemian, N.; Zhou, T.; Chalasani, N.; Narayan, A.; Cedeño Laurent, J.G.; Olvera Alvarez, H.A.; Pakpour, S. Long-Term Impact of Childhood Adversity on the Gut Microbiome of Nursing Students. Int. J. Environ. Res. Public Health 2024, 21, 68. [Google Scholar] [CrossRef] [PubMed]
- Reininghaus, E.Z.; Platzer, M.; Kohlhammer-Dohr, A.; Hamm, C.; Mörkl, S.; Bengesser, S.A.; Fellendorf, F.T.; Lahousen-Luxenberger, T.; Leitner-Afschar, B.; Schöggl, H.; et al. PROVIT: Supplementary Probiotic Treatment and Vitamin B7 in Depression-A Randomized Controlled Trial. Nutrients 2020, 12, 3422. [Google Scholar] [CrossRef] [PubMed]
- Clapp, M.; Aurora, N.; Herrera, L.; Bhatia, M.; Wilen, E.; Wakefield, S. Gut microbiota’s effect on mental health: The gut-brain axis. Clin. Pract. 2017, 7, 987. [Google Scholar] [CrossRef]
- Lee, S.H.; Yoon, S.H.; Jung, Y.; Kim, N.; Min, U.; Chun, J.; Choi, I. Emotional well-being and gut microbiome profiles by enterotype. Sci. Rep. 2020, 10, 20736. [Google Scholar] [CrossRef]
- Bastiaanssen, T.F.S.; Cryan, J.F. The Microbiota-Gut-Brain Axis in Mental Health and Medication Response: Parsing Directionality and Causality. Int. J. Neuropsychopharmacol. 2021, 24, 216–220. [Google Scholar] [PubMed]
- Liu, L.; Huh, J.R.; Shah, K. Microbiota and the gut-brain-axis: Implications for new therapeutic design in the CNS. eBioMedicine 2022, 77, 103908. [Google Scholar]
- Rogers, G.B.; Keating, D.J.; Young, R.L.; Wong, M.L.; Licinio, J.; Wesselingh, S. From gut dysbiosis to altered brain function and mental illness: Mechanisms and pathways. Mol. Psychiatry 2016, 21, 738–748. [Google Scholar] [PubMed]
- Bastiaanssen, T.F.S.; Cussotto, S.; Claesson, M.J.; Clarke, G.; Dinan, T.G.; Cryan, J.F. Gutted! Unraveling the Role of the Microbiome in Major Depressive Disorder. Harv. Rev. Psychiatry 2020, 28, 26–39. [Google Scholar]
- Van Ameringen, M.; Turna, J.; Patterson, B.; Pipe, A.; Mao, R.Q.; Anglin, R.; Surette, M.G. The gut microbiome in psychiatry: A primer for clinicians. Depress. Anxiety 2019, 36, 1004–1025. [Google Scholar] [PubMed]
- Zhao, H.; Jin, K.; Jiang, C.; Pan, F.; Wu, J.; Luan, H.; Zhao, Z.; Chen, J.; Mou, T.; Wang, Z.; et al. A pilot exploration of multi-omics research of gut microbiome in major depressive disorders. Transl. Psychiatry 2022, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Wu, X.; Hu, X.; Wang, T.; Jin, F. Recognizing Depression from the Microbiota-Gut-Brain Axis. Int. J. Mol. Sci. 2018, 19, 1592. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Yang, C.; Ren, Q.; Ma, M.; Dong, C.; Hashimoto, K. Comparison of (R)-ketamine and lanicemine on depression-like phenotype and abnormal composition of gut microbiota in a social defeat stress model. Sci. Rep. 2017, 7, 15725. [Google Scholar]
- Luo, Y.; Zeng, B.; Zeng, L.; Du, X.; Li, B.; Huo, R.; Liu, L.; Wang, H.; Dong, M.; Pan, J.; et al. Gut microbiota regulates mouse behaviors through glucocorticoid receptor pathway genes in the hippocampus. Transl. Psychiatry 2018, 8, 187. [Google Scholar] [CrossRef]
- Cheung, S.G.; Goldenthal, A.R.; Uhlemann, A.C.; Mann, J.J.; Miller, J.M.; Sublette, M.E. Systematic Review of Gut Microbiota and Major Depression. Front. Psychiatry 2019, 10, 34. [Google Scholar]
- Bilenduke, E.; Sterrett, J.D.; Ranby, K.W.; Borges, V.F.; Grigsby, J.; Carr, A.L.; Kilbourn, K.; Lowry, C.A. Impacts of breast cancer and chemotherapy on gut microbiome, cognitive functioning, and mood relative to healthy controls. Sci. Rep. 2022, 12, 19547. [Google Scholar]
- McGuinness, A.J.; O’Hely, M.; Stupart, D.; Watters, D.; Dawson, S.L.; Hair, C.; Berk, M.; Mohebbi, M.; Loughman, A.; Guest, G.; et al. Depressive Symptoms and Gut Microbiota after Bowel Preparation and Colonoscopy: A Pre-Post Intervention Study. Microorganisms 2024, 12, 1960. [Google Scholar] [CrossRef] [PubMed]
- Nohesara, S.; Abdolmaleky, H.M.; Zhou, J.R.; Thiagalingam, S. Microbiota-Induced Epigenetic Alterations in Depressive Disorders Are Targets for Nutritional and Probiotic Therapies. Genes 2023, 14, 2217. [Google Scholar] [CrossRef]
- Park, S.S.; Kim, T.W.; Kim, B.K.; Kim, S.H.; Park, J.S.; Shin, M.S. The effects of exercise and diet on mental status, insulin signaling pathway, and microbiome in obese mice. J. Exerc. Rehabil. 2022, 18, 171–178. [Google Scholar]
- Taylor, B.C.; Sheikh Andalibi, M.; Wandro, S.; Weldon, K.C.; Sepich-Poore, G.D.; Carpenter, C.S.; Fraraccio, S.; Franklin, D.; Iudicello, J.E.; Letendre, S.; et al. Signatures of HIV and Major Depressive Disorder in the Plasma Microbiome. Microorganisms 2023, 11, 1022. [Google Scholar] [CrossRef]
