
Not Just an Alternative Energy Source: Diverse Biological Functions of Ketone Bodies and Relevance of HMGCS2 to Health and Disease

 ,
,
Abstract
:1. Introduction
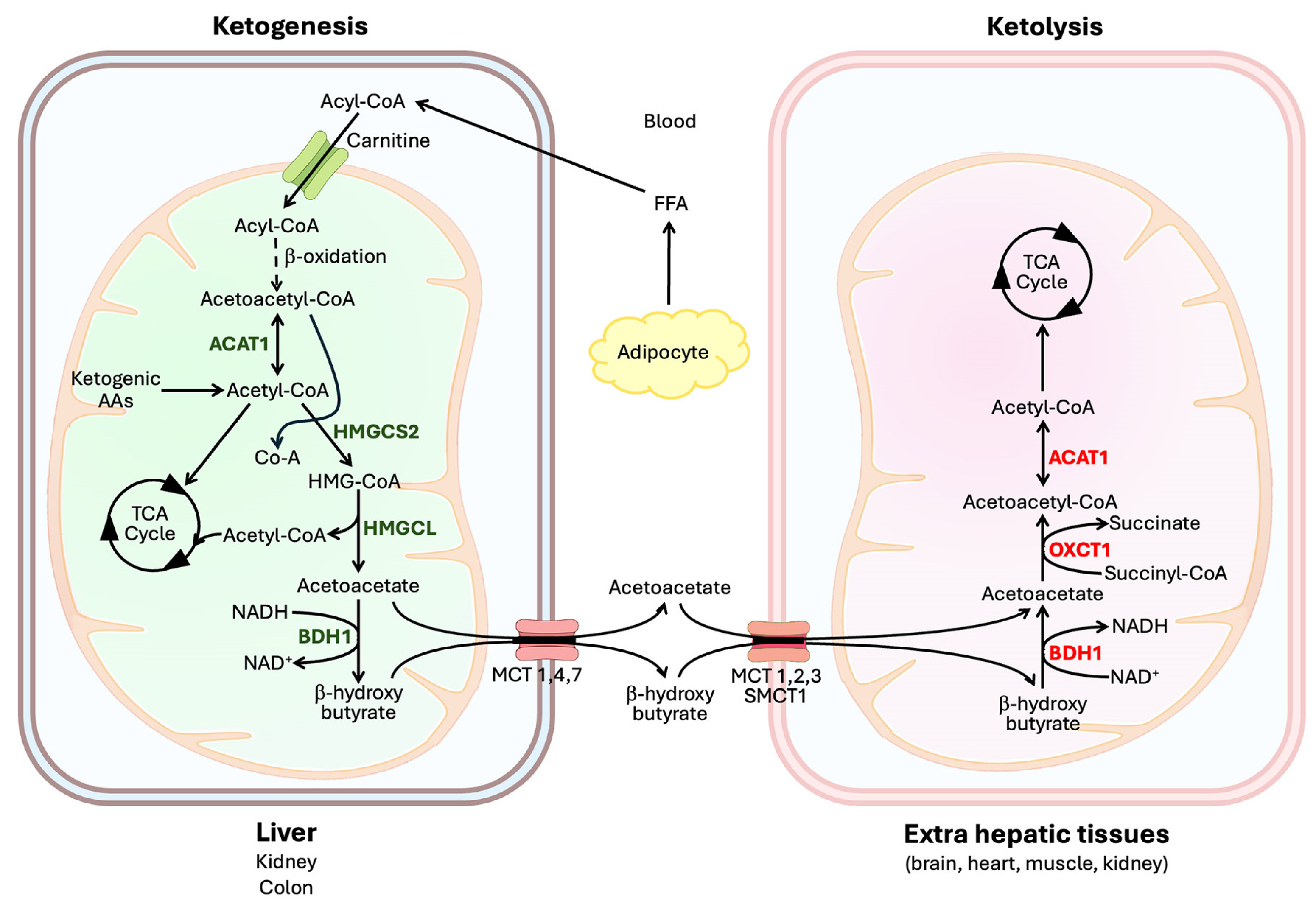
2. Enzymatic Reactions Involved in Ketogenesis and Ketone Utilization
2.1. Ketogenesis
2.2. Ketone Utilization
3. Ketogenesis in Different Organs
3.1. Liver
3.2. Kidney
3.3. Small Intestine and Colon
3.4. Retina
3.5. Astrocytes
4. Transporters for Ketone Bodies
4.1. Influx Transporters
4.2. Efflux Transporters
5. Biological Functions of Ketone Bodies
5.1. Alternative Energy Substrate During Limited Glucose Availability
5.2. Hormone-like Signaling Functions via Cell-Surface G-Protein-Coupled Receptors (GPRs)
5.3. Epigenetic Modulation via Inhibition of Class-I/IIa Histone Deacetylases
5.4. Post-Translational Modification via β-Hydroxybutyrylation
6. 3-Hydroxy-3-Methylglutaryl-CoA Synthase-2 (HMGCS2)
6.1. HMGCS2 Versus HMGCS1
6.2. Human HMGCS2: Gene, Protein, and Catalytic Mechanism
6.3. Small-Molecule Inhibitors of HMGCS2
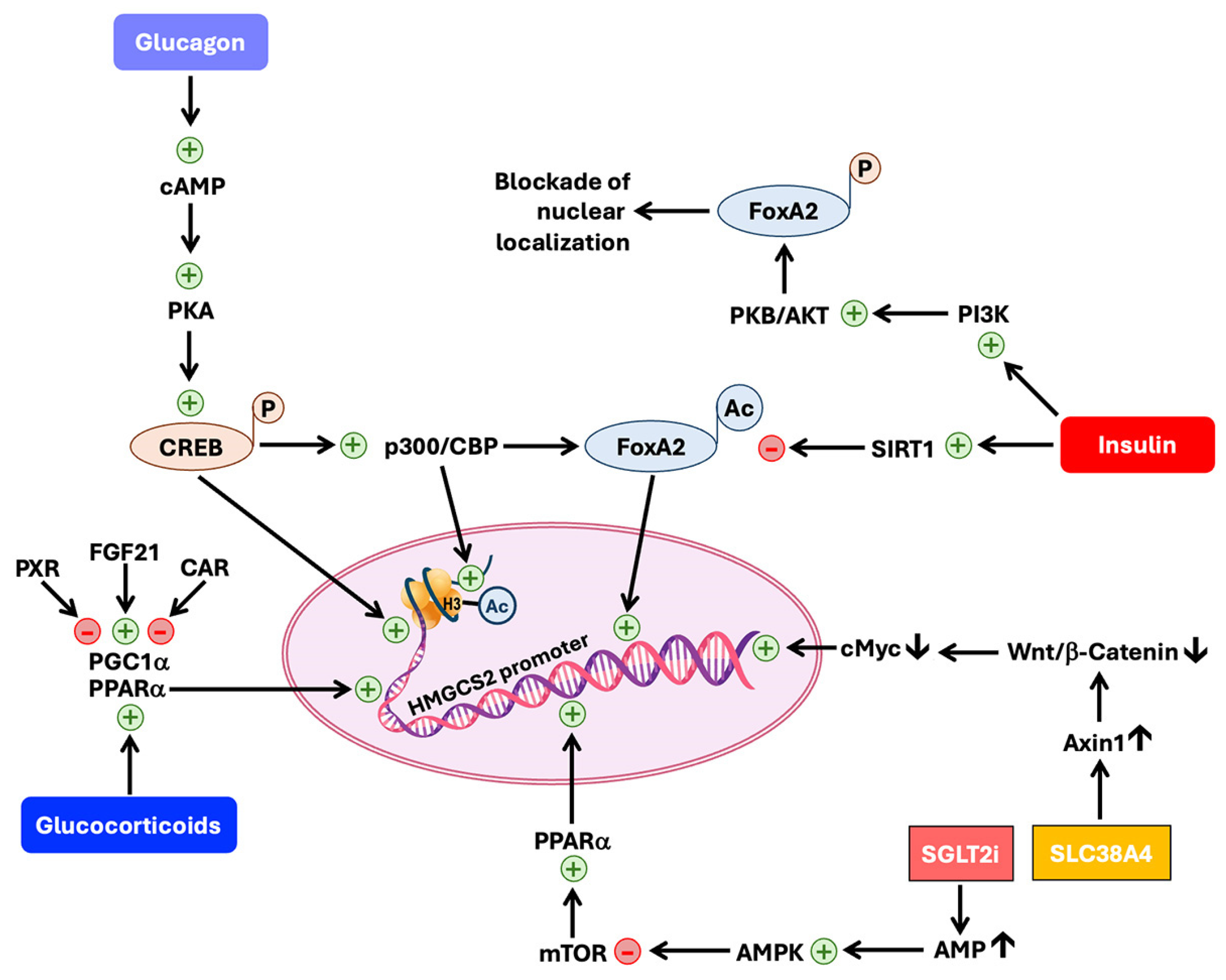
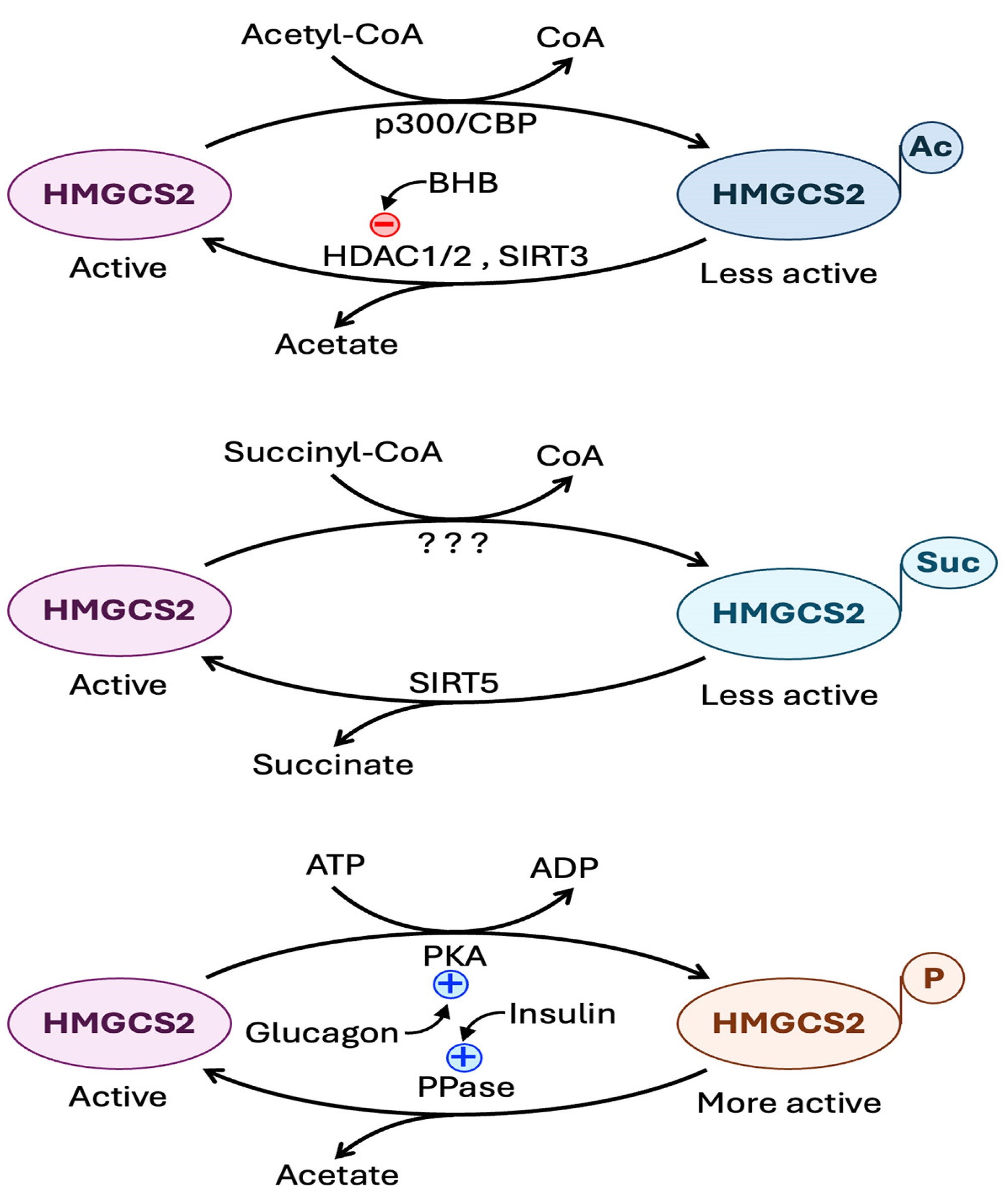
6.4. Regulation of HMGCS2 Expression and Activity
6.4.1. Regulation at the Level of Transcription of the HMGCS2 Gene
6.4.2. Regulation at the Level of Post-Translational Modification of HMGCS2 Protein
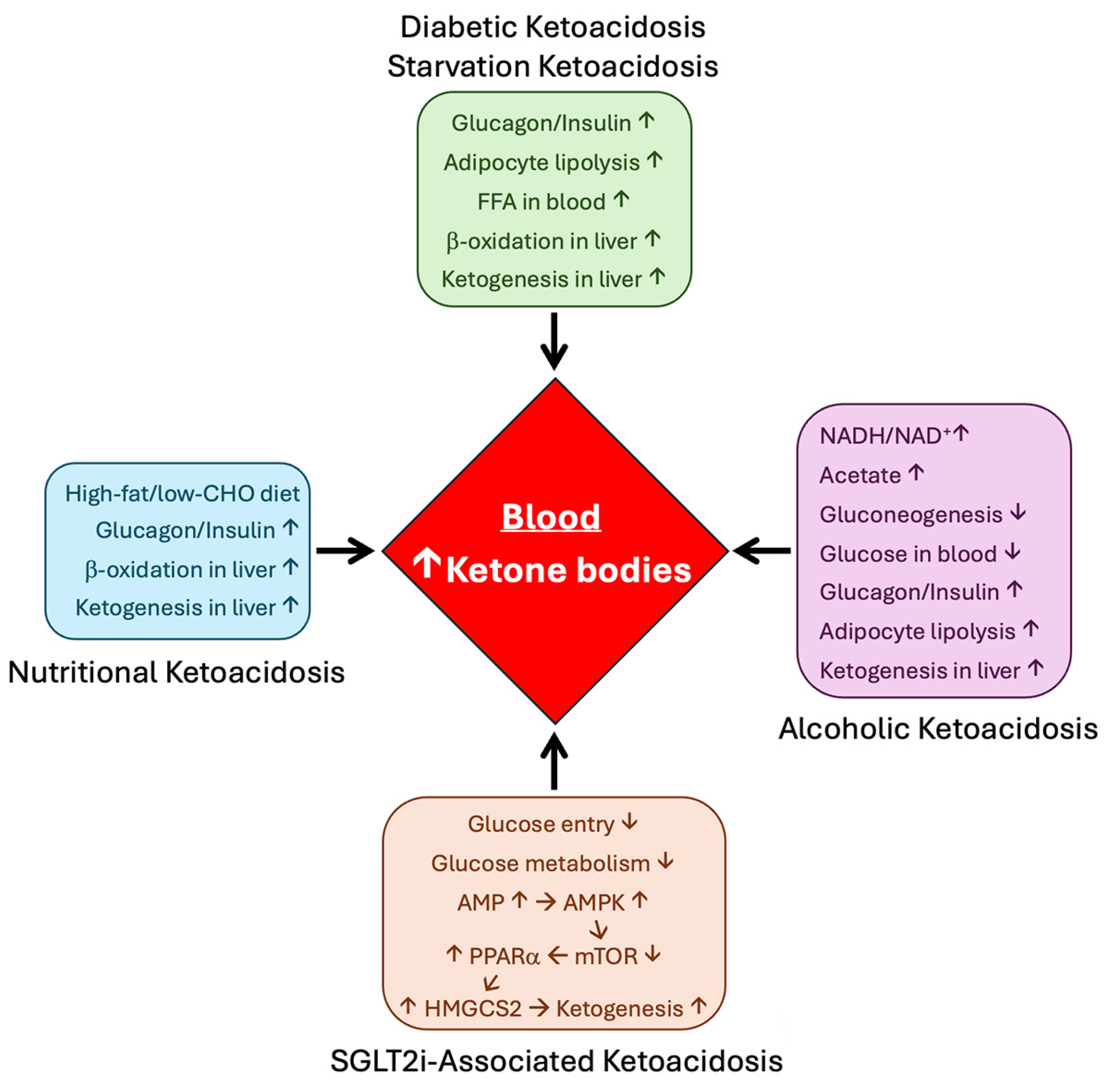
7. HMGCS2 in Pathological Conditions
7.1. Diabetes
7.2. Inflammation
7.3. Cancer
7.4. Neurodegeneration
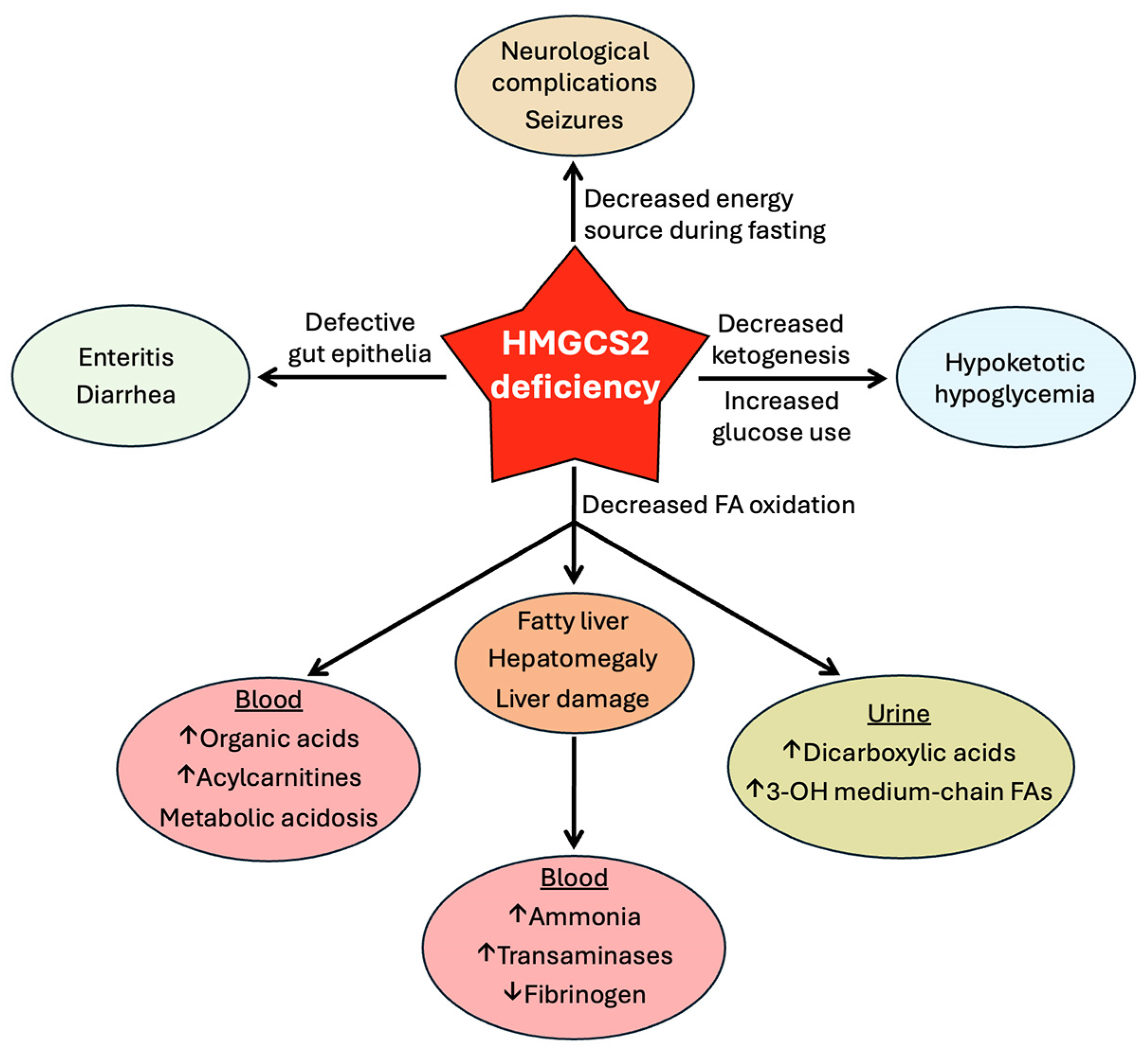
8. HMGCS2 Deficiency
8.1. Loss-of-Function Mutations in HMGCS2
8.2. Clinical Consequences of HMGCS2 Deficiency
9. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HMG-CoA | 3-Hydroxy-3-methylglutaryl-coenzyme A |
| HMGCS1 | 3-Hydroxy-3-methylglutaryl-coenzyme A synthase 1 |
| HMGCS2 | 3-Hydroxy-3-methylglutaryl-coenzyme A synthase 2 |
| OXCT1 | 3-Oxoacid CoA transferase 1 |
| SCOT1 | Succinyl-CoA:3-ketoacid CoA transferase 1 |
| SUCLA2 | Succinyl-CoA ligase 2 |
| ACAT1 | Acetyl CoA acetyl transferase 1 |
| MAPK | Mitogen-activated protein kinase |
| FGF21 | Fibroblast growth factor 21 |
| MCT | Monocarboxylate transporter |
| SMCT | Sodium-coupled monocarboxylate transporter |
| GLUT1 | Glucose transporter 1 |
| SGLT2 | Sodium-coupled glucose transporter 2 |
| SGLT2i | Sodium-coupled glucose transporter 2 inhibitor |
| SLC5A2 | Solute carrier, gene family 5, subfamily A, member 2 |
| SLC5A8 | Solute carrier, gene family 5, subfamily A, member 8 |
| SLC16A1 | Solute carrier, gene family 16, subfamily A, member 1 |
| SLC16A3 | Solute carrier, gene family 16, subfamily A, member 3 |
| SLC16A6 | Solute carrier, gene family 16, subfamily A, member 6 |
