
Outlook for Implementation of Genomics-Based Selection in Public Cotton Breeding Programs

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
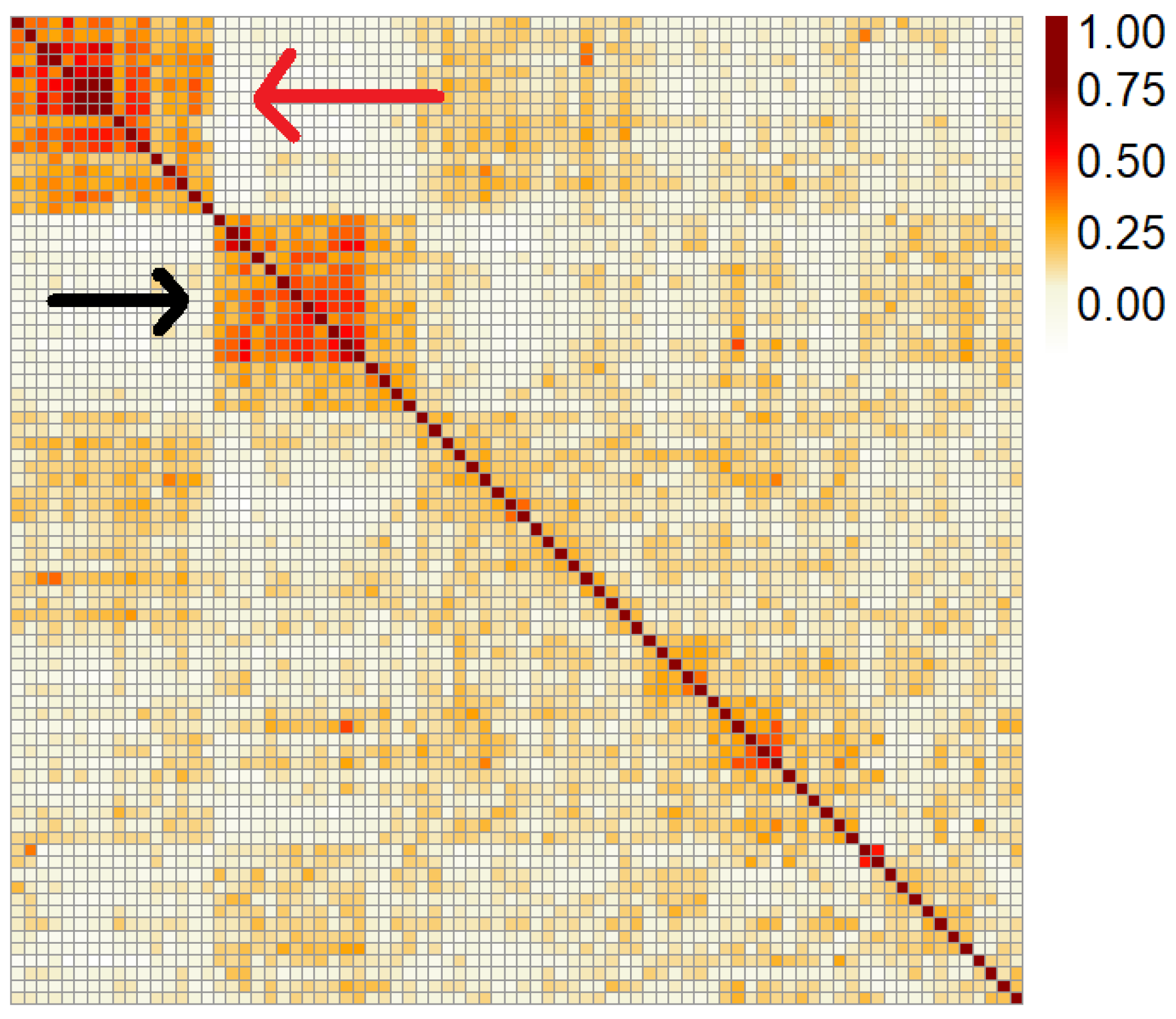
2.1. Genomic Relationships between Lines
2.2. Trait Heritabilities