- Mörkl, S.; Butler, M.I.; Lackner, S. Advances in the gut microbiome and mood disorders. Curr. Opin. Psychiatry 2023, 36, 1–7. [Google Scholar]
- Tomizawa, Y.; Kurokawa, S.; Ishii, D.; Miyaho, K.; Ishii, C.; Sanada, K.; Fukuda, S.; Mimura, M.; Kishimoto, T. Effects of Psychotropics on the Microbiome in Patients with Depression and Anxiety: Considerations in a Naturalistic Clinical Setting. Int. J. Neuropsychopharmacol. 2021, 24, 97–107. [Google Scholar] [CrossRef]
- Zhao, H.; Shi, Y.; Luo, X.; Peng, L.; Yang, Y.; Zou, L. The Effect of Fecal Microbiota Transplantation on a Child with Tourette Syndrome. Case Rep. Med. 2017, 2017, 6165239. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.; Sánchez, J.; Aucay, J. The role of the intestine as a target with a therapeutic role in people with neurodegenerative diseases. Salud Cienc. Tecnol. 2023, 3, 310. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, X.; Yue, Q.; Zhu, S.; Guo, L.; Li, G.; Zhou, Q.; Xiang, Y.; Chen, G.; Yin, W.; et al. Exploring the role and mechanism of gut microbiota in methamphetamine addiction using antibiotic treatment followed by fecal microbiota transplantation. Anat. Rec. 2023, 306, 1149–1164. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Sun, Q.; Zheng, H.; Zhang, Y.; Wang, Y.; Liu, S.; Duan, L. Roseburia hominis Alleviates Neuroinflammation via Short-Chain Fatty Acids through Histone Deacetylase Inhibition. Mol. Nutr. Food Res. 2022, 66, e2200164. [Google Scholar] [CrossRef]
- Ayten, Ş.; Bilici, S. Modulation of Gut Microbiota Through Dietary Intervention in Neuroinflammation and Alzheimer’s and Parkinson’s Diseases. Curr. Nutr. Rep. 2024, 13, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhou, G.; Shao, B.; Zhou, H.; Xu, C.; Yan, F.; Wang, L.; Chen, G.; Li, J.; Fu, X. Gut Microbiota Dysbiosis Induced by Intracerebral Hemorrhage Aggravates Neuroinflammation in Mice. Front. Microbiol. 2021, 12, 647304. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, H.; Yu, T.; Fu, X.; Qian, C.; Feng, X. Berberine mitigates intracerebral hemorrhage-induced neuroinflammation in a gut microbiota-dependent manner in mice. Aging 2023, 15, 2705–2720. [Google Scholar] [CrossRef]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef]
- Wu, S.; Zhu, Z.; Chen, M.; Huang, A.; Xie, Y.; Hu, H.; Zhang, J.; Wu, Q.; Wang, J.; Ding, Y. Comparison of Neuroprotection and Regulating Properties on Gut Microbiota between Selenopeptide Val-Pro-Arg-Lys-Leu-SeMet and Its Native Peptide Val-Pro-Arg-Lys-Leu-Met In Vitro and In Vivo. J. Agric. Food Chem. 2023, 71, 12203–12215. [Google Scholar] [CrossRef]
- Liu, P.; Liu, Z.; Wang, J.; Wang, J.; Gao, M.; Zhang, Y.; Yang, C.; Zhang, A.; Li, G.; Li, X.; et al. Immunoregulatory role of the gut microbiota in inflammatory depression. Nat. Commun. 2024, 15, 3003. [Google Scholar] [CrossRef]
- Shukla, P.K.; Delotterie, D.F.; Xiao, J.; Pierre, J.F.; Rao, R.; McDonald, M.P.; Khan, M.M. Alterations in the Gut-Microbial-Inflammasome-Brain Axis in a Mouse Model of Alzheimer’s Disease. Cells 2021, 10, 779. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.Y.; Jiang, W.Y.; Yin, T.; Xu, H.; Lyu, Y.T.; Chen, Y.Y.; Chen, M.; Geng, J.; Jiang, Z.X.; Shan, Q.J. Intestinal Barrier Dysfunction Exacerbates Neuroinflammation via the TLR4 Pathway in Mice with Heart Failure. Front. Physiol. 2021, 12, 712338. [Google Scholar] [CrossRef] [PubMed]
- Bairamian, D.; Sha, S.; Rolhion, N.; Sokol, H.; Dorothée, G.; Lemere, C.A.; Krantic, S. Microbiota in neuroinflammation and synaptic dysfunction: A focus on Alzheimer’s disease. Mol. Neurodegener. 2022, 17, 19. [Google Scholar] [PubMed]
- Bo, T.B.; Zhang, X.Y.; Wen, J.; Deng, K.; Qin, X.W.; Wang, D.H. The microbiota-gut-brain interaction in regulating host metabolic adaptation to cold in male Brandt’s voles (Lasiopodomys brandtii). ISME J. 2019, 13, 3037–3053. [Google Scholar]
- Mittal, R.; Debs, L.H.; Patel, A.P.; Nguyen, D.; Patel, K.; O’Connor, G.; Grati, M.; Mittal, J.; Yan, D.; Eshraghi, A.A.; et al. Neurotransmitters: The Critical Modulators Regulating Gut-Brain Axis. J. Cell Physiol. 2017, 232, 2359–2372. [Google Scholar] [PubMed]
- Gurow, K.; Joshi, D.C.; Gwasikoti, J.; Joshi, N. Gut Microbial Control of Neurotransmitters and Their Relation to Neurological Disorders: A Comprehensive Review. Horm. Metab. Res. 2025, 12. [Google Scholar] [CrossRef]
- Linhares, G.; Lima, E.; Perico, C.; Filho, A.; Pierri, C.; Raittz, R.; Marchaukoski, J. How the endocrine system and microbiota explain the emotions biochemistry and their impact in health. ARACE 2024, 6, 7087–7110. [Google Scholar]