| SLC16A7 | Solute carrier, gene family 16, subfamily A, member 7 |
| SLC16A8 | Solute carrier, gene family 16, subfamily A, member 8 |
| SLC38A4 | Solute carrier, gene family 38, subfamily A, member 4 |
| PPARα | Peroxisome proliferator-activated receptor α |
| PPRE | Peroxisome proliferator-activated receptor responsive element |
| PGC1α | Peroxisome proliferator-activated receptor gamma coactivator 1α |
| AhR | Aryl hydrocarbon receptor |
| PXR | Pregnane X receptor |
| CAR | Constitutive androstane receptor |
| NAFLD | Nonalcoholic fatty liver disease |
| MASLD | Metabolic dysfunction-associated steatotic liver disease |
| NLRP3 | NOD-like receptor family pyrin domain containing protein 3 |
| HDAC | Histone deacetylase |
| SIRT | Silent mating type information regulation 2 homolog |
| GPR | G-protein-coupled receptor |
| AMPK | AMP-activated protein kinase |
| PI3K | Phosphatidyl inositol-3 kinase |
| CBP | CREB-binding protein |
| CREB | cAMP-responsive element binding protein |
| FOXA2 | Forkhead box protein A2 |
| BDH1 | β-Hydroxybutyrate dehydrogenase 1 |
| FFA3 | Free fatty acid receptor 3 |
| PUMA-G | Protein upregulated in macrophages in response to interferon γ |
References
- Robinson, A.M.; Williamson, D.H. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Foster, D.W. Regulation of Hepatic Fatty Acid Oxidation and Ketone Body Production. Annu. Rev. Biochem. 1980, 49, 395–420. [Google Scholar] [CrossRef]
- Cahill, G.F., Jr. Fuel Metabolism in Starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef]
- Koeslag, J.H.; Noakes, T.D.; Sloan, A.W. Post-exercise ketosis. J. Physiol. 1980, 301, 79–90. [Google Scholar] [CrossRef]
- Torrens, S.L.; Robergs, R.A.; Curry, S.C.; Nalos, M. The Computational Acid–Base Chemistry of Hepatic Ketoacidosis. Metabolites 2023, 13, 803. [Google Scholar] [CrossRef]
- Goudarzi, A. The recent insights into the function of ACAT1: A possible anti-cancer therapeutic target. Life Sci. 2019, 232, 116592. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. β-Hydroxybutyrate: A signaling metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef]
- Nissen, S.L.; Abumrad, N.N. Nutritional role of the leucine metabolite β-hydroxy β-methylbutyrate (HMB). Nutr. Biochem. 1997, 8, 300–311. [Google Scholar] [CrossRef]
- Ikon, N.; Ryan, R.O. On the origin of 3-methylglutaconic acid in disorders of mitochondrial energy metabolism. J. Inherit. Metab. Dis. 2016, 39, 749–756. [Google Scholar] [CrossRef]
- Abdelkreem, E.; Harijan, R.K.; Yamaguchi, S.; Wierenga, R.K.; Fukao, T. Mutation update on ACAT1 variants associated with mitochondrial acetoacetyl-CoA thiolase (T2) deficiency. Hum. Mutat. 2019, 40, 1641–1663. [Google Scholar] [CrossRef]
