1. Introduction
Neutrophils play a crucial role in the innate immune response as they are the first to encounter and neutralize invaders [
1,
2]. They make ~60–70% of all white blood cells and use different strategies to fight pathogens, including phagocytosis, oxidative burst and neutrophil extracellular trap (NET) formation [
3,
4]. Numerous studies have demonstrated that when neutrophils interact with certain inflammatory stimuli (e.g., phorbol 12-myristate 13-acetate (PMA), lipopolysaccharide (LPS), A23187, ionomycin), they undergo a unique form of programmed cell death that results in releasing decondensed chromatin coated with cytotoxic peptides in a form of web-like structures called NETs [
5,
6]. It is well-known that during NET formation (NETosis), neutrophils generate high levels of reactive oxygen species (ROS) that activate specific kinase pathways, which results in promoter melting, transcriptional firing, chromatin decondensation and finally, NET release [
7,
8,
9]. There are currently two major pathways that explain how NETs are formed: NOX-dependent pathway which requires high cytosolic ROS levels and NOX-independent pathways that depends on mitochondrial ROS (mROS) production [
6]. Also, some known granular enzymes, such as myeloperoxidase (MPO) and neutrophil elastase, cover the decondensed chromatin during the final stages of NET formation [
7].
Lately, attention has been directed towards the relevance of histone modifications in delineating the molecular mechanism of NET formation. Studies reported that citrullination of histones has the potential to decondense chromatin during NET formation [
7,
10]. It is well known that increased calcium influx activates peptidylarginine deiminase 4 (PAD4), which converts the arginine present on histones (e.g., H3) into citrulline. Citrullination of histone H3 (CitH3) results in histone relaxation and was considered as a hallmark of NOX-independent NETosis [
11]. We have recently shown that another epigenetic modification—histone acetylation (e.g., acetylated histone H4, AcH4)—promotes both types of NET formation [
12]. Due to the loss of positively charged lysine, AcH4 results in increased levels of histone decondensation and gene transcription which eventually promoted NETosis by ~20% without altering intracellular ROS production by either NOX or mitochondria. To achieve increased levels of AcH4, histone deacetylase complex (HDAC) inhibitors within their half maximal inhibitory concentrations (IC50) were used because primary neutrophils express 18 different HDACs [
13,
14]. These inhibitors (HDACis) can act on most HDACs in the form of pan-deacetylase (pan-DAC) inhibitors and three of them are approved for clinical use, including belinostat and panobinostat. In fact, some clinical studies have shown some adverse effects of using HDACis in treating diseases. For example, a phase 2 study showed that treating myelodysplastic syndrome with belinostat resulted in 19 and 48 % of patients with grade 2 and 3/4 neutropenia, respectively [
15]. Also, despite that belinostat and panobinostat are used in a large range of concentrations depending on their applications, the relevance of these HDAC is in inducing different cytotoxic effects in neutrophils is not yet known [
16]. Therefore, treating neutrophils with increasing concentrations of either belinostat or panobinostat could result in neutrophil apoptosis or NET formation.
We hypothesized that treating neutrophils with increasing concentrations of HDACis would promote apoptosis and suppress NETosis. To examine this, we utilized belinostat and panobinostat on primary neutrophils of healthy donors and tested histone acetylation, ROS production and NETosis at baseline and with the use of agonists for NOX-dependent and -independent pathways. Our studies show that increasing concentrations of HDACis promote histone acetylation while suppressing spontaneous NET formation in a dose-dependent manner. Also, HDACis significantly inhibits NETosis in the presence of NETotic agonists. Nevertheless, a significant increase in apoptosis was accompanied with the inhibition of NET formation, particularly in non-activated cells. In addition, HDACis significantly increased the cytosolic but not mitochondrial, ROS levels, which could promote both apoptosis and NETosis. Therefore, the data presented in this study show that HDACis have a biphasic effect on NET formation. For the first time, we show direct evidence that belinostat and panobinostat have both NETotic and apoptotic effects on neutrophils. These findings would be useful in improving our understanding of the molecular mechanism of NETosis and clinical use of HDAC inhibitors for treating conditions including autoimmune diseases (e.g., lupus) and cancers [
17,
18,
19,
20].
2. Materials and Methods
2.1. Research Ethics Board Approval
The study protocol for using human blood samples was approved by the ethics committee of The Hospital for Sick Children (No. 1000020217). The ethics committee guidelines were followed while performing all procedures, including healthy human volunteer recruitment for blood donation. All the volunteers participated in this study signed informed consents prior to the blood donation.