- Wu, M.; Tian, T.; Mao, Q.; Zou, T.; Zhou, C.J.; Xie, J.; Chen, J.J. Associations between disordered gut microbiota and changes of neurotransmitters and short-chain fatty acids in depressed mice. Transl. Psychiatry 2020, 10, 350. [Google Scholar]
- Strandwitz, P. Neurotransmitter modulation by the gut microbiota. Brain Res. 2018, 1693 Pt B, 128–133. [Google Scholar] [CrossRef]
- Mhanna, A.; Martini, N.; Hmaydoosh, G.; Hamwi, G.; Jarjanazi, M.; Zaifah, G.; Kazzazo, R.; Haji Mohamad, A.; Alshehabi, Z. The correlation between gut microbiota and both neurotransmitters and mental disorders: A narrative review. Medicine 2024, 103, e37114. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [PubMed]
- Hoyles, L.; Snelling, T.; Umlai, U.K.; Nicholson, J.K.; Carding, S.R.; Glen, R.C.; McArthur, S. Microbiome-host systems interactions: Protective effects of propionate upon the blood-brain barrier. Microbiome 2018, 6, 55. [Google Scholar]
- Yang, H.L.; Li, M.M.; Zhou, M.F.; Xu, H.S.; Huan, F.; Liu, N.; Gao, R.; Wang, J.; Zhang, N.; Jiang, L. Links Between Gut Dysbiosis and Neurotransmitter Disturbance in Chronic Restraint Stress-Induced Depressive Behaviours: The Role of Inflammation. Inflammation 2021, 44, 2448–2462. [Google Scholar] [PubMed]
- Huang, F.; Wu, X. Brain Neurotransmitter Modulation by Gut Microbiota in Anxiety and Depression. Front. Cell Dev. Biol. 2021, 9, 649103. [Google Scholar]
- Swann, J.R.; Spitzer, S.O.; Diaz Heijtz, R. Developmental Signatures of Microbiota-Derived Metabolites in the Mouse Brain. Metabolites 2020, 10, 172. [Google Scholar] [CrossRef]
- Ortega, M.A.; Alvarez-Mon, M.A.; García-Montero, C.; Fraile-Martinez, O.; Guijarro, L.G.; Lahera, G.; Monserrat, J.; Valls, P.; Mora, F.; Rodríguez-Jiménez, R.; et al. Gut Microbiota Metabolites in Major Depressive Disorder-Deep Insights into Their Pathophysiological Role and Potential Translational Applications. Metabolites 2022, 12, 50. [Google Scholar] [CrossRef]
- Jaglin, M.; Rhimi, M.; Philippe, C.; Pons, N.; Bruneau, A.; Goustard, B.; Daugé, V.; Maguin, E.; Naudon, L.; Rabot, S. Indole, a Signaling Molecule Produced by the Gut Microbiota, Negatively Impacts Emotional Behaviors in Rats. Front. Neurosci. 2018, 12, 216. [Google Scholar] [CrossRef]
- Vuong, H.E.; Pronovost, G.N.; Williams, D.W.; Coley, E.J.L.; Siegler, E.L.; Qiu, A.; Kazantsev, M.; Wilson, C.J.; Rendon, T.; Hsiao, E.Y. The maternal microbiome modulates fetal neurodevelopment in mice. Nature 2020, 586, 281–286. [Google Scholar]
- Pessa-Morikawa, T.; Husso, A.; Kärkkäinen, O.; Koistinen, V.; Hanhineva, K.; Iivanainen, A.; Niku, M. Maternal microbiota-derived metabolic profile in fetal murine intestine, brain and placenta. BMC Microbiol. 2022, 22, 46. [Google Scholar]
- Colosimo, D.A.; Kohn, J.A.; Luo, P.M.; Piscotta, F.J.; Han, S.M.; Pickard, A.J.; Rao, A.; Cross, J.R.; Cohen, L.J.; Brady, S.F. Mapping Interactions of Microbial Metabolites with Human G-Protein-Coupled Receptors. Cell Host Microbe 2019, 26, 273–282.e7. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.Y.; Kim, S.; Kim, P.; Jo, M.J.; Park, S.-E.; Choi, Y.; Jung, S.M.; Kang, H.J. G-Protein-Coupled Receptor (GPCR) Signaling and Pharmacology in Metabolism: Physiology, Mechanisms, and Therapeutic Potential. Biomolecules 2025, 15, 291. [Google Scholar] [CrossRef]
- Han, M.; Dong, Y.; Wang, S.; Huang, X.; Bai, C.; Gai, Z. Regulation of gut microbiota and serum neurotransmitters in mice by Streptococcus thermophilus GA8- and Lacticaseibacillus rhamnosus HAO9-fermented milk containing high levels of gamma-aminobutyric acid. J. Sci. Food Agric. 2024, 104, 8050–8058. [Google Scholar] [CrossRef] [PubMed]
- Stopińska, K.; Radziwoń-Zaleska, M.; Domitrz, I. The Microbiota-Gut-Brain Axis as a Key to Neuropsychiatric Disorders: A Mini Review. J. Clin. Med. 2021, 10, 4640. [Google Scholar] [CrossRef] [PubMed]
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F.; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 2015, 29, 1395–1403. [Google Scholar] [PubMed]
- Hellmann-Regen, J.; Clemens, V.; Grözinger, M.; Kornhuber, J.; Reif, A.; Prvulovic, D.; Goya-Maldonado, R.; Wiltfang, J.; Gruber, O.; Schüle, C.; et al. Effect of Minocycline on Depressive Symptoms in Patients with Treatment-Resistant Depression: A Randomized Clinical Trial. JAMA Netw. Open 2022, 5, e2230367. [Google Scholar] [CrossRef]
- Majeed, M.; Nagabhushanam, K.; Mundkur, L. A standardized Ashwagandha root extract alleviates stress, anxiety, and improves quality of life in healthy adults by modulating stress hormones: Results from a randomized, double-blind, placebo-controlled study. Medicine 2023, 102, e35521. [Google Scholar] [CrossRef] [PubMed]