- Hori, T.; Yamaguchi, S.; Shinkaku, H.; Horikawa, R.; Shigematsu, Y.; Takayanagi, M.; Fukao, T. Inborn errors of ketone body utilization. Pediatr. Int. 2015, 57, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Shafqat, N.; Kavanagh, K.L.; Sass, J.O.; Christensen, E.; Fukao, T.; Lee, W.H.; Oppermann, U.; Yue, W.W. A structural mapping of mutations causing succinyl-CoA:3-ketoacid CoA transferase (SCOT) deficiency. J. Inherit. Metab. Dis. 2013, 36, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Adeva-Andany, M.M.; Funcasta-Calderon, R.; Fernandez-Fernandez, C.; Castro-Quintela, E.; Carneiro-Freire, N. Metabolic effects of glucagon in humans. J. Clin. Transl. Endocrinol. 2018, 15, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Ragolia, L.; Begum, N. Protein phosphatase-1 and insulin action. Mol. Cell. Biochem. 1998, 182, 49–58. [Google Scholar] [CrossRef]
- Woo, Y.C.; Xu, A.; Wang, Y.; Lam, K.S.L. Fibroblast Growth Factor 21 as an emerging metabolic regulator: Clinical perspectives. Clin. Endocrinol. 2012, 78, 489–496. [Google Scholar] [CrossRef]
- Barros, D.R.; Hegele, R.A. Fibroblast growth factor 21: Update on genetics and molecular biology. Curr. Opin. Infect. Dis. 2024, 36, 88–95. [Google Scholar] [CrossRef]
- Weidemann, M.J.; Krebs, H.A. The fuel of respiration of rat kidney cortex. Biochem. J. 1969, 112, 149–166. [Google Scholar] [CrossRef]
- Takagi, A.; Kume, S.; Kondo, M.; Nakazawa, J.; Chin-Kanasaki, M.; Araki, H.; Araki, S.-I.; Koya, D.; Haneda, M.; Chano, T.; et al. Mammalian autophagy is essential for hepatic and renal ketogenesis during starvation. Sci. Rep. 2016, 6, 18944. [Google Scholar] [CrossRef]
- Venable, A.H.; Lee, L.E.; Feola, K.; Santoyo, J.; Broomfield, T.; Huen, S.C. Fasting-induced HMGCS2 expression in the kidney does not contribute to circulating ketones. Am. J. Physiol. Physiol. 2022, 322, F460–F467. [Google Scholar] [CrossRef]
- Zhang, D.; Yang, H.; Kong, X.; Wang, K.; Mao, X.; Yan, X.; Wang, Y.; Liu, S.; Zhang, X.; Li, J.; et al. Proteomics analysis reveals diabetic kidney as a ketogenic organ in type 2 diabetes. Am. J. Physiol. Metab. 2011, 300, E287–E295. [Google Scholar] [CrossRef]
- Nakatani, T.; Sakamoto, Y.; Ando, H.; Kobayashi, K. Enhanced ketogenesis in the kidney during hepatic inflow occlusion with the administration of Ringer’s acetate solution. Surgery 1996, 119, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E.; Felig, P.; Morgan, A.P.; Wahren, J.; Cahill, G.F. Liver and kidney metabolism during prolonged starvation. J. Clin. Investig. 1969, 48, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://v24.proteinatlas.org/ENSG00000134240-HMGCS2/tissue (accessed on 27 February 2025).