2.2. Human Peripheral Blood Neutrophil Isolation
In this study, neutrophils from healthy male donors were used. However, data were excluded if donors had eosinophils or either too low or too high neutrophil count. A volume of 40–60 mL of peripheral blood, which usually yields 1–1.5 million neutrophils per millilitre of blood, was drawn from healthy donors into K2-EDTA (ethylenediaminetetraacetic acid) blood collection tubes (Becton, Dickinson and Co., Franklin Lakes, NJ, USA) at the nursing station of the hospital. Neutrophils were purified from peripheral blood by using PolymorphPrepTM (Axis-Shield, Oslo, Norway) by following the manufacturer’s instructions but with minor modifications. Blood was layered over PolymorphPrepTM solution in a 1:1 ratio and was then centrifuged at 600× g for 35 min without any brakes. Then, the polymorphonuclear neutrophil layer was collected and washed with 0.425% (w/v) NaCl with 10 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) to eliminate residues of PolymorphPrepTM. A hypotonic solution (0.2% (w/v) NaCl) was used twice for 30 s to purify neutrophils from the residual red blood cells (RBCs). To obtain isotonic condition, neutrophils were immediately mixed with an equal volume of 1.6% (w/v) NaCl solution with 20 mM HEPES buffer. Next, the cells were washed twice to eliminate RBC debris and soluble components. Neutrophils were re-suspended by using Roswell Park Memorial Institute (RPMI) 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10 mM HEPES buffer. A haemocytometer was used to quantify cell density and Cytospin preparations were used to check the purity of the neutrophils. Neutrophil preparations with >95–98% were used in all of the experiments.
2.3. Sytox Green NETosis Assay
To estimate NETosis kinetics, we used a cell-impermeable DNA binding dye called Sytox Green (Life Technologies, Carlsbad, CA, USA). A concentration of 5 µM Sytox Green were added in 100 µL media (RPMI 1640 medium supplemented with 10 mM HEPES) containing 50,000 neutrophils and then seeded into 96-well black clear-bottom plates. A volume of 5 µL of HDACis (final concentrations: 0.5, 1, 2, 4, 5, 10, 20 and 40 µM belinostat (PXD-101, Selleckchem); 0.08, 0.2, 0.4, 0.8, 1.6, 3.2 and 6.4 µM panobinostat (LBH589, Selleckchem) was added to respective wells with controls (RMPI + neutrophils only) for 30 min at 37 °C and 5% (v/v) CO2. A volume of 5 µL agonists (final concentrations: 25 nM PMA; 4 μM A23187; 5 μg/mL LPS (from E. coli 0128); 5 μM ionomycin, (unless otherwise stated)) was then added and placed at 37 °C and 5% (v/v) CO2 incubator. By using a fluorescence plate reader, the Sytox Green fluorescence intensities were measured every 60 min for up to 4 h (504 nm excitation, 523 nm emission, POLARstar OMEGA, BMG Labtech, Guelph, ON, Canada). Finally, the NETotic index (% of Sytox Green accessible total DNA) was calculated by subtracting the baseline green fluorescence at time 0-min from the fluorescence at each time point and was then divided by the fluorescence values of 100% NET formation obtained by lysing the cells with 0.5% (v/v) Triton X-100 (240 min time point).
2.4. DHR123 and MitoSOX Plate Reader Assays (NOX- and Mitochondrial-Mediated ROS Analyses)
A concentration of 20 μM dihydrorhodamine 123 (DHR123, Thermo Fisher Scientific, Waltham, MA, USA) was added in media containing 100,000 neutrophils and were then incubated at 37 °C and 5% (v/v) CO2 incubator for 15 min. Neutrophils were centrifuged for 10 min (400× g; with nine acceleration and nine deceleration ramps) and were washed with an equal amount of media. A volume of 100 μL of the neutrophil-containing solution was seeded into 96-well black clear-bottom plates and HDACis (belinostat and panobinostat) were added to respective wells with controls (RMPI + neutrophils only) for 30 min at 37 °C and 5% (v/v) CO2. A volume of 5 µL agonists (final concentrations: 25 nM PMA; 4 μM A23187; 5 μg/mL LPS from E. coli 0128; 5 μM ionomycin) were then added and incubated at 37 °C and 5% (v/v) CO2. A fluorescence plate reader was used to measure the fluorescence (507 nm excitation, 529 nm emission, POLARstar OMEGA, BMG Labtech) every 10 min in the first 30 min and then every 30 min up until 90 min. To calculate the relative fluorescence units (RFU), the fluorescence at each time point was subtracted from the baseline fluorescence at time 0-min.