- Yaikwawong, M.; Jansarikit, L.; Jirawatnotai, S.; Chuengsamarn, S. Curcumin Reduces Depression in Obese Patients with Type 2 Diabetes: A Randomized Controlled Trial. Nutrients 2024, 16, 2414. [Google Scholar] [CrossRef]
- Zandifar, A.; Panahi, M.; Badrfam, R.; Qorbani, M. Efficacy of empagliflozin as adjunctive therapy to citalopram in major depressive disorder: A randomized double-blind, placebo-controlled clinical trial. BMC Psychiatry 2024, 24, 163. [Google Scholar] [CrossRef]
- Berk, M.; Mohebbi, M.; Dean, O.M.; Cotton, S.M.; Chanen, A.M.; Dodd, S.; Ratheesh, A.; Amminger, G.P.; Phelan, M.; Weller, A.; et al. Youth Depression Alleviation with Anti-inflammatory Agents (YoDA-A): A randomised clinical trial of rosuvastatin and aspirin. BMC Med. 2020, 18, 16. [Google Scholar]
- Berk, M.; Woods, R.L.; Nelson, M.R.; Shah, R.C.; Reid, C.M.; Storey, E.; Fitzgerald, S.; Lockery, J.E.; Wolfe, R.; Mohebbi, M.; et al. Effect of Aspirin vs Placebo on the Prevention of Depression in Older People: A Randomized Clinical Trial. JAMA Psychiatry 2020, 77, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Fourrier, C.; Sampson, E.; Mills, N.T.; Baune, B.T. Anti-inflammatory treatment of depression: Study protocol for a randomised controlled trial of vortioxetine augmented with celecoxib or placebo. Trials 2018, 19, 447. [Google Scholar] [CrossRef] [PubMed]
- Plank, J.R.; Glover, S.C.; Moloney, B.D.; Hoeh, N.R.; Sundram, F.; Sumner, R.L.; Muthukumaraswamy, S.; Lin, J.C. A randomized, double-blind, placebo-controlled, hybrid parallel-arm study of low-dose naltrexone as an adjunctive anti-inflammatory treatment for major depressive disorder. Trials 2022, 23, 822. [Google Scholar] [PubMed]
- Yang, C.; Bosker, F.J.; Li, J.; Schoevers, R.A. N-acetylcysteine as add-on to antidepressant medication in therapy refractory major depressive disorder patients with increased inflammatory activity: Study protocol of a double-blind randomized placebo-controlled trial. BMC Psychiatry 2018, 18, 279. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.Y.; Liu, Y.Z.; Li, J.M.; Ruan, Y.M.; Yan, W.J.; Zhong, S.Y.; Zhang, T.; Liu, L.L.; Wu, R.; Wang, B.; et al. Glycyrrhizic acid as an adjunctive treatment for depression through anti-inflammation: A randomized placebo-controlled clinical trial. J. Affect. Disord. 2020, 265, 247–254. [Google Scholar] [CrossRef]
- Kim, K.Y.; Shin, K.Y.; Chang, K.-A. Potential Inflammatory Biomarkers for Major Depressive Disorder Related to Suicidal Behaviors: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 13907. [Google Scholar] [CrossRef]
- Burton, T.C.J.; Lv, N.; Tsai, P.; Peñalver Bernabé, B.; Tussing-Humphreys, L.; Xiao, L.; Pandey, G.N.; Wu, Y.; Ajilore, O.A.; Ma, J. Associations between fecal short-chain fatty acids, plasma inflammatory cytokines, and dietary markers with depression and anxiety: Post hoc analysis of the ENGAGE-2 pilot trial. Am. J. Clin. Nutr. 2023, 117, 717–730. [Google Scholar]
- Kim, J.; Suh, Y.H.; Chang, K.A. Interleukin-17 induced by cumulative mild stress promoted depression-like behaviors in young adult mice. Mol. Brain 2021, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, H.; Sakakibara, H.; Minamida, Y.; Tsujiguchi, H.; Matsunaga, M.; Hara, A.; Nakamura, H. Elevated Levels of Serum IL-17A in Community-Dwelling Women with Higher Depressive Symptoms. Behav. Sci. 2018, 8, 102. [Google Scholar] [CrossRef]
- Saraykar, S.; Cao, B.; Barroso, L.S.; Pereira, K.S.; Bertola, L.; Nicolau, M.; Ferreira, J.D.; Dias, N.S.; Vieira, E.L.; Teixeira, A.L.; et al. Plasma IL-17A levels in patients with late-life depression. Braz. J. Psychiatry 2017, 40, 212–215. [Google Scholar] [CrossRef]
- Bliźniewska-Kowalska, K.; Szewczyk, B.; Gałecka, M.; Su, K.P.; Maes, M.; Szemraj, J.; Gałecki, P. Is Interleukin 17 (IL-17) Expression A Common Point in the Pathogenesis of Depression and Obesity? J. Clin. Med. 2020, 9, 4018. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, C.; Lan, X.; Li, H.; Chao, Z.; Ning, Y. Plasma inflammatory cytokines and treatment-resistant depression with comorbid pain: Improvement by ketamine. J. Neuroinflamm. 2021, 18, 200. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Chou, Y.C.; Chen, H.C.; Lu, C.C.; Chang, D.M. Interleukin-6 and interleukin-17 are related to depression in patients with rheumatoid arthritis. Int. J. Rheum. Dis. 2019, 22, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Lorkiewicz, P.; Waszkiewicz, N. Biomarkers of Post-COVID Depression. J. Clin. Med. 2021, 10, 4142. [Google Scholar] [CrossRef]
- Roohi, E.; Jaafari, N.; Hashemian, F. On inflammatory hypothesis of depression: What is the role of IL-6 in the middle of the chaos? J. Neuroinflamm. 2021, 18, 45. [Google Scholar] [CrossRef] [PubMed]