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Wallenius, V.; Elias, E.; Elebring, E.; Haisma, B.; Casselbrant, A.; Larraufie, P.; Spak, E.; Reimann, F.; le Roux, C.W.; Docherty, N.G.; et al. Suppression of enteroendocrine cell glucagon-like peptide (GLP)-1 release by fat-induced small intestinal ketogenesis: A mechanism targeted by Roux-en-Y gastric bypass surgery but not by preoperative very-low-calorie diet. Gut 2019, 69, 1423–1431. [Google Scholar] [CrossRef]
- Kim, J.T.; Napier, D.L.; Kim, J.; Li, C.; Lee, E.Y.; Weiss, H.L.; Wang, Q.; Evers, B.M. Ketogenesis alleviates TNFα-induced apoptosis and inflammatory responses in intestinal cells. Free. Radic. Biol. Med. 2021, 172, 90–100. [Google Scholar] [CrossRef]
- Helenius, T.O.; Misiorek, J.O.; Nyström, J.H.; Fortelius, L.E.; Habtezion, A.; Liao, J.; Asghar, M.N.; Zhang, H.; Azhar, S.; Omary, M.B.; et al. Keratin 8 absence down-regulates colonocyte HMGCS2 and modulates colonic ketogenesis and energy metabolism. Mol. Biol. Cell 2015, 26, 2298–2310. [Google Scholar] [CrossRef]
- Bass, K.; Sivaprakasam, S.; Dharmalingam-Nandagopal, G.; Thangaraju, M.; Ganapathy, V. Colonic ketogenesis, a microbiota-regulated process, contributes to blood ketones and protects against colitis in mice. Biochem. J. 2024, 481, 295–312. [Google Scholar] [CrossRef]
- Gebert, N.; Cheng, C.-W.; Kirkpatrick, J.M.; Di Fraia, D.; Yun, J.; Schädel, P.; Pace, S.; Garside, G.B.; Werz, O.; Rudolph, K.L.; et al. Region-Specific Proteome Changes of the Intestinal Epithelium during Aging and Dietary Restriction. Cell Rep. 2020, 31, 107565. [Google Scholar] [CrossRef]
- Kim, J.T.; Li, C.; Weiss, H.L.; Zhou, Y.; Liu, C.; Wang, Q.; Evers, B.M. Regulation of Ketogenic Enzyme HMGCS2 by Wnt/β-catenin/PPARγ Pathway in Intestinal Cells. Cells 2019, 8, 1106. [Google Scholar] [CrossRef]
- Békési, A.; Williamson, D.H. An Explanation for Ketogenesis by the Intestine of the Suckling Rat: The Presence of an Active Hydroxymethylglutaryl-Coenzyme A Pathway. Neonatology 1990, 58, 160–165. [Google Scholar] [CrossRef]
- Serra, D.; Asins, G.; Hegardt, F. Ketogenic Mitochondrial 3-Hydroxy 3-Methylglutaryl-CoA Synthase Gene Expression in Intestine and Liver of Suckling Rats. Arch. Biochem. Biophys. 1993, 301, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Serra, D.; Bellido, D.; Asins, G.; Arias, G.; Vilaro, S.; Hegardt, F.G. The expression of mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme-A synthase in neonatal rat intestine and liver is under transcriptional control. Eur. J. Biochem. 1996, 237, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-W.; Biton, M.; Haber, A.L.; Gunduz, N.; Eng, G.; Gaynor, L.T.; Tripathi, S.; Calibasi-Kocal, G.; Rickelt, S.; Butty, V.L.; et al. Ketone Body Signaling Mediates Intestinal Stem Cell Homeostasis and Adaptation to Diet. Cell 2019, 178, 1115–1131.e15. [Google Scholar] [CrossRef] [PubMed]
- Terranova, C.J.; Stemler, K.M.; Barrodia, P.; Jeter-Jones, S.L.; Ge, Z.; Bonilla, M.d.l.C.; Raman, A.; Cheng, C.-W.; Allton, K.L.; Arslan, E.; et al. Reprogramming of H3K9bhb at regulatory elements is a key feature of fasting in the small intestine. Cell Rep. 2021, 37, 110044. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, Y.; Rychahou, P.; Fan, T.W.-M.; Lane, A.N.; Weiss, H.L.; Evers, B.M. Ketogenesis contributes to intestinal cell differentiation. Cell Death Differ. 2017, 24, 458–468. [Google Scholar] [CrossRef]
- Adijanto, J.; Du, J.; Moffat, C.; Seifert, E.L.; Hurley, J.B.; Philp, N.J. The Retinal Pigment Epithelium Utilizes Fatty Acids for Ketogenesis. J. Biol. Chem. 2014, 289, 20570–20582. [Google Scholar] [CrossRef]
- Zhang, Q.; Presswalla, F.; Calton, M.; Charniga, C.; Stern, J.; Temple, S.; Vollrath, D.; Zacks, D.N.; Ali, R.R.; Thompson, D.A.; et al. Highly Differentiated Human Fetal RPE Cultures Are Resistant to the Accumulation and Toxicity of Lipofuscin-Like Material. Investig. Opthalmology Vis. Sci. 2019, 60, 3468–3479. [Google Scholar] [CrossRef]
- Gulette, G.A.; Hass, D.T.; Pandey, K.; Zhang, Q.; Han, J.Y.; Engel, A.; Chao, J.R.; Philp, N.J.; Hurley, J.B.; Miller, J.M. Reassessing retinal pigment epithelial ketogenesis: Enzymatic assays for ketone body levels provide inaccurate results. Exp. Eye Res. 2024, 245, 109966. [Google Scholar] [CrossRef]
- Reyes-Reveles, J.; Dhingra, A.; Alexander, D.; Bragin, A.; Philp, N.J.; Boesze-Battaglia, K. Phagocytosis-dependent ketogenesis in retinal pigment epithelium. J. Biol. Chem. 2017, 292, 8038–8047. [Google Scholar] [CrossRef]
- Guzmán, M.; Blázquez, C. Ketone body synthesis in the brain: Possible neuroprotective effects. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 287–292. [Google Scholar] [CrossRef]
- Auestad, N.; Korsak, R.A.; Morrow, J.W.; Edmond, J. Fatty Acid Oxidation and Ketogenesis by Astrocytes in Primary Culture. J. Neurochem. 1991, 56, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Iizumi, T.; Mashima, K.; Abe, T.; Suzuki, N. Roles and Regulation of Ketogenesis in Cultured Astroglia and Neurons Under Hypoxia and Hypoglycemia. ASN Neuro 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, C.; Woods, A.; De Ceballos, M.L.; Carling, D.; Guzmán, M. The AMP-Activated Protein Kinase Is Involved in the Regulation of Ketone Body Production by Astrocytes. J. Neurochem. 1999, 73, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.; Mantha, O.L.; Schor, J.; Pascual, A.; Plaçais, P.-Y.; Pavlowsky, A.; Preat, T. Glia fuel neurons with locally synthesized ketone bodies to sustain memory under starvation. Nat. Metab. 2022, 4, 213–224. [Google Scholar] [CrossRef]
- McMullen, E.; Hertenstein, H.; Strassburger, K.; Deharde, L.; Brankatschk, M.; Schirmeier, S. Glycolytically impaired Drosophila glial cells fuel neural metabolism via β-oxidation. Nat. Commun. 2023, 14, 2996. [Google Scholar] [CrossRef]
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and Inflammatory Adaptation of Reactive Astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538. [Google Scholar] [CrossRef]
- Poole, R.C.; Halestrap, A.P. Transport of lactate and other monocarboxylates across mammalian plasma membranes. Am. J. Physiol. Physiol. 1993, 264, C761–C782. [Google Scholar] [CrossRef]
- Ganapathy, V.; Thangaraju, M.; Gopal, E.; Martin, P.M.; Itagaki, S.; Miyauchi, S.; Prasad, P.D. Sodium-coupled Monocarboxylate Transporters in Normal Tissues and in Cancer. AAPS J. 2008, 10, 193–199. [Google Scholar] [CrossRef]
- Sivaprakasam, S.; Bhutia, Y.D.; Yang, S.; Ganapathy, V. Short-Chain Fatty Acid Transporters: Role in Colonic Homeostasis. Compr. Physiol. 2017, 8, 299–314. [Google Scholar] [CrossRef]
- Felmlee, M.A.; Jones, R.S.; Rodriguez-Cruz, V.; Follman, K.E.; Morris, M.E. Monocarboxylate Transporters (SLC16): Function, Regulation, and Role in Health and Disease. Pharmacol. Rev. 2020, 72, 466–485. [Google Scholar] [CrossRef]
- Miyauchi, S.; Gopal, E.; Fei, Y.-J.; Ganapathy, V. Functional Identification of SLC5A8, a Tumor Suppressor Down-regulated in Colon Cancer, as a Na+-coupled Transporter for Short-chain Fatty Acids. J. Biol. Chem. 2004, 279, 13293–13296. [Google Scholar] [CrossRef] [PubMed]
- Gopal, E.; Fei, Y.-J.; Sugawara, M.; Miyauchi, S.; Zhuang, L.; Martin, P.; Smith, S.B.; Prasad, P.D.; Ganapathy, V. Expression of slc5a8 in Kidney and Its Role in Na+-coupled Transport of Lactate. J. Biol. Chem. 2004, 279, 44522–44532. [Google Scholar] [CrossRef] [PubMed]
- Gopal, E.; Fei, Y.-J.; Miyauchi, S.; Zhuang, L.; Prasad, P.D.; Ganapathy, V. Sodium-coupled and electrogenic transport of B-complex vitamin nicotinic acid by slc5a8, a member of the Na/glucose co-transporter gene family. Biochem. J. 2005, 388, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.M.; Gopal, E.; Ananth, S.; Zhuang, L.; Itagaki, S.; Prasad, B.M.; Smith, S.B.; Prasad, P.D.; Ganapathy, V. Identity of SMCT1 (SLC5A8) as a neuron-specific Na+-coupled transporter for active uptake of L-lactate and ketone bodies in the brain. J. Neurochem. 2006, 98, 279–288. [Google Scholar] [CrossRef]
- Pierre, K.; Pellerin, L. Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J. Neurochem. 2005, 94, 1–14. [Google Scholar] [CrossRef]
- Hosoya, K.; Kondo, T.; Tomi, M.; Takanaga, H.; Ohtsuki, S.; Terasaki, T. MCT1-Mediated Transport of L-Lactic Acid at the Inner Blood–Retinal Barrier: A Possible Route for Delivery of Monocarboxylic Acid Drugs to the Retina. Pharm. Res. 2001, 18, 1669–1676. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Lisanti, M.P.; Sotgia, F. Ketone bodies and two-compartment tumor metabolism: Stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle 2012, 11, 3956–3963. [Google Scholar] [CrossRef]
- Philp, N.J.; Wang, D.; Yoon, H.; Hjelmeland, L.M. Polarized Expression of Monocarboxylate Transporters in Human Retinal Pigment Epithelium and ARPE-19 Cells. Investig. Opthalmology Vis. Sci. 2003, 44, 1716–1721. [Google Scholar] [CrossRef]
- Pierre, K.; Magistretti, P.J.; Pellerin, L. MCT2 is a Major Neuronal Monocarboxylate Transporter in the Adult Mouse Brain. J. Cereb. Blood Flow Metab. 2002, 22, 586–595. [Google Scholar] [CrossRef]
- Hugo, S.E.; Cruz-Garcia, L.; Karanth, S.; Anderson, R.M.; Stainier, D.Y.; Schlegel, A. A monocarboxylate transporter required for hepatocyte secretion of ketone bodies during fasting. Genes Dev. 2012, 26, 282–293. [Google Scholar] [CrossRef]
- Uebanso, T.; Fukui, M.; Naito, C.; Shimohata, T.; Mawatari, K.; Takahashi, A. SLC16a6, mTORC1, and Autophagy Regulate Ketone Body Excretion in the Intestinal Cells. Biology 2023, 12, 1467. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, K.; Sugiyama, K.; Tomabechi, R.; Kishimoto, H.; Inoue, K. Mammalian monocarboxylate transporter 7 (MCT7/Slc16a6) is a novel facilitative taurine transporter. J. Biol. Chem. 2022, 298, 101800. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.proteinatlas.org/ENSG00000108932-SLC16A6/tissue (accessed on 27 February 2025).