The MitoSOX Red (Thermo Fisher Scientific) plate reader assays were performed similarly to the DHR123 assay with the following exceptions: 5 μM MitoSOX red were incubated at 37 °C and 5% (v/v) CO2 incubator for 15 min (no washing) and measured by fluorescence plate reader (510 nm excitation, 580 nm emission).
2.5. Fluorescence Confocal Imaging
A volume of 100 µL of media containing 100,000 neutrophils were seeded into 12-well chamber slides. A volume of 5 µL of HDACis (0.5, 2, 5 and 20 µM belinostat; 0.08, 0.4, 0.8 and 3.2 µM panobinostat) and/or NETotic agonists (25 nM PMA; 4 μM A23187; 5 μg/mL LPS from E. coli 0128; 5 μM ionomycin) were then added to respective wells with controls (RMPI + neutrophils only) and incubated for 120 min at 37 °C and 5% (v/v) CO2. As a positive control for apoptosis, neutrophils were treated with ultraviolet (UV) irradiation (0.24 J/cm2). To fix neutrophils and NETs, paraformaldehyde (4%, w/v) was used for 30 min and cells were then washed and permeabilized with 0.1% Triton-X 100 for 15 min at room temperature. Unspecific antigens were then blocked by using bovine serum albumin (BSA) (5%, w/v) for 60 min at room temperature. The cells were washed with phosphate-buffered saline (PBS) and incubated with primary antibodies: mouse anti- myeloperoxidase (MPO) antibody (ab25989, Abcam; 1:500 dilution) was used for staining MPO (with secondary antibody conjugated with a green fluorescence Alexa Fluor 488 dye; 1:1000 dilution; Thermo Fisher Scientific), while rabbit anti-CitH3 antibody (ab5103, Abcam; 1:500 dilution) or rabbit anti-histone H4 (acetyl K5) antibody (ab51997, Abcam, 1:1000) was used for detecting the presence of CitH3 or acetylated H4K5 (H4K5ac), respectively (with secondary antibody conjugated with a far-red fluorescence dye Alexa Fluor 647; 1:1000dilution; Thermo Fisher Scientific). The 4′,6-diamidino-2-phenylindole (DAPI) (1:100 dilution) was used to stain DNA. After treating with the secondary antibody, slides were washed and mounted by glass coverslips (Fisher Scientific) with anti-fade fluorescent mounting medium (Dako, Carpinteria, CA, USA). To acquire images, Olympus IX81 inverted fluorescence microscope was used with a Hamamatsu C9100-13 back-thinned EM-CCD camera and Yokogawa CSU × 1 spinning disk confocal scan head (Olympus Canada Inc., Richmond Hill, ON, Canada) with Spectral Aurora Borealis upgrade, four separate diode-pumped solid-state laser lines (405, 491, 561 and 642 nm; Spectral Applied Research, Richmond Hill, ON, Canada). The images were taken at 20 × /0.75 and 40 × /0.95 magnification and processed by Volocity software (version 6.3, Cell Imaging Perkin-Elmer; Quorum Technologies Inc., Puslinch, ON, Canada).