- Osimo, E.F.; Pillinger, T.; Rodriguez, I.M.; Khandaker, G.M.; Pariante, C.M.; Howes, O.D. Inflammatory markers in depression: A meta-analysis of mean differences and variability in 5,166 patients and 5,083 controls. Brain Behav. Immun. 2020, 87, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Himmerich, H.; Patsalos, O.; Lichtblau, N.; Ibrahim, M.A.A.; Dalton, B. Cytokine Research in Depression: Principles, Challenges, and Open Questions. Front. Psychiatry 2019, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Foley, É.M.; Slaney, C.; Donnelly, N.A.; Kaser, M.; Ziegler, L.; Khandaker, G.M. A novel biomarker of interleukin 6 activity and clinical and cognitive outcomes in depression. Psychoneuroendocrinology 2024, 164, 107008. [Google Scholar] [CrossRef]
- Moriarity, D.P.; Kautz, M.M.; Mac Giollabhui, N.; Klugman, J.; Coe, C.L.; Ellman, L.M.; Abramson, L.Y.; Alloy, L.B. Bidirectional Associations Between Inflammatory Biomarkers and Depressive Symptoms in Adolescents: Potential Causal Relationships. Clin. Psychol. Sci. 2020, 8, 690–703. [Google Scholar] [CrossRef]
- Neupane, S.P.; Virtej, A.; Myhren, L.E.; Bull, V.H. Biomarkers common for inflammatory periodontal disease and depression: A systematic review. Brain Behav. Immun.-Health 2022, 21, 100450. [Google Scholar] [CrossRef]
- Young, J.J.; Silber, T.; Bruno, D.; Galatzer-Levy, I.R.; Pomara, N.; Marmar, C.R. Is there Progress? An Overview of Selecting Biomarker Candidates for Major Depressive Disorder. Front. Psychiatry 2016, 7, 72. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K. Inflammatory Biomarkers as Differential Predictors of Antidepressant Response. Int. J. Mol. Sci. 2015, 16, 7796–7801. [Google Scholar] [CrossRef] [PubMed]
- Yeager, M.P.; Pioli, P.A.; Guyre, P.M. Cortisol exerts bi-phasic regulation of inflammation in humans. Dose Response 2011, 9, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.A.; Bankier, S.; Altmaier, E.; Barnes, C.L.K.; Clark, D.W.; Ermel, R.; Friedrich, N.; van der Harst, P.; Joshi, P.K.; Karhunen, V.; et al. Variation in the SERPINA6/SERPINA1 locus alters morning plasma cortisol, hepatic corticosteroid binding globulin expression, gene expression in peripheral tissues, and risk of cardiovascular disease. J. Hum. Genet. 2021, 66, 625–636. [Google Scholar] [CrossRef]
- Prete, A.; Yan, Q.; Al-Tarrah, K.; Akturk, H.K.; Prokop, L.J.; Alahdab, F.; Foster, M.A.; Lord, J.M.; Karavitaki, N.; Wass, J.A.; et al. The cortisol stress response induced by surgery: A systematic review and meta-analysis. Clin. Endocrinol. 2018, 89, 554–567. [Google Scholar] [CrossRef]
- Qin, D.D.; Rizak, J.; Feng, X.L.; Yang, S.C.; Lü, L.B.; Pan, L.; Yin, Y.; Hu, X.T. Prolonged secretion of cortisol as a possible mechanism underlying stress and depressive behaviour. Sci. Rep. 2016, 6, 30187. [Google Scholar] [CrossRef] [PubMed]
- Jarcho, M.R.; Slavich, G.M.; Tylova-Stein, H.; Wolkowitz, O.M.; Burke, H.M. Dysregulated diurnal cortisol pattern is associated with glucocorticoid resistance in women with major depressive disorder. Biol. Psychol. 2013, 93, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Limanaqi, F.; Busceti, C.L.; Biagioni, F.; Fornai, F.; Puglisi-Allegra, S. Autophagy-Based Hypothesis on the Role of Brain Catecholamine Response During Stress. Front. Psychiatry 2020, 11, 569248. [Google Scholar] [CrossRef]
- Williams, M.A.; Li, C.; Kash, T.L.; Matthews, R.T.; Winder, D.G. Excitatory drive onto dopaminergic neurons in the rostral linear nucleus is enhanced by norepinephrine in an α1 adrenergic receptor-dependent manner. Neuropharmacology 2014, 86, 116–124. [Google Scholar] [CrossRef]
- Sinclair, D.; Purves-Tyson, T.D.; Allen, K.M.; Weickert, C.S. Impacts of stress and sex hormones on dopamine neurotransmission in the adolescent brain. Psychopharmacology 2014, 231, 1581–1599. [Google Scholar] [CrossRef]
- Nehls, M. Unified theory of Alzheimer’s disease (UTAD): Implications for prevention and curative therapy. J. Mol. Psychiatry 2016, 4, 3. [Google Scholar] [PubMed]
- Younge, J.O.; Wester, V.L.; van Rossum, E.F.C.; Gotink, R.A.; Wery, M.F.; Utens, E.M.W.J.; Hunink, M.G.M.; Roos-Hesselink, J.W. Cortisol levels in scalp hair of patients with structural heart disease. Int. J. Cardiol. 2015, 184, 1–78. [Google Scholar] [CrossRef]
- Maninger, N.; Capitanio, J.P.; Mason, W.A.; Ruys, J.D.; Mendoza, S.P. Acute and chronic stress increase DHEAS concentrations in rhesus monkeys. Psychoneuroendocrinology 2010, 35, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Jauregui-Huerta, F.; Ruvalcaba-Delgadillo, Y.; Gonzalez-Castañeda, R.; Garcia-Estrada, J.; Gonzalez-Perez, O.; Luquin, S. Responses of glial cells to stress and glucocorticoids. Curr. Immunol. Rev. 2010, 6, 195–204. [Google Scholar] [CrossRef]