- Matsuura, T.R.; Puchalska, P.; Crawford, P.A.; Kelly, D.P. Ketones and the Heart: Metabolic Principles and Therapeutic Implications. Circ. Res. 2023, 132, 882–898. [Google Scholar] [CrossRef] [PubMed]
- Shahtaghi, N.R.; Soni, B.; Bakrey, H.; Bigdelitabar, S.; Jain, S.K. Beta-Hydroxybutyrate: A Supplemental Molecule for Various Diseases. Curr. Drug Targets 2024, 25, 919–933. [Google Scholar] [CrossRef]
- Jang, J.; Kim, S.R.; Lee, J.E.; Lee, S.; Son, H.J.; Choe, W.; Yoon, K.-S.; Kim, S.S.; Yeo, E.-J.; Kang, I. Molecular Mechanisms of Neuroprotection by Ketone Bodies and Ketogenic Diet in Cerebral Ischemia and Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 25, 124. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Röhling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone bodies: From enemy to friend and guardian angel. BMC Med. 2021, 19, 1–15. [Google Scholar] [CrossRef]
- Tsuruta, H.; Yamahara, K.; Yasuda-Yamahara, M.; Kume, S. Emerging Pathophysiological Roles of Ketone Bodies. Physiology 2024, 39, 167–177. [Google Scholar] [CrossRef]
- NA Dong, Y.; Mesaros, C.; Xu, P.; Mercado-Ayón, E.; Halawani, S.; Ngaba, L.V.; Warren, N.; Sleiman, P.; Rodden, L.N.; A Schadt, K.; et al. Frataxin controls ketone body metabolism through regulation of OXCT1. PNAS Nexus 2022, 1, pgac142. [Google Scholar] [CrossRef]
- Ma, W.; Sun, Y.; Yan, R.; Zhang, P.; Shen, S.; Lu, H.; Zhou, Z.; Jiang, Z.; Ye, L.; Mao, Q.; et al. OXCT1 functions as a succinyltransferase, contributing to hepatocellular carcinoma via succinylating LACTB. Mol. Cell 2024, 84, 538–551.e7. [Google Scholar] [CrossRef]
- Guo, D.; Yu, Q.; Tong, Y.; Qian, X.; Meng, Y.; Ye, F.; Jiang, X.; Wu, L.; Yang, Q.; Li, S.; et al. OXCT1 succinylation and activation by SUCLA2 promotes ketolysis and liver tumor growth. Mol. Cell 2025, 85, 843–856.e6. [Google Scholar] [CrossRef]
- Spigoni, V.; Cinquegrani, G.; Iannozzi, N.T.; Frigeri, G.; Maggiolo, G.; Maggi, M.; Parello, V.; Cas, A.D. Activation of G protein-coupled receptors by ketone bodies: Clinical implication of the ketogenic diet in metabolic disorders. Front. Endocrinol. 2022, 13, 972890. [Google Scholar] [CrossRef] [PubMed]
- Offermanns, S. Hydroxy-Carboxylic Acid Receptor Actions in Metabolism. Trends Endocrinol. Metab. 2017, 28, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Thangaraju, M.; Prasad, P.D.; Martin, P.M.; Singh, N. Transporters and receptors for short-chain fatty acids as the molecular link between colonic bacteria and the host. Curr. Opin. Pharmacol. 2013, 13, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Taggart, A.K.; Kero, J.; Gan, X.; Cai, T.-Q.; Cheng, K.; Ippolito, M.; Ren, N.; Kaplan, R.; Wu, K.; Wu, T.-J.; et al. (d)-β-Hydroxybutyrate Inhibits Adipocyte Lipolysis via the Nicotinic Acid Receptor PUMA-G. J. Biol. Chem. 2005, 280, 26649–26652. [Google Scholar] [CrossRef]
- Thangaraju, M.; Cresci, G.A.; Liu, K.; Ananth, S.; Gnanaprakasam, J.P.; Browning, D.D.; Mellinger, J.D.; Smith, S.B.; Digby, G.J.; Lambert, N.A.; et al. GPR109A Is a G-protein–Coupled Receptor for the Bacterial Fermentation Product Butyrate and Functions as a Tumor Suppressor in Colon. Cancer Res. 2009, 69, 2826–2832. [Google Scholar] [CrossRef]
- Won, Y.J.; Lu, V.B.; Puhl, H.L., III; Ikeda, S.R. β-Hydroxybutyrate modulates N-type calcium channels in rat sympathetic neurons by acting as an agonist for the G-protein-coupled receptor FFA3. J. Neurosci. 2013, 33, 19314–19325. [Google Scholar] [CrossRef]
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035. [Google Scholar] [CrossRef]
- Miyamoto, J.; Ohue-Kitano, R.; Mukouyama, H.; Nishida, A.; Watanabe, K.; Igarashi, M.; Irie, J.; Tsujimoto, G.; Satoh-Asahara, N.; Itoh, H.; et al. Ketone body receptor GPR43 regulates lipid metabolism under ketogenic conditions. Proc. Natl. Acad. Sci. USA 2019, 116, 23813–23821. [Google Scholar] [CrossRef]
- Mårtensson, J.; Björkman, L.; Lind, S.; Viklund, M.B.; Zhang, L.; Gutierrez, S.; Dahlgren, C.; Sundqvist, M.; Xie, X.; Forsman, H. The ketone body acetoacetate activates human neutrophils through FFAR2. J. Leukoc. Biol. 2023, 113, 577–587. [Google Scholar] [CrossRef]
- Zou, X.; Meng, J.; Li, L.; Han, W.; Li, C.; Zhong, R.; Miao, X.; Cai, J.; Zhang, Y.; Zhu, D. Acetoacetate Accelerates Muscle Regeneration and Ameliorates Muscular Dystrophy in Mice. J. Biol. Chem. 2016, 291, 2181–2195. [Google Scholar] [CrossRef]
- Wu, X.-J.; Shu, Q.-Q.; Wang, B.; Dong, L.; Hao, B. Acetoacetate Improves Memory in Alzheimer’s Mice via Promoting Brain-Derived Neurotrophic Factor and Inhibiting Inflammation. Am. J. Alzheimer’s Dis. Other Dementiasr 2022, 37. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.B.; Queathem, E.D.; Puchalska, P.; Crawford, P.A. Metabolic Messengers: Ketone bodies. Nat. Metab. 2023, 5, 2062–2074. [Google Scholar] [CrossRef] [PubMed]
- Andersen, O.E.; Poulsen, J.V.; Farup, J.; de Morree, A. Regulation of adult stem cell function by ketone bodies. Front. Cell Dev. Biol. 2023, 11, 1246998. [Google Scholar] [CrossRef] [PubMed]
- Moller, N. Ketone body, 3-hydroxybutyrate: Minor metabolite—major medical manifestations. J. Clin. Endocrinol. Metab. 2020, 105, dgaa370. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Neuman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Sleiman, S.F.; Henry, J.; Al-Haddad, R.; El Hayek, L.; Abou Haidar, E.; Stringer, T.; Ulja, D.; Karuppagounder, S.S.; Holson, E.B.; Ratan, R.R.; et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β-hydroxybutyrate. eLife 2016, 5, e15092. [Google Scholar] [CrossRef]
- Tanegashima, K.; Sato-Miyata, Y.; Funakoshi, M.; Nishito, Y.; Aigaki, T.; Hara, T. Epigenetic regulation of the glucose transporter gene Slc2a1 by β-hydroxybutyrate underlies preferential glucose supply to the brain of fasted mice. Genes Cells 2016, 22, 71–83. [Google Scholar] [CrossRef]
- Tognini, P.; Murakami, M.; Liu, Y.; Eckel-Mahan, K.L.; Newman, J.C.; Verdin, E.; Baldi, P.; Sassone-Corsi, P. Distinct Circadian Signatures in Liver and Gut Clocks Revealed by Ketogenic Diet. Cell Metab. 2017, 26, 523–538.e5. [Google Scholar] [CrossRef]
- Wang, X.; Wu, X.; Liu, Q.; Kong, G.; Zhou, J.; Jiang, J.; Wu, X.; Huang, Z.; Su, W.; Zhu, Q. Ketogenic Metabolism Inhibits Histone Deacetylase (HDAC) and Reduces Oxidative Stress After Spinal Cord Injury in Rats. Neuroscience 2017, 366, 36–43. [Google Scholar] [CrossRef]
- Li, B.; Yu, Y.; Liu, K.; Zhang, Y.; Geng, Q.; Zhang, F.; Li, Y.; Qi, J. β-Hydroxybutyrate inhibits histone deacetylase 3 to promote claudin-5 generation and attenuate cardiac microvascular hyperpermeability in diabetes. Diabetologia 2020, 64, 226–239. [Google Scholar] [CrossRef]
- Oka, S.-I.; Tang, F.; Chin, A.; Ralda, G.; Xu, X.; Hu, C.; Yang, Z.; Abdellatif, M.; Sadoshima, J. β-Hydroxybutyrate, a Ketone Body, Potentiates the Antioxidant Defense via Thioredoxin 1 Upregulation in Cardiomyocytes. Antioxidants 2021, 10, 1153. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Ma, K.; Yan, Z. Rescue of histone hypoacetylation and social deficits by ketogenic diet in a Shank3 mouse model of autism. Neuropsychopharmacology 2021, 47, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.I.; Both, P.; Benjamin, J.S.; Nutter, C.W.; Tan, J.H.; Kang, J.; Machado, L.A.; Klein, J.D.; de Morree, A.; Kim, S.; et al. Fasting induces a highly resilient deep quiescent state in muscle stem cells via ketone body signaling. Cell Metab. 2022, 34, 902–918.e6. [Google Scholar] [CrossRef]
- Chriett, S.; Dabek, A.; Wojtala, M.; Vidal, H.; Balcerczyk, A.; Pirola, L. Prominent action of butyrate over β-hydroxybutyrate as histone deacetylase inhibitor, transcriptional modulator and anti-inflammatory molecule. Sci. Rep. 2019, 9, 742. [Google Scholar] [CrossRef]
- Zhou, T.; Cheng, X.; He, Y.; Xie, Y.; Xu, F.; Xu, Y.; Huang, W. Function and mechanism of histone β-hydroxybutyrylation in health and disease. Front. Immunol. 2022, 13, 981285. [Google Scholar] [CrossRef]
- He, Y.; Cheng, X.; Zhou, T.; Li, D.; Peng, J.; Xu, Y.; Huang, W. β-Hydroxybutyrate as an epigenetic modifier: Underlying mechanisms and implications. Heliyon 2023, 9, e21098. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, D.; Weng, Y.; Delaney, K.; Tang, Z.; Yan, C.; Qi, S.; Peng, C.; Cole, P.A.; Roeder, R.G.; et al. The regulatory enzymes and protein substrates for the lysine β-hydroxybutyrylation pathway. Sci. Adv. 2021, 7, eabe2771. [Google Scholar] [CrossRef]
- Li, R.; Liu, Y.; Wu, J.; Chen, X.; Lu, Q.; Xia, K.; Liu, C.; Sui, X.; Liu, Y.; Wang, Y.; et al. Adaptive Metabolic Responses Facilitate Blood-Brain Barrier Repair in Ischemic Stroke via BHB-Mediated Epigenetic Modification of ZO-1 Expression. Adv. Sci. 2024, 11, e2400426. [Google Scholar] [CrossRef]
- Available online: https://v24.proteinatlas.org/ENSG00000112972-HMGCS1/tissue (accessed on 27 February 2025).