2.6. Western Blotting
A volume of 5 µL of HDACis (0.5, 2, 5 and 20 µM belinostat; 0.08, 0.8 and 3.2 µM panobinostat), NETotic agonists (25 nM PMA; 4 μM A23187; 5 μg/mL LPS from E. coli 0128; 5 μM ionomycin) and/or ultraviolet (UV) irradiation (0.24 J/cm2) were added to tubes containing 1 × 106 cells and incubated for 90 min at 37 °C and 5% (v/v) CO2. After being cooled on ice for 10 min, radioimmunoprecipitation assay (RIPA) lysis buffer (Millipore, Etobicoke, ON, Canada) containing 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM sodium orthovanadate, 1 mM sodium fluoride, 1 mg/mL aprotinin, 1 mg/mL leupeptin, 1 mg/mL pepstatin, protease inhibitor cocktail tablet per 5 mL (Roche Diagnostics, Laval, QC, Canada), DNase I (Invitrogen) and a phosphatase inhibitor cocktail tablet per 10 mL (Roche) was used to lyse neutrophils. Samples were vortexed for 10 s to mix the solution and incubated for 30 min at 37 °C and 5% (v/v) CO2 and then sonicated three times for 30 s intervals. The bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific) was used to ensure the same amount of total protein of each sample was used. A reducing sample loading buffer (20% (v/v) glycerol, 2% (v/v) beta-mercaptoethanol, 4% (w/v) sodium dodecyl sulfate (SDS), 0.130 M Tris, bromophenol blue (1 mg/100 mL), pH 6.8) was added to each sample and were then heated at 99 °C for 5 min prior to loading into the SDS-PAGE (polyacrylamide gel electrophoresis). Samples were run on a 4–20% Mini-PROTEAN® TGX™ precast protein gels (Bio-RAD Laboratories, Mississauga, ON, Canada) at 120 V for 60 min to separate protein by size. Wet transfer was performed to transfer proteins to nitrocellulose membranes. Unspecific binding on the membrane were blocked with 5% (w/v) BSA in TBST (Tris-buffered saline with 0.1% (v/v) Tween 20) for 1 h at room temperature. All primary antibodies were dissolved in 1% (w/v) BSA in 0.1% (v/v) TBST and incubated with membranes overnight at 4 °C followed by three washes with 0.1% PBST for 30 min. Primary antibodies used were: anti-GADPH (2118, Cell Signalling Technology, Whitby, ON, Canada) rabbit mAb at 1:1000; anti-histone H4K5ac (ab51997, Abcam) rabbit mAb at 1:1000; anti-cleaved caspase-3 (Asp175, Cell Signalling) rabbit mAb at 1:500. Then, the membranes were incubated in the secondary antibody solution (1% (w/v) BSA in 0.1% (v/v) TBST) for 60 min and then washed three times with 0.1% (v/v) PBST for 30 min. The secondary antibody used was: anti-rabbit IgG-HRP (Cell Signalling) at 1:2000. Enhanced chemiluminescent reagents were used to determine the protein intensity and the blots were imaged in Li-Cor Odyssey FC Imaging System and densitometry analysis of the images performed using Image Studio software (LI-COR Biotechnology, Lincoln, NE, USA). The H4K5ac and cleaved caspase 3 protein bands were normalized to the GAPDH bands.
2.7. Statistical Analyses
All data are presented as mean ± standard error of the mean (SEM) in line graphs. A two-way analysis of variance (ANOVA) with Dunnett test was performed to analyse line graphs and box plots. In box plots, each data point is presented. The mean is indicated with “+” and the full data spread is indicated with lines and boxes are marked with median and upper and lower interquartile ranges. Best fit linear regression analysis was performed and the equations, r2 values and p-values are indicated on each panel. The statistical analyses by One-way ANOVA with a Dunnett or Tukey test, where appropriate, were performed by using Prism GraphPad statistical analysis software (Version 8.0.2 for Windows, San Diego, CA, USA). A Dunnett post-test was used for comparing a fixed value of 1 with respective treatment conditions. A mean difference with a p-value of ≤0.05 was considered to be statistically significant.