- Abercrombie, H.C.; Frost, C.P.; Walsh, E.C.; Hoks, R.M.; Cornejo, M.D.; Sampe, M.C.; Gaffey, A.E.; Plante, D.T.; Ladd, C.O.; Birn, R.M. Neural Signaling of Cortisol, Childhood Emotional Abuse, and Depression-Related Memory Bias. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2018, 3, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Feier, A.M.; Portan, D.; Manu, D.R.; Kostopoulos, V.; Kotrotsos, A.; Strnad, G.; Dobreanu, M.; Salcudean, A.; Bataga, T. Primary MSCs for Personalized Medicine: Ethical Challenges, Isolation and Biocompatibility Evaluation of 3D Electrospun and Printed Scaffolds. Biomedicines 2022, 10, 1563. [Google Scholar] [CrossRef]
- Tilinca, M.C.; Antal, C.; Sălcudean, A.; Abălașei, B.L.; Fărcaș, R.M.; Groșan, A. New directions in pharmacological treatment with SGLT-2 inhibitor molecules in the light of current guidelines for diabetes mellitus, heart failure and kidney disease. Farmacia 2023, 71, 686–696. [Google Scholar] [CrossRef]
- Brunoni, A.R.; Machado-Vieira, R.; Zarate, C.A.; Valiengo, L.; Vieira, E.L.; Benseñor, I.M.; Lotufo, P.A.; Gattaz, W.F.; Teixeira, A.L. Cytokines plasma levels during antidepressant treatment with sertraline and transcranial direct current stimulation (tDCS): Results from a factorial, randomized, controlled trial. Psychopharmacology 2014, 231, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Hanamsagar, R.; Torres, V.; Kielian, T. Inflammasome activation and IL-1β/IL-18 processing are influenced by distinct pathways in microglia. J. Neurochem. 2011, 119, 736–748. [Google Scholar] [CrossRef]
- Ősz, B.E.; Jîtcă, G.; Sălcudean, A.; Rusz, C.M.; Vari, C.E. Benzydamine-An Affordable Over-the-Counter Drug with Psychoactive Properties-From Chemical Structure to Possible Pharmacological Properties. Pharmaceuticals 2023, 16, 566. [Google Scholar] [CrossRef]
- Tripathi, A.; Whitehead, C.; Surrao, K.; Pillai, A.; Madeshiya, A.; Li, Y.; Khodadadi, H.; Ahmed, A.O.; Turecki, G.; Baban, B.; et al. Type 1 interferon mediates chronic stress-induced neuroinflammation and behavioral deficits via complement component 3-dependent pathway. Mol. Psychiatry 2021, 26, 3043–3059. [Google Scholar] [CrossRef] [PubMed]
- Sălcudean, A.; Nan, A.G.; Bodo, C.R.; Cosma, M.C.; Strete, E.G.; Lica, M.M. Association between Childhood Onset Inflammatory Bowel Disease and Psychiatric Comorbidities in Adulthood. Diagnostics 2023, 13, 1868. [Google Scholar] [CrossRef]
- Wang, Q.; Zhong, Y.; Chen, N.; Chen, J. From the immune system to mood disorders especially induced by Toxoplasma gondii: CD4+ T cell as a bridge. Front. Cell Infect. Microbiol. 2023, 13, 1078984. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Xia, D.; Cai, J.; Gu, B.; Liu, X.; Friedman, V.; Liu, Q.S.; Zhao, L. Treadmill Exercise Reduces Neuroinflammation, Glial Cell Activation and Improves Synaptic Transmission in the Prefrontal Cortex in 3 × Tg-AD Mice. Int. J. Mol. Sci. 2022, 23, 12655. [Google Scholar] [CrossRef]
- Bull, C.; Syed, W.A.; Minter, S.C.; Bowers, M.S. Differential response of glial fibrillary acidic protein-positive astrocytes in the rat prefrontal cortex following ethanol self-administration. Alcohol. Clin. Exp. Res. 2015, 39, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Herlo, L.-F.; Salcudean, A.; Sirli, R.; Iurciuc, S.; Herlo, A.; Nelson-Twakor, A.; Alexandrescu, L.; Dumache, R. Gut Microbiota Signatures in Colorectal Cancer as a Potential Diagnostic Biomarker in the Future: A Systematic Review. Int. J. Mol. Sci. 2024, 25, 7937. [Google Scholar] [CrossRef]
- Kalinovic, R.; Pascariu, A.; Vlad, G.; Nitusca, D.; Sălcudean, A.; Sirbu, I.O.; Marian, C.; Enatescu, V.R. Involvement of the Expression of G Protein-Coupled Receptors in Schizophrenia. Pharmaceuticals 2024, 17, 85. [Google Scholar] [CrossRef]
- Salcudean, A.; Lica, M.M. The Role of Systemic Family Psychotherapy in Glycemic Control for Children with Type 1 Diabetes. Children 2024, 11, 104. [Google Scholar] [CrossRef]
- Gold, S.M.; Kern, K.C.; O’Connor, M.F.; Montag, M.J.; Kim, A.; Yoo, Y.S.; Giesser, B.S.; Sicotte, N.L. Smaller cornu ammonis 2-3/dentate gyrus volumes and elevated cortisol in multiple sclerosis patients with depressive symptoms. Biol. Psychiatry 2010, 68, 553–559. [Google Scholar] [CrossRef]
- Gold, S.M.; O’Connor, M.F.; Gill, R.; Kern, K.C.; Shi, Y.; Henry, R.G.; Pelletier, D.; Mohr, D.C.; Sicotte, N.L. Detection of altered hippocampal morphology in multiple sclerosis-associated depression using automated surface mesh modeling. Hum. Brain Mapp. 2014, 35, 30–37. [Google Scholar] [CrossRef]