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Royo, T.; Pedragosa, M.J.; Ayte, J.; Gil-Gomez, G.; Vilaro, S.; Hegardt, F.G. Testis and ovary express the gene for the ketogenic mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase. J. Lipid Res. 1993, 34, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Pignataro, O.P.; Radicella, J.P.; Calvo, J.C.; Charreau, E.H. Mitochondrial biosynthesis of cholesterol in leydig cells from rat testis. Mol. Cell. Endocrinol. 1983, 33, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer-Polokoff, R.; von Gunten, C.; Logel, J.; Torget, R.; Sinensky, M. Isolation and characterization of a mammalian cell mutant defective in 3-hydroxy-3-methylglutaryl coenzyme A synthase. J. Biol. Chem. 1982, 257, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Fan, J.; Xia, S.; Pan, Y.; Liu, S.; Qian, G.; Qian, Z.; Kang, H.-B.; Arbiser, J.L.; Pollack, B.P.; et al. HMG-CoA synthase 1 is a synthetic lethal partner of BRAFV600E in human cancers. J. Biol. Chem. 2017, 292, 10142–10152. [Google Scholar] [CrossRef]
- Montgomery, C.; Pei, Z.; Watkins, P.A.; Miziorko, H.M. Identification and Characterization of an Extramitochondrial Human 3-Hydroxy-3-methylglutaryl-CoA Lyase. J. Biol. Chem. 2012, 287, 33227–33236. [Google Scholar] [CrossRef]
- Arnedo, M.; Latorre-Pellicer, A.; Lucia-Campos, C.; Gil-Salvador, M.; Antoñanzas-Peréz, R.; Gómez-Puertas, P.; Bueno-Lozano, G.; Puisac, B.; Pié, J. More Than One HMG-CoA Lyase: The Classical Mitochondrial Enzyme Plus the Peroxisomal and the Cytosolic Ones. Int. J. Mol. Sci. 2019, 20, 6124. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Suk, F.-M.; Liao, Y.-J. Loss of HMGCS2 Enhances Lipogenesis and Attenuates the Protective Effect of the Ketogenic Diet in Liver Cancer. Cancers 2020, 12, 1797. [Google Scholar] [CrossRef]
- Ayté, J.; Gil-Gómez, G.; Haro, D.; Marrero, P.F.; Hegardt, F.G. Rat mitochondrial and cytosolic 3-hydroxy-3-methylglutaryl-CoA synthases are encoded by two different genes. Proc. Natl. Acad. Sci. USA 1990, 87, 3874–3878. [Google Scholar] [CrossRef]
- Mascaro, C.; Buesa, C.; Ortiz, J.A.; Haro, D.; Hegardt, F.G. Molecular cloning and tissue expression of human mitochnondrial 3-hydroxy-3-methylglutaryl-C0A synthase. Arch. Biochem. Biophys. 1995, 317, 385–390. [Google Scholar] [CrossRef]
- Hegardt, F.G. Transcriptional regulation of mitochondrial HMG-CoA synthase in the control of ketogenesis. Biochimie 1998, 80, 803–806. [Google Scholar] [CrossRef]
- Hegardt, F.G. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: A control enzyme in ketogenesis. Biochem. J. 1999, 338, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Boukaftane, Y.; A Mitchell, G. Cloning and characterization of the human mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase gene. Gene 1997, 195, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Greenspan, M.D.; Yudkovitz, J.B.; Lo, C.Y.; Chen, J.S.; Alberts, A.W.; Hunt, V.M.; Chang, M.N.; Yang, S.S.; Thompson, K.L.; Chiang, Y.C.; et al. Inhibition of hydroxymethylglutaryl-coenzyme A synthase by L-659,699. Proc. Natl. Acad. Sci. USA 1987, 84, 7488–7492. [Google Scholar] [CrossRef] [PubMed]
- Greenspan, M.D.; Bull, H.G.; Yudkovitz, J.B.; Hanf, D.P.; Alberts, A.W. Inhibition of 3-hydroxy-3-methylglutaryl-CoA synthase and cholesterol biosynthesis by β-lactone inhibitors and binding of these inhibitors to the enzyme. Biochem. J. 1993, 289, 889–895. [Google Scholar] [CrossRef]
- Tomoda, H.; Ohbayashi, N.; Morikawa, Y.; Kumagai, H.; Ōmura, S. Binding site for fungal β-lactone hymeglusin on cytosolic 3-hydroxy-3-methylglutaryl coenzyme A synthase. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2004, 1636, 22–28. [Google Scholar] [CrossRef]
- Tomoda, H.; Ohbayashi, N.; Kumagai, H.; Hashizume, H.; Sunazuka, T.; Ōmura, S. Differential Inhibition of HMG-CoA Synthase and Pancreatic Lipase by the Specific Chiral Isomers of β-Lactone DU-6622. Biochem. Biophys. Res. Commun. 1999, 265, 536–540. [Google Scholar] [CrossRef]
- Shamsi, A.; Furkan, M.; Khan, M.S.; Yadav, D.K.; Shahwan, M. Computational Screening of Repurposed Drugs for HMG-CoA Synthase 2 in Alzheimer’s Disease. J. Alzheimer’s Dis. 2024, 100, 475–485. [Google Scholar] [CrossRef]
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of Ketone Body Metabolism and the Role of PPARα. Int. J. Mol. Sci. 2016, 17, 2093. [Google Scholar] [CrossRef]
- Hwang, C.Y.; Choe, W.; Yoon, K.-S.; Ha, J.; Kim, S.S.; Yeo, E.-J.; Kang, I. Molecular Mechanisms for Ketone Body Metabolism, Signaling Functions, and Therapeutic Potential in Cancer. Nutrients 2022, 14, 4932. [Google Scholar] [CrossRef]
- Ruppert, P.M.; Kersten, S. Mechanisms of hepatic fatty acid oxidation and ketogenesis during fasting. Trends Endocrinol. Metab. 2023, 35, 107–124. [Google Scholar] [CrossRef]
- Ristic, B.; Bhutia, Y.D.; Ganapathy, V. Cell-surface G-protein-coupled receptors for tumor-associated metabolites: A direct link to mitochondrial dysfunction in cancer. Biochim. Biophys. Acta (BBA)-Bioenerg. 2017, 1868, 246–257. [Google Scholar] [CrossRef] [PubMed]
- A Kliewer, S.; Mangelsdorf, D.J. Fibroblast growth factor 21: From pharmacology to physiology. Am. J. Clin. Nutr. 2010, 91, 254S–257S. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Weiskirchen, R. What does the “AKT” stand for in the name “AKT kinase”? Some historical comments. Front. Oncol. 2020, 10, 1329. [Google Scholar] [CrossRef] [PubMed]
- von Meyenn, F.; Porstmann, T.; Gasser, E.; Selevsek, N.; Schmidt, A.; Aebersold, R.; Stoffel, M. Glucagon-Induced Acetylation of Foxa2 Regulates Hepatic Lipid Metabolism. Cell Metab. 2013, 17, 436–447. [Google Scholar] [CrossRef]
- Crooks, D.R.; Natarajan, T.G.; Jeong, S.Y.; Chen, C.; Park, S.Y.; Huang, H.; Ghosh, M.C.; Tong, W.H.; Haller, R.G.; Wu, C.; et al. Elevated FGF21, PGC-1α and ketogenic enzyme expression are hallmarks of iron-sulfur cluster depletion in human skeletal muscle. Hum. Mol. Genet. 2014, 23, 24–39. [Google Scholar] [CrossRef]
- Wang, C.; Xu, C.-X.; Krager, S.L.; Bottum, K.M.; Liao, D.-F.; Tischkau, S.A. Aryl Hydrocarbon Receptor Deficiency Enhances Insulin Sensitivity and Reduces PPAR-α Pathway Activity in Mice. Environ. Health Perspect. 2011, 119, 1739–1744. [Google Scholar] [CrossRef]
- Sivaprakasam, S.; Bhutia, Y.D.; Ramachandran, S.; Ganapathy, V. Cell-Surface and Nuclear Receptors in the Colon as Targets for Bacterial Metabolites and Its Relevance to Colon Health. Nutrients 2017, 9, 856. [Google Scholar] [CrossRef]
- Wong, J.M.W.; de Souza, R.; Kendall, C.W.C.; Emam, A.; Jenkins, D.J.A. Colonic Health: Fermentation and Short Chain Fatty Acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef]
- Meertens, L.M.; Miyata, K.S.; Cechetto, J.D.; Rachubinski, R.A.; Capone, J.P. A mitochondrial ketogenic enzyme regulates its gene expression by association with the nuclear hormone receptor PPARα. EMBO J. 1998, 17, 6972–6978. [Google Scholar] [CrossRef]
- Kostiuk, M.A.; Keller, B.O.; Berthiaume, L.G. Palmitoylation of ketogenic enzyme HMGCS2 enhances its interaction with PPARα and transcription at the Hmgcs2 PPRE. FASEB J. 2010, 24, 1914–1924. [Google Scholar] [CrossRef]
- Li, J.; Li, M.-H.; Wang, T.-T.; Liu, X.-N.; Zhu, X.-T.; Dai, Y.-Z.; Zhai, K.-C.; Liu, Y.-D.; Lin, J.-L.; Ge, R.-L.; et al. SLC38A4 functions as a tumour suppressor in hepatocellular carcinoma through modulating Wnt/β-catenin/MYC/HMGCS2 axis. Br. J. Cancer 2021, 125, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, M.; Nakanishi, T.; Fei, Y.-J.; Martindale, R.G.; Ganapathy, M.E.; Leibach, F.H.; Ganapathy, V. Structure and function of ATA3, a new subtype of amino acid transport system A, primarily expressed in the liver and skeletal muscle. Biochim. Biophys. Acta (BBA)-Biomembr. 2000, 1509, 7–13. [Google Scholar] [CrossRef]