4. Discussion
Several studies have indicated the important role of histone modification in mediating NET formation. Our previous studies showed that citrullination of histone H3 plays a role in unwinding the chromatin exclusively during calcium-mediated NOX-independent NETosis [
7,
10]. We have recently demonstrated the importance of histone acetylation in NETosis, as belinostat and panobinostat within the IC50 concentrations were able to promote both types of NET formation [
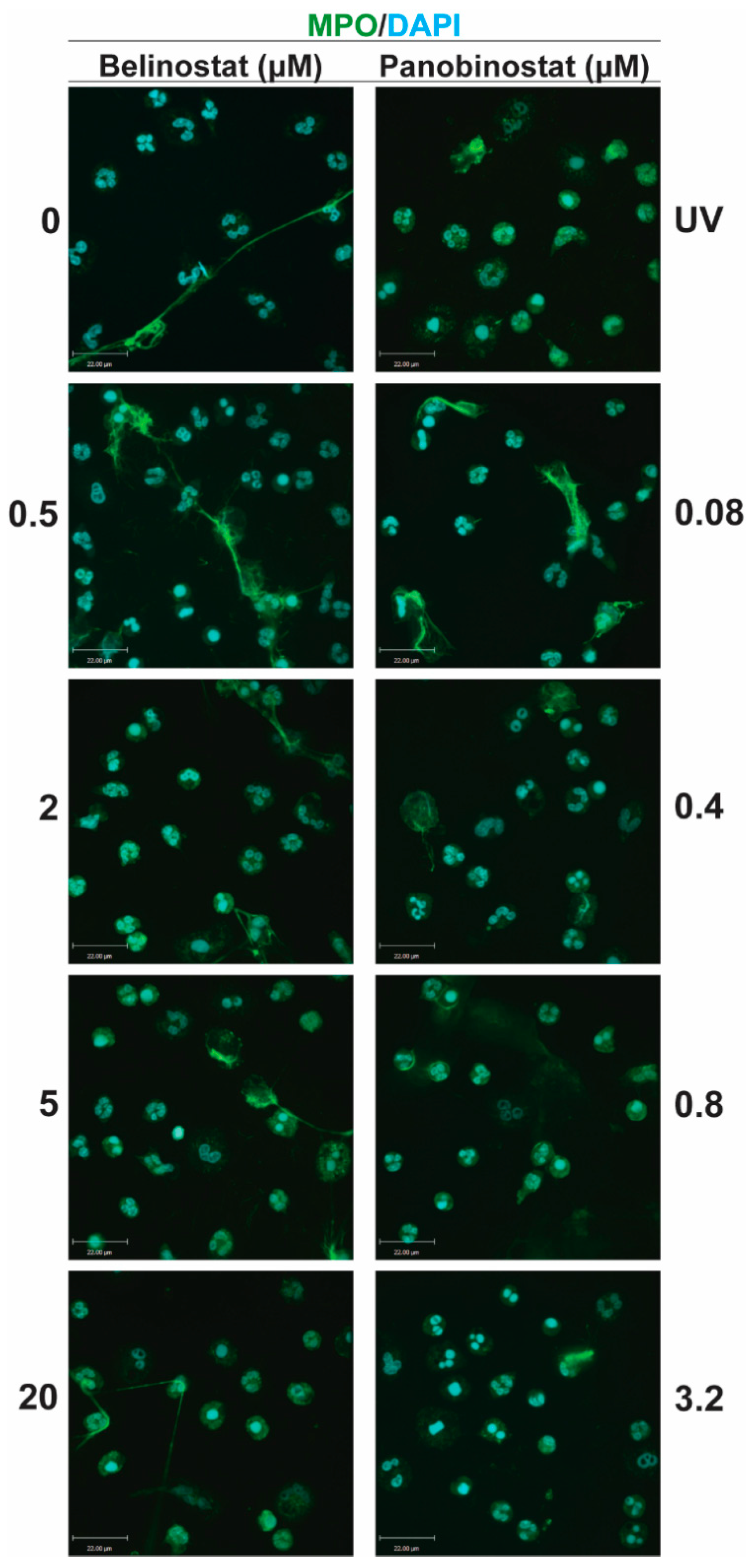
12]. Yet, the effect of increasing concentrations of belinostat and panobinostat on neutrophils are still not clearly understood. In this study, we showed that histone hyperacetylation is found in both NETotic and apoptotic cells that are treated with HDACis, either with belinostat or panobinostat (
Figure 1 and
Figure 2). Increasing higher concentrations of both belinostat and panobinostat significantly inhibited spontaneous, NOX-dependent and -independent NETosis (
Figure 3,
Figure 4 and
Figure 5;
Supplementary Figures S3 and S4). Also, we demonstrated that HDACis significantly increased apoptosis levels at baseline (
Figure 5,
Figure 6 and
Figure 7). However, HDACis-treated neutrophils resulted in a significant decrease in intact cells, while similar levels of cells undergoing NETosis were observed when compared to the control. On the other hand, the presence of NETotic agonists partially prevented HDACis-mediated cell death as the number of intact neutrophils significantly increased while apoptotic and NETotic levels were lower than neutrophils treated only with HDAC is (
Figure 8). In addition, our results show that HDACis significantly increased NOX— but not mitochondrial—derived ROS production (
Figure 9;
Supplementary Figures S6 and S7). Collectively, these data show that treating human neutrophils with increasing concentrations of belinostat and panobinostat inhibit NETosis and promote apoptotic effects but prevent cell death in the presence of NETotic agonists.