- Chen, L.Q.; Lv, X.J.; Guo, Q.H.; Lv, S.S.; Lv, N.; Xu, W.D.; Yu, J.; Zhang, Y.Q. Asymmetric activation of microglia in the hippocampus drives anxiodepressive consequences of trigeminal neuralgia in rodents. Br. J. Pharmacol. 2023, 180, 1090–1113. [Google Scholar] [PubMed]
- Răchită, A.; Strete, G.E.; Suciu, L.M.; Ghiga, D.V.; Sălcudean, A.; Mărginean, C. Psychological Stress Perceived by Pregnant Women in the Last Trimester of Pregnancy. Int. J. Environ. Res. Public Health 2022, 19, 8315. [Google Scholar]
- Tilinca, M.C.; Antal, C.; Balint, A.; Salcudean, A.; Varga, A. The newest therapeutically approach of “diabesity” using GLP-1 RA molecules: Impact of the oral formulation. Farmacia 2023, 71, 1–10. [Google Scholar] [CrossRef]
- Bodo, C.R.; Salcudean, A.; Nirestean, A.; Lukacs, E.; Lica, M.M.; Muntean, D.L.; Anculia, R.C.; Popovici, R.A.; Neda Stepan, O.; Enătescu, V.R.; et al. Association between Chronic Misophonia-Induced Stress and Gastrointestinal Pathology in Children—A Hypothesis. Children 2024, 11, 699. [Google Scholar] [CrossRef] [PubMed]
- Sălcudean, A.; Osz, B.E.; Bodo, C.R.; Muntean, D.L. Serum serotonin level can be used as a predictive marker for depression in patients with type 2 diabetes mellitus. True or false? Farmacia 2024, 72, 1283–1289. [Google Scholar] [CrossRef]
- Sălcudean, A.; Truşculescu, L.M.; Popovici, R.A.; Şerb, N.; Paşca, C.; Bodo, C.R.; Crăciun, R.E.; Olariu, I. Dental anxiety—A psychosocial cause affecting the quality of life—A systematic review. Rom. J. Oral. Rehab 2024, 4, 471–483. [Google Scholar]
- Xiao, K.; Luo, Y.; Liang, X.; Tang, J.; Wang, J.; Xiao, Q.; Qi, Y.; Li, Y.; Zhu, P.; Yang, H.; et al. Beneficial effects of running exercise on hippocampal microglia and neuroinflammation in chronic unpredictable stress-induced depression model rats. Transl. Psychiatry 2021, 11, 461. [Google Scholar] [CrossRef] [PubMed]
- Sălcudean, A.; Strete, G.; Enatescu, V.R.; Tilinca, M.C.; Sasu, A.B.; Ceana, D.E.; Dumache, R.; Kiss, M.; Ősz, B.E.; Teodorescu, D.R.; et al. Pilot Study to Evaluate Adherence to Treatment Using the Mars Scale on A Sample of Romanian Patients with Cardiovascular Diseases. Farmacia 2024, 72, 66–77. [Google Scholar] [CrossRef]
- Tilinca, M.C.; Merlan, I.; Salcudean, A.; Tilea, I.; Nemes-Nagy, E. Oxidative stress and cytokines’ involvement in the occurence and progression of diabetic complications in the COVID-19 pandemic context. Farmacia 2021, 69, 635–641. [Google Scholar]
- Ito, N.; Hirose, E.; Ishida, T.; Hori, A.; Nagai, T.; Kobayashi, Y.; Kiyohara, H.; Oikawa, T.; Hanawa, T.; Odaguchi, H. Kososan, a Kampo medicine, prevents a social avoidance behavior and attenuates neuroinflammation in socially defeated mice. J. Neuroinflamm. 2017, 14, 98. [Google Scholar]
- Ashraf, A.; Mahmoud, P.A.; Reda, H.; Mansour, S.; Helal, M.H.; Michel, H.E.; Nasr, M. Silymarin and silymarin nanoparticles guard against chronic unpredictable mild stress induced depressive-like behavior in mice: Involvement of neurogenesis and NLRP3 inflammasome. J. Psychopharmacol. 2019, 33, 615–631. [Google Scholar] [PubMed]
- Arab, H.H.; Khames, A.; Mohammad, M.K.; Alsufyani, S.E.; Ashour, A.M.; El-Sheikh, A.A.K.; Darwish, H.W.; Gad, A.M. Meloxicam Targets COX-2/NOX1/NOX4/Nrf2 Axis to Ameliorate the Depression-like Neuropathology Induced by Chronic Restraint Stress in Rats. Pharmaceuticals 2023, 16, 848. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Huang, H.; Li, C.; Li, S.; Gan, J.; Lian, C.; Ling, Y. The antidepressive mechanism of Longya Lilium combined with Fluoxetine in mice with depression-like behaviors. NPJ Syst. Biol. Appl. 2024, 10, 5. [Google Scholar]
- Sciolino, N.R.; Dishman, R.K.; Holmes, P.V. Voluntary exercise offers anxiolytic potential and amplifies galanin gene expression in the locus coeruleus of the rat. Behav. Brain Res. 2012, 233, 191–200. [Google Scholar]
- Guo, L.; Stormmesand, J.; Fang, Z.; Zhu, Q.; Balesar, R.; van Heerikhuize, J.; Sluiter, A.; Swaab, D.; Bao, A.M. Quantification of Tyrosine Hydroxylase and ErbB4 in the Locus Coeruleus of Mood Disorder Patients Using a Multispectral Method to Prevent Interference with Immunocytochemical Signals by Neuromelanin. Neurosci. Bull. 2019, 35, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Tomassini, A.; Hezemans, F.H.; Ye, R.; Tsvetanov, K.A.; Wolpe, N.; Rowe, J.B. Prefrontal Cortical Connectivity Mediates Locus Coeruleus Noradrenergic Regulation of Inhibitory Control in Older Adults. J. Neurosci. 2022, 42, 3484–3493. [Google Scholar] [CrossRef]