- Hatanaka, T.; Huang, W.; Ling, R.; Prasad, P.D.; Sugawara, M.; Leibach, F.H.; Ganapathy, V. Evidence for the transport of neutral as well as cationic amino acids by ATA3, a novel and liver-specific subtype of amino acid transport system A. Biochim. Biophys. Acta (BBA)-Biomembr. 2001, 1510, 10–17. [Google Scholar] [CrossRef]
- Morace, C.; Lorello, G.; Bellone, F.; Quartarone, C.; Ruggeri, D.; Giandalia, A.; Mandraffino, G.; Minutoli, L.; Squadrito, G.; Russo, G.T.; et al. Ketoacidosis and SGLT2 Inhibitors: A Narrative Review. Metabolites 2024, 14, 264. [Google Scholar] [CrossRef]
- Mahfooz, R.S.; Khan, M.K.; Al Hennawi, H.; Khedr, A. SGLT-2 Inhibitor-Associated Euglycemic Diabetic Ketoacidosis: A Case Report and a Literature Review. Cureus 2022, 14, e26267. [Google Scholar] [CrossRef]
- Vallon, V. How can inhibition of glucose and sodium transport in the early proximal tubule protect the cardiorenal system? Nephrol. Dial. Transplant. 2024, 39, 1565–1573. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, M.; Kim, S.H.; Kim, S.R.; Lee, B.; Kang, E.S.; Cha, B.; Cho, J.W.; Lee, Y. Sodium-glucose cotransporter 2 inhibitors regulate ketone body metabolism via inter-organ crosstalk. Diabetes Obes. Metab. 2018, 21, 801–811. [Google Scholar] [CrossRef]
- Cai, W.; Chong, K.; Huang, Y.; Huang, C.; Yin, L. Empagliflozin improves mitochondrial dysfunction in diabetic cardiomyopathy by modulating ketone body metabolism and oxidative stress. Redox Biol. 2023, 69, 103010. [Google Scholar] [CrossRef]
- Dutka, M.; Bobiński, R.; Francuz, T.; Garczorz, W.; Zimmer, K.; Ilczak, T.; Ćwiertnia, M.; Hajduga, M.B. SGLT-2 Inhibitors in Cancer Treatment—Mechanisms of Action and Emerging New Perspectives. Cancers 2022, 14, 5811. [Google Scholar] [CrossRef]
- Mooli, R.G.R.; Ramakrishnan, S.K. Emerging Role of Hepatic Ketogenesis in Fatty Liver Disease. Front. Physiol. 2022, 13, 946474. [Google Scholar] [CrossRef]
- Stram, A.R.; Payne, R.M. Post-translational modifications in mitochondria: Protein signaling in the powerhouse. Cell. Mol. Life Sci. 2016, 73, 4063–4073. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Hua, L.; Dittenhafer-Reed, K.E.; Schwer, B.; Lombard, D.B.; Li, Y.; Bunkenborg, J.; Alt, F.W.; Denu, J.M.; et al. SIRT3 Deacetylates Mitochondrial 3-Hydroxy-3-Methylglutaryl CoA Synthase 2 and Regulates Ketone Body Production. Cell Metab. 2010, 12, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Quant, P.A.; Tubbs, P.K.; Brand, M.D. Glucagon activates mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase in vivo by decreasing the extent of succinylation of the enzyme. Eur. J. Biochem. 1990, 187, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Rardin, M.J.; He, W.; Nishida, Y.; Newman, J.C.; Carrico, C.; Danielson, S.R.; Guo, A.; Gut, P.; Sahu, A.K.; Li, B.; et al. SIRT5 Regulates the Mitochondrial Lysine Succinylome and Metabolic Networks. Cell Metab. 2013, 18, 920–933. [Google Scholar] [CrossRef]
- Grimsrud, P.A.; Carson, J.J.; Hebert, A.S.; Hubler, S.L.; Niemi, N.M.; Bailey, D.J.; Jochem, A.; Stapleton, D.S.; Keller, M.P.; Westphall, M.S.; et al. A Quantitative Map of the Liver Mitochondrial Phosphoproteome Reveals Posttranslational Control of Ketogenesis. Cell Metab. 2012, 16, 672–683. [Google Scholar] [CrossRef]
- Hughes, M.M.; O’Neill, L.A. Metabolic regulation of NLRP3. Immunol. Rev. 2017, 281, 88–98. [Google Scholar] [CrossRef]
- Neudorf, H.; Little, J.P. Impact of fasting & ketogenic interventions on the NLRP3 inflammasome: A narrative review. Biomed. J. 2023, 47, 100677. [Google Scholar] [CrossRef]
- Graff, E.C.; Fang, H.; Wanders, D.; Judd, R.L. Anti-inflammatory effects of the hydroxycarboxylic acid receptor 2. Metabolism 2016, 65, 102–113. [Google Scholar] [CrossRef]
- Sivaprakasam, S.; Prasad, P.D.; Singh, N. Benefits of short-chain fatty acids and their receptors in inflammation and carcinogenesis. Pharmacol. Ther. 2016, 164, 144–151. [Google Scholar] [CrossRef]
- Martín-Adrados, B.; Wculek, S.K.; Fernández-Bravo, S.; Torres-Ruiz, R.; Valle-Noguera, A.; Gomez-Sánchez, M.J.; Hernández-Walias, J.C.; Ferreira, F.M.; Corraliza, A.M.; Sancho, D.; et al. Expression of HMGCS2 in intestinal epithelial cells is downregulated in inflammatory bowel disease associated with endoplasmic reticulum stress. Front. Immunol. 2023, 14, 1185517. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; Lisanti, M.P. Ketone body utilization drives tumor growth and metastasis. Cell Cycle 2012, 11, 3964–3971. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Park, S.; Kim, J.H.; Bang, S.-B.; Kim, H.-J.; Ka, N.-L.; Ko, Y.; Kim, S.-S.; Lim, G.Y.; Lee, S.; et al. Targeting HMG-CoA synthase 2 suppresses tamoxifen-resistant breast cancer growth by augmenting mitochondrial oxidative stress-mediated cell death. Life Sci. 2023, 328, 121827. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Jin, C.; Kumar, P.; Yu, X.; Lenahan, C.; Sheng, J. Ketogenic Diets and Hepatocellular Carcinoma. Front. Oncol. 2022, 12, 879205. [Google Scholar] [CrossRef]
- Su, S.-G.; Yang, M.; Zhang, M.-F.; Peng, Q.-Z.; Li, M.-Y.; Liu, L.-P.; Bao, S.-Y. miR-107-mediated decrease of HMGCS2 indicates poor outcomes and promotes cell migration in hepatocellular carcinoma. Int. J. Biochem. Cell Biol. 2017, 91, 53–59. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Liu, C.-L.; Chiu, W.-C.; Twu, Y.-C.; Liao, Y.-J. HMGCS2 Mediates Ketone Production and Regulates the Proliferation and Metastasis of Hepatocellular Carcinoma. Cancers 2019, 11, 1876. [Google Scholar] [CrossRef]
- Ding, R.; Chen, T.; Zhang, Y.; Chen, X.; Zhuang, L.; Yang, Z. HMGCS2 in metabolic pathways was associated with overall survival in hepatocellular carcinoma: A LASSO-derived study. Sci. Prog. 2021, 104. [Google Scholar] [CrossRef]
- Camarero, N.; Mascaró, C.; Mayordomo, C.; Vilardell, F.; Haro, D.; Marrero, P.F. Ketogenic HMGCS2 Is a c-Myc Target Gene Expressed in Differentiated Cells of Human Colonic Epithelium and Down-Regulated in Colon Cancer. Mol. Cancer Res. 2006, 4, 645–653. [Google Scholar] [CrossRef]
- Suk, F.-M.; Wu, C.-Y.; Chiu, W.-C.; Chien, C.-Y.; Chen, T.-L.; Liao, Y.-J. HMGCS2 Mediation of Ketone Levels Affects Sorafenib Treatment Efficacy in Liver Cancer Cells. Molecules 2022, 27, 8015. [Google Scholar] [CrossRef]
- Suk, F.-M.; Wu, C.-Y.; Fang, C.-C.; Chen, T.-L.; Liao, Y.-J. β-HB treatment reverses sorafenib resistance by shifting glycolysis–lactate metabolism in HCC. Biomed. Pharmacother. 2023, 166, 115293. [Google Scholar] [CrossRef]
- Kim, Y.; Shin, S.-Y.; Jeung, J.; Kim, Y.; Kang, Y.-W.; Lee, S.; Oh, C.-M. Integrative analysis of mitochondrial metabolic reprogramming in early-stage colon and liver cancer. Front. Oncol. 2023, 13, 1218735. [Google Scholar] [CrossRef]
- Zou, K.; Hu, Y.; Li, M.; Wang, H.; Zhang, Y.; Huang, L.; Xie, Y.; Li, S.; Dai, X.; Xu, W.; et al. Potential Role of HMGCS2 in Tumor Angiogenesis in Colorectal Cancer and Its Potential Use as a Diagnostic Marker. Can. J. Gastroenterol. Hepatol. 2019, 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mathew, M.; Nguyen, N.T.; Bhutia, Y.D.; Sivaprakasam, S.; Ganapathy, V. Metabolic Signature of Warburg Effect in Cancer: An Effective and Obligatory Interplay between Nutrient Transporters and Catabolic/Anabolic Pathways to Promote Tumor Growth. Cancers 2024, 16, 504. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-T.; Chang, C.-C.; Li, Y.-J.; Chen, S.-T.; Lin, I.-C.; Kok, S.-H.; Cheng, S.-J.; Lee, J.-J.; Wu, T.-S.; Kuo, M.-L.; et al. HMGCS2 enhances invasion and metastasis via direct interaction with PPARα to activate Src signaling in colorectal cancer and oral cancer. Oncotarget 2016, 8, 22460–22476. [Google Scholar] [CrossRef]
- Wan, S.; Xi, M.; Zhao, H.-B.; Hua, W.; Liu, Y.-L.; Zhou, Y.-L.; Zhuo, Y.-J.; Liu, Z.-Z.; Cai, Z.-D.; Wan, Y.-P.; et al. HMGCS2 functions as a tumor suppressor and has a prognostic impact in prostate cancer. Pathol.-Res. Pract. 2019, 215, 152464. [Google Scholar] [CrossRef]