We recently provided evidence that HDACis have the potential to promote histone acetylation in neutrophils [
12]. Also, previous studies have reported that HDACis—for example, SAHA and belinostat—induces AcH4 in oligodendrocyte precursor cells (OPC) and in human promyelocytic leukaemia NB4 and HL-60 cells [
27,
28]. By performing immunofluorescence imaging and Western blotting, we found consistent results suggesting that belinostat and panobinostat significantly induce histone acetylation in a dose-dependent manner (
Figure 1 and
Figure 2). We previously showed that HDACis promote AcH4 in neutrophils in both types of NETosis [
12]. Therefore, our findings suggest that increasing doses of belinostat and panobinostat can alter their effects on neutrophils.
To better understand the mechanism, we used two ROS probes, DHR123 and MitoSOX, to measure the cytosolic and mitochondrial ROS, respectively. We previously showed that belinostat and panobinostat, within the IC50 values, induce NETosis without altering intracellular ROS levels [
12]. In fact, such a finding is supported by the fact that apoptotic microparticles, which have the potential to stimulate numerous immune reactions, were shown to drive ROS-independent NET formation in systemic lupus erythematosus (SLE) [
29]. Our current study demonstrates that increasing doses of belinostat and panobinostat results in altering intracellular ROS levels, as DHR123 assays recorded a dose-dependent.
NOX-derived ROS production increased when HDACis were used in the presence and absence of NETotic agonists (
Figure 9;
Supplementary Figures S6 and S7). In fact, our data are consistent with the previous studies as it was reported that 10 µM belinostat significantly induced intracellular ROS levels when incubated with pancreatic cancer cells [
30]. Another study has shown that belinostat also induces a dose-dependent increase in NOX-derived ROS production in thyroid cancer cells [
31].
Previous studies also show that HDACis-induced histone hyperacetylation results in increased intracellular ROS levels and apoptosis. Dincman et al. (2016) studied the cytotoxicity of HDACi on OPC where they demonstrated that SAHA-induced histone acetylation is associated with the decreased viability of OPCs [
27]. Another study showed that treating pancreatic cancer cells with 10 µM belinostat resulted in both increased levels of intracellular ROS production and apoptosis [
30]. Indeed, our studies showed that increasing concentrations of belinostat and panobinostat result in significant inhibition of NETosis (
Figure 3,
Figure 4,
Figure 5 and
Figure 7;
Supplementary Figures S3 and S4) and increased levels of cytosolic ROS and apoptosis (
Figure 5,
Figure 6,
Figure 7 and
Figure 8). Linear regression analyses demonstrate a positive correlation between AcH4 and apoptosis but negatively between NETosis and apoptosis (
Figure 5 and
Figure 6). Consistent with previous studies, promyelocytic leukaemia cells showed a significant increase in cell death when treated with increasing concentrations of belinostat [
28]. Similarly, Jiang et al. (2012) studied the effects of panobinostat on HL-60 and acute myelogenous leukaemia (AML) primary cells and found that cells became less viable with increasing concentrations of panobinostat [
32].
A decrease in ~10–40% NET release could have a significant impact on diseases. Previous studies have demonstrated the post-translational modification of NET histones in lupus and found that induced NET release results in increased levels of autoantibodies that target NETs [
33,
34]. Our studies may be helpful for understanding the correct application behind using HDACis in treating SLE, as our results show that using high doses of belinostat and panobinostat can inhibit NET release. Also, previous studies mentioned above demonstrate the clinical application of using belinostat and panobinostat in managing numerous types of cancers, such as thyroid and pancreatic cancers [
30,
31].
In summary, increasing concentrations of belinostat and panobinostat significantly induce neutrophil death while inhibiting baseline, NOX-dependent and -independent NETosis. These HDACis have the potential to induce histone acetylation in a dose-dependent manner which was positively correlated with apoptosis but negatively with NETosis (at higher concentrations of the drugs). Our results demonstrated that increasing concentrations of HDACis was accompanied with increased cytosolic but not mitochondrial, ROS production. We conclude that belinostat and panobinostat have a biphasic effect on neutrophils; they induce NETosis within the IC50 concentrations but have apoptosis-inducing effects at higher doses. In the presence of NETotic agonists, high concentrations of HDACis prevent neutrophil death. Hence, these points should be considered when interpreting the effect of HDACis on neutrophils death.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}