- Matchett, B.J.; Grinberg, L.T.; Theofilas, P.; Murray, M.E. The mechanistic link between selective vulnerability of the locus coeruleus and neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2021, 141, 631–650. [Google Scholar] [CrossRef]
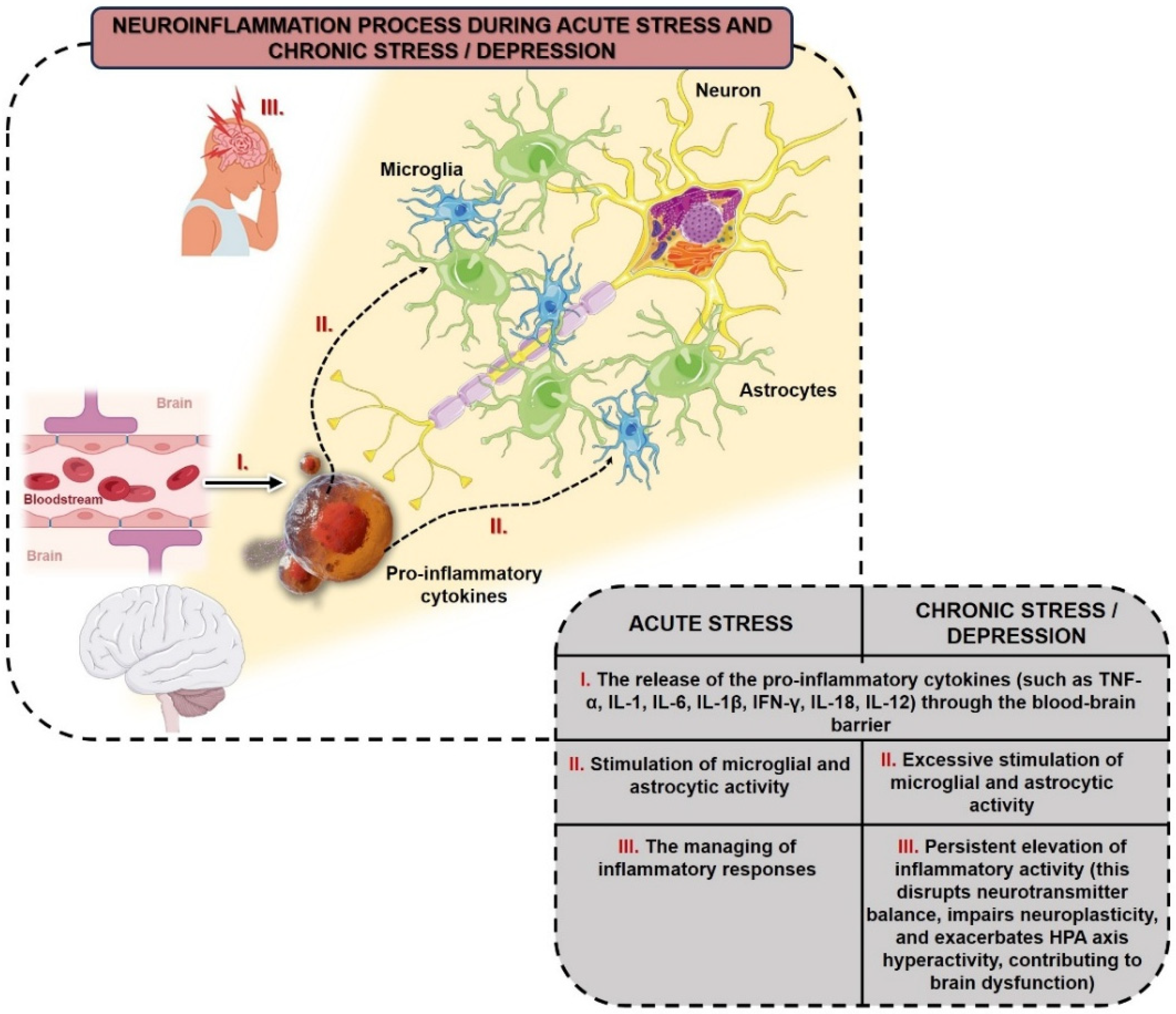
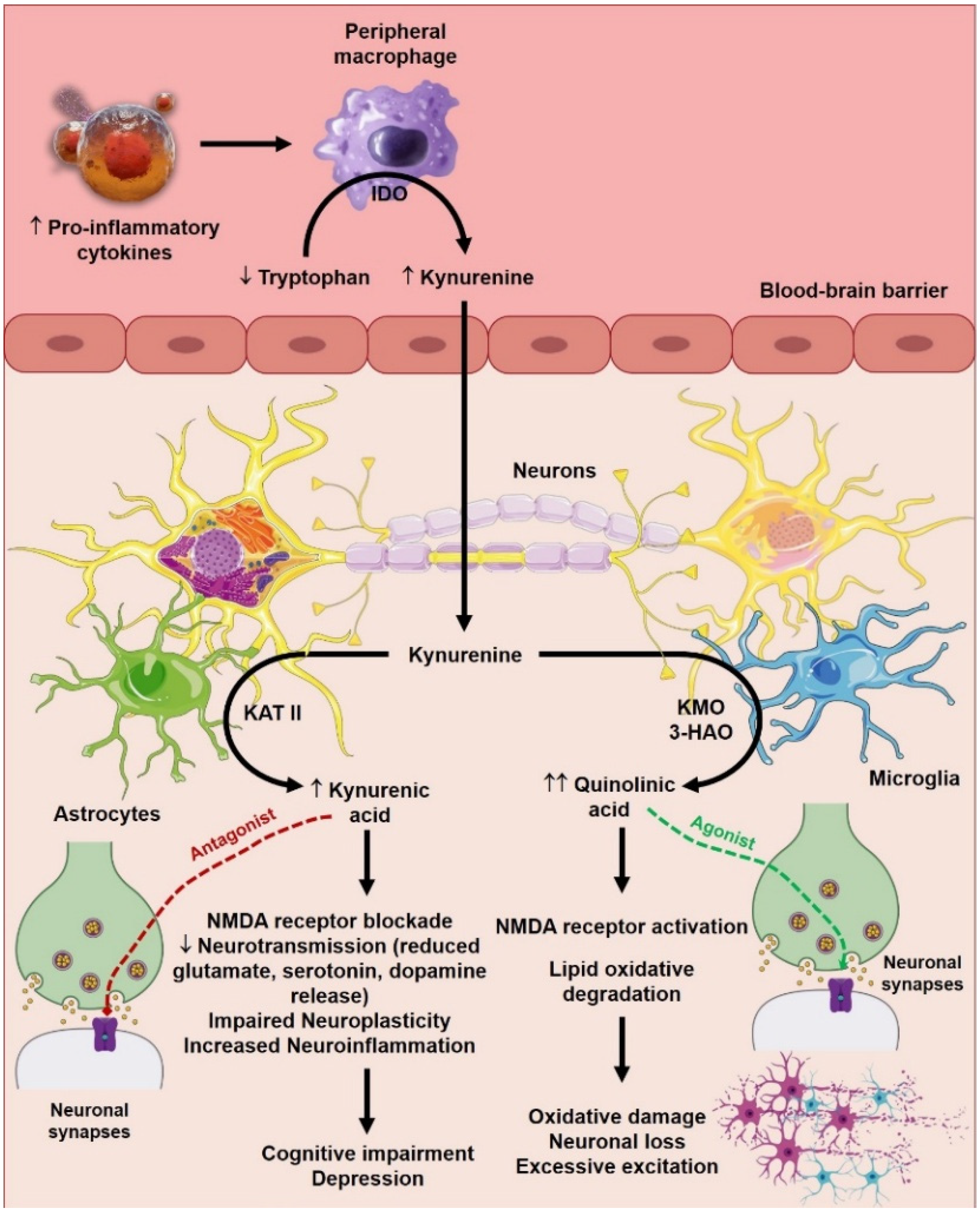

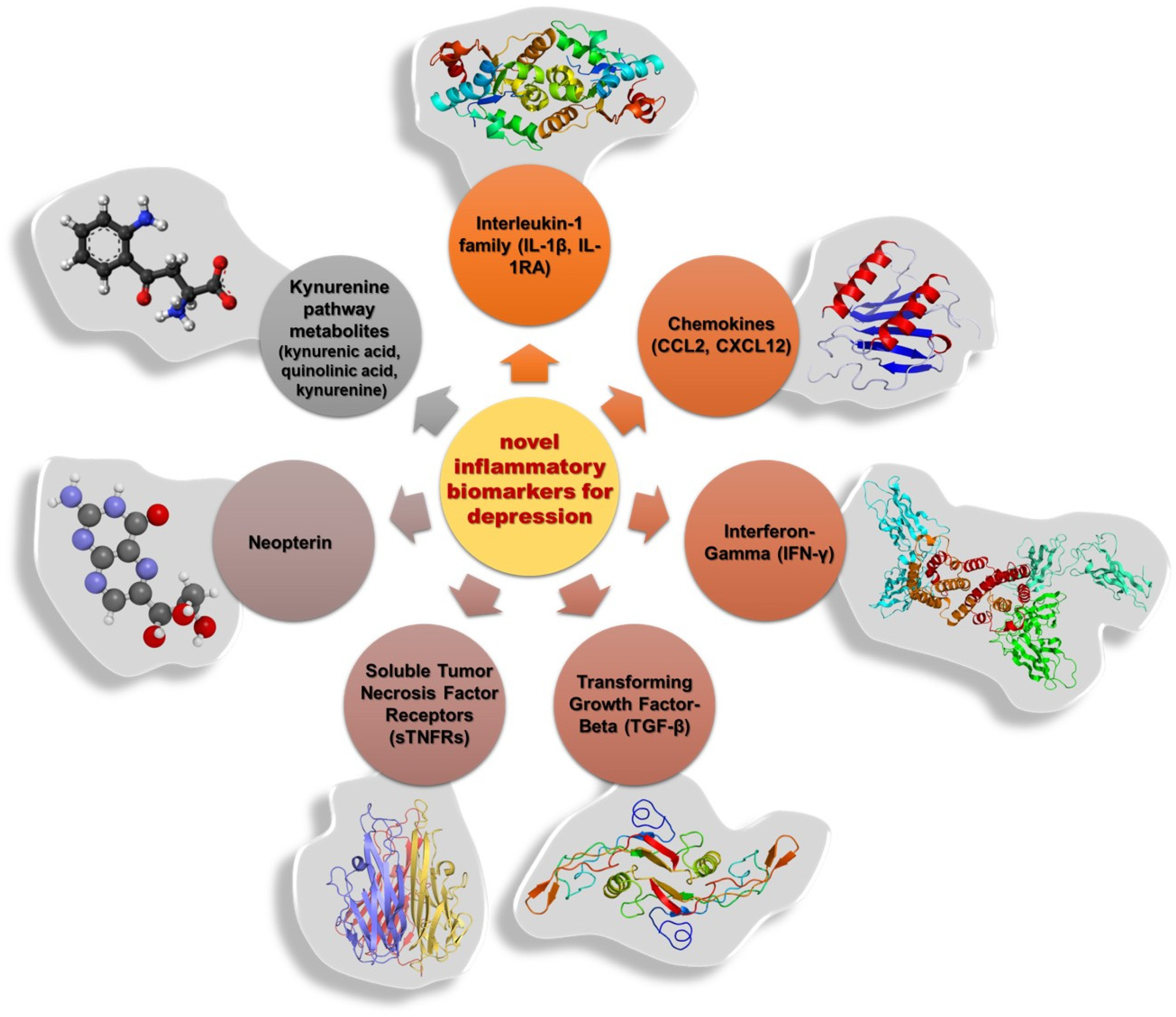
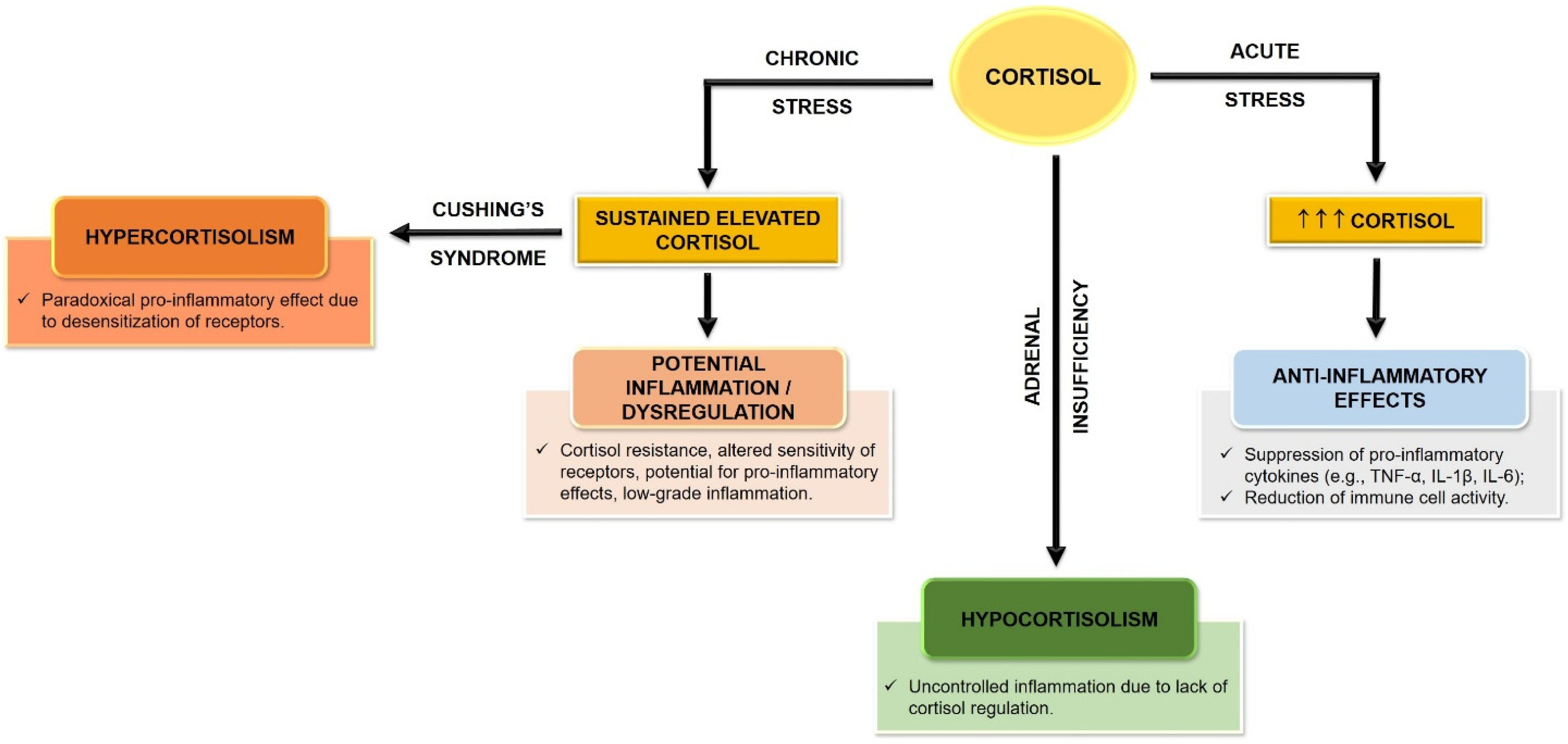

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Study | Subjects Included in the Study | Treatment | Key Findings | References |
|---|---|---|---|---|
| Randomized Clinical Trial | Minocycline group (n = 24), Placebo group (n = 26); Patients with treatment-resistant depression | Minocycline (200 mg daily) vs. Placebo | Minocycline significantly improved depressive symptoms compared to placebo. | [277] |
| Randomized, Double-Blind, Placebo-Controlled Study | Healthy adults experiencing stress and anxiety (n = 54) | Ashwagandha root extract (500 mg daily) vs. Placebo | Ashwagandha reduced stress and anxiety, increased serotonin, and lowered cortisol levels. | [278] |
| Randomized Controlled Trial | Obese patients with type 2 diabetes and depression (n = 227) | Curcumin (dose not specified) vs. Placebo | Curcumin improved depression severity and reduced inflammatory markers in patients with type 2 diabetes. | [279] |
| Randomized, Double-Blind, Placebo-Controlled Trial | Patients with moderate to severe depression (n = 90) | Empagliflozin (10 mg daily) + Citalopram vs. Citalopram + Placebo | Empagliflozin significantly reduced depression severity over time compared to placebo. | [280] |
| Randomized Clinical Trial | Young adults with moderate to severe MDD (n = 130) | Aspirin (100 mg daily), Rosuvastatin (10 mg daily) vs. Placebo | Neither aspirin nor rosuvastatin showed significant improvement in depression; rosuvastatin showed some secondary benefits. | [281] |
| Randomized Clinical Trial | Older adults (n = 19,114) | Aspirin (100 mg daily) vs. Placebo | Aspirin did not reduce the risk of depression in older adults. | [282] |
| Randomized Controlled Trial | Patients with high and low inflammation MDD (n = 200) | Vortioxetine + Celecoxib vs. Vortioxetine + Placebo | Vortioxetine + Celecoxib showed potential for greater improvement in inflammation-related depression. | [283] |
| Randomized, Double-Blind, Placebo-Controlled Study | Patients with major depressive disorder (n = 48) | Low-dose Naltrexone vs. Placebo | Naltrexone improved depressive symptoms, particularly in those with elevated inflammation markers. | [284] |
| Randomized, Double-Blind, Placebo-Controlled Trial | TRD patients with increased inflammatory activity (n = unknown) | N-acetylcysteine + Antidepressants vs. Placebo + Antidepressants | N-acetylcysteine showed promise in TRD patients with inflammation, improving depression symptoms. | [285] |
| Randomized Placebo-Controlled Clinical Trial | Patients with SSRI treatment (n = 56) | Glycyrrhizic Acid (GZA) + SSRI vs. SSRI + Placebo | GZA significantly improved depressive symptoms, with better response in patients with higher baseline inflammation. | [286] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sălcudean, A.; Bodo, C.-R.; Popovici, R.-A.; Cozma, M.-M.; Păcurar, M.; Crăciun, R.-E.; Crisan, A.-I.; Enatescu, V.-R.; Marinescu, I.; Cimpian, D.-M.; et al. Neuroinflammation—A Crucial Factor in the Pathophysiology of Depression—A Comprehensive Review. Biomolecules 2025, 15, 502. https://doi.org/10.3390/biom15040502
Sălcudean A, Bodo C-R, Popovici R-A, Cozma M-M, Păcurar M, Crăciun R-E, Crisan A-I, Enatescu V-R, Marinescu I, Cimpian D-M, et al. Neuroinflammation—A Crucial Factor in the Pathophysiology of Depression—A Comprehensive Review. Biomolecules. 2025; 15(4):502. https://doi.org/10.3390/biom15040502
Chicago/Turabian StyleSălcudean, Andreea, Cristina-Raluca Bodo, Ramona-Amina Popovici, Maria-Melania Cozma, Mariana Păcurar, Ramona-Elena Crăciun, Andrada-Ioana Crisan, Virgil-Radu Enatescu, Ileana Marinescu, Dora-Mihaela Cimpian, and et al. 2025. "Neuroinflammation—A Crucial Factor in the Pathophysiology of Depression—A Comprehensive Review" Biomolecules 15, no. 4: 502. https://doi.org/10.3390/biom15040502
APA StyleSălcudean, A., Bodo, C.-R., Popovici, R.-A., Cozma, M.-M., Păcurar, M., Crăciun, R.-E., Crisan, A.-I., Enatescu, V.-R., Marinescu, I., Cimpian, D.-M., Nan, A.-G., Sasu, A.-B., Anculia, R.-C., & Strete, E.-G. (2025). Neuroinflammation—A Crucial Factor in the Pathophysiology of Depression—A Comprehensive Review. Biomolecules, 15(4), 502. https://doi.org/10.3390/biom15040502