- Saraon, P.; Cretu, D.; Musrap, N.; Karagiannis, G.S.; Batruch, I.; Drabovich, A.P.; van der Kwast, T.; Mizokami, A.; Morrissey, C.; Jarvi, K.; et al. Quantitative Proteomics Reveals That Enzymes of the Ketogenic Pathway Are Associated with Prostate Cancer Progression. Mol. Cell. Proteom. 2013, 12, 1589–1601. [Google Scholar] [CrossRef]
- Neuwirt, H.; Bouchal, J.; Kharaishvili, G.; Ploner, C.; Jöhrer, K.; Pitterl, F.; Weber, A.; Klocker, H.; Eder, I.E. Cancer-associated fibroblasts promote prostate tumor growth and progression through upregulation of cholesterol and steroid biosynthesis. Cell Commun. Signal. 2020, 18, 1–18. [Google Scholar] [CrossRef]
- Zhang, H.; Tao, Y.; Leng, S.X. Ketogenic Diet: An Effective Treatment Approach for Neurodegenerative Diseases. Curr. Neuropharmacol. 2022, 20, 2303–2319. [Google Scholar] [CrossRef]
- Włodarek, D. Role of Ketogenic Diets in Neurodegenerative Diseases (Alzheimer’s Disease and Parkinson’s Disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef]
- Sridharan, B.; Lee, M.-J. Ketogenic Diet: A Promising Neuroprotective Composition for Managing Alzheimer’s Diseases and its Pathological Mechanisms. Curr. Mol. Med. 2022, 22, 640–656. [Google Scholar] [CrossRef]
- Dyńka, D.; Kowalcze, K.; Paziewska, A. The Role of Ketogenic Diet in the Treatment of Neurological Diseases. Nutrients 2022, 14, 5003. [Google Scholar] [CrossRef]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Polito, R.; La Torre, M.E.; Moscatelli, F.; Cibelli, G.; Valenzano, A.; Panaro, M.A.; Monda, M.; Messina, A.; Monda, V.; Pisanelli, D.; et al. The Ketogenic Diet and Neuroinflammation: The Action of Beta-Hydroxybutyrate in a Microglial Cell Line. Int. J. Mol. Sci. 2023, 24, 3102. [Google Scholar] [CrossRef]
- Lanthaler, B.; Wieser, S.; Deutschmann, A.; Schossig, A.; Fauth, C.; Zschocke, J.; Witsch-Baumgartner, M. Genotype-based databases for variants causing rare diseases. Gene 2014, 550, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://databases.lovd.nl/shared/genes (accessed on 27 February 2025).
- Ramos, M.; Menao, S.; Arnedo, M.; Puisac, B.; Gil-Rodríguez, M.C.; Teresa-Rodrigo, M.E.; Hernández-Marcos, M.; Pierre, G.; Ramaswami, U.; Baquero-Montoya, C.; et al. New case of mitochondrial HMG-CoA synthase deficiency. Functional analysis of eight mutations. Eur. J. Med Genet. 2013, 56, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Puisac, B.; Marcos-Alcalde, I.; Hernández-Marcos, M.; Morlana, P.T.; Levtova, A.; Schwahn, B.C.; DeLaet, C.; Lace, B.; Gómez-Puertas, P.; Pié, J. Human Mitochondrial HMG-CoA Synthase Deficiency: Role of Enzyme Dimerization Surface and Characterization of Three New Patients. Int. J. Mol. Sci. 2018, 19, 1010. [Google Scholar] [CrossRef]
- Zhang, P.; Hu, X.; Guo, R.; Guo, J.; Li, W.; Qian, S.; Hao, C.; Liu, J. Novel HMGCS2 pathogenic variants in a Chinese family with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency. Pediatr. Investig. 2019, 3, 86–90. [Google Scholar] [CrossRef]
- Lee, T.; Takami, Y.; Yamada, K.; Kobayashi, H.; Hasegawa, Y.; Sasai, H.; Otsuka, H.; Takeshima, Y.; Fukao, T. A Japanese case of mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency who presented with severe metabolic acidosis and fatty liver without hypoglycemia. JIMD Rep. 2019, 48, 19–25. [Google Scholar] [CrossRef]
- Kilic, M.; Dorum, S.; Topak, A.; Yazici, M.U.; Ezgu, F.S.; Coskun, T. Expanding the clinical spectrum of mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency with Turkish cases harboring novel HMGCS2 gene mutations and literature review. Am. J. Med. Genet. A 2020, 182, 1608–1614. [Google Scholar] [CrossRef]
- Ago, Y.; Otsuka, H.; Sasai, H.; Abdelkreem, E.; Nakama, M.; Aoyama, Y.; Matsumoto, H.; Fujiki, R.; Ohara, O.; Akiyama, K.; et al. Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: In vitro functional analysis of five novel HMGCS2 mutations. Exp. Ther. Med. 2020, 20, 39. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, Y.-L.; Liu, M.; Chen, J.-J.; Li, X.-Q.; Cao, B.-Y.; Gong, C.-X. Clinical, biochemical, molecular and therapeutic characteristics of four new patients of mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency. Clin. Chim. Acta 2020, 509, 83–90. [Google Scholar] [CrossRef]
- Rojnueangnit, K.; Maneechai, P.; Thaweekul, P.; Piriyanon, P.; Khositseth, S.; Ittiwut, C.; Chetruengchai, W.; Kamolvisit, W.; Theerapanon, T.; Suphapeetiporn, K.; et al. Expanding phenotypic and mutational spectra of mitochondrial HMG-CoA synthase deficiency. Eur. J. Med. Genet. 2020, 63, 104086. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Shen, L.; Chen, Q.; Gong, C.; Yang, Y.; Wei, H.; Cao, B.; Chen, Y. Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients. Front. Genet. 2022, 12, 816779. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, D.; El-Karaksy, H.; Wali, Y.; Youssry, I. Mitochondrial 3-hydroxymethylglutaryl-CoA synthase-2 (HMGCS2) deficiency: A rare case with bicytopenia and coagulopathy. BMJ Case Rep. 2023, 16, e257011. [Google Scholar] [CrossRef] [PubMed]
- Decru, B.; Lys, M.; Truijens, K.; Mercier, N.; Papadopoulos, J.; Rymen, D.; Roland, D.; Dewulf, J.P.; Vermeersch, P. Mitochondrial HMG-CoA synthase deficiency. Mol. Genet. Metab. 2024, 144, 109007. [Google Scholar] [CrossRef]
- Tomita, I.; Tsuruta, H.; Yasuda-Yamahara, M.; Yamahara, K.; Kuwagata, S.; Tanaka-Sasaki, Y.; Chin-Kanasaki, M.; Fujita, Y.; Nishi, E.; Katagiri, H.; et al. Ketone bodies: A double-edged sword for mammalian life span. Aging Cell 2023, 22, e13833. [Google Scholar] [CrossRef]
- Desrochers, S.; David, F.; Garneau, M.; Jetté, M.; Brunengraber, H. Metabolism of R- and S-1,3-butanediol in perfused livers from meal-fed and starved rats. Biochem. J. 1992, 285, 647–653. [Google Scholar] [CrossRef]
- Thompson, G.N.; Hsu, B.Y.; Pitt, J.J.; Treacy, E.; Stanley, C.A. Fasting Hypoketotic Coma in a Child with Deficiency of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase. N. Engl. J. Med. 1997, 337, 1203–1207. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Expression Level | HMGCS1 | HMGCS2 |
|---|---|---|
| High | Esophagus Stomach Small intestine Large intestine Liver Testis | Urinary bladder Gallbladder Small intestine Large intestine Liver Kidney |
| Moderate | Nasopharynx Bronchus Lung Oral mucosa Gallbladder Pancreas Kidney Urinary bladder Epididymis Seminal vesicle Prostate Vagina Endometrium Cervix Placenta Skin | Stomach Testis Mammary gland |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suresh, V.V.; Sivaprakasam, S.; Bhutia, Y.D.; Prasad, P.D.; Thangaraju, M.; Ganapathy, V. Not Just an Alternative Energy Source: Diverse Biological Functions of Ketone Bodies and Relevance of HMGCS2 to Health and Disease. Biomolecules 2025, 15, 580. https://doi.org/10.3390/biom15040580
Suresh VV, Sivaprakasam S, Bhutia YD, Prasad PD, Thangaraju M, Ganapathy V. Not Just an Alternative Energy Source: Diverse Biological Functions of Ketone Bodies and Relevance of HMGCS2 to Health and Disease. Biomolecules. 2025; 15(4):580. https://doi.org/10.3390/biom15040580
Chicago/Turabian StyleSuresh, Varshini V., Sathish Sivaprakasam, Yangzom D. Bhutia, Puttur D. Prasad, Muthusamy Thangaraju, and Vadivel Ganapathy. 2025. "Not Just an Alternative Energy Source: Diverse Biological Functions of Ketone Bodies and Relevance of HMGCS2 to Health and Disease" Biomolecules 15, no. 4: 580. https://doi.org/10.3390/biom15040580
APA StyleSuresh, V. V., Sivaprakasam, S., Bhutia, Y. D., Prasad, P. D., Thangaraju, M., & Ganapathy, V. (2025). Not Just an Alternative Energy Source: Diverse Biological Functions of Ketone Bodies and Relevance of HMGCS2 to Health and Disease. Biomolecules, 15(4), 580. https://doi.org/10.3390/biom15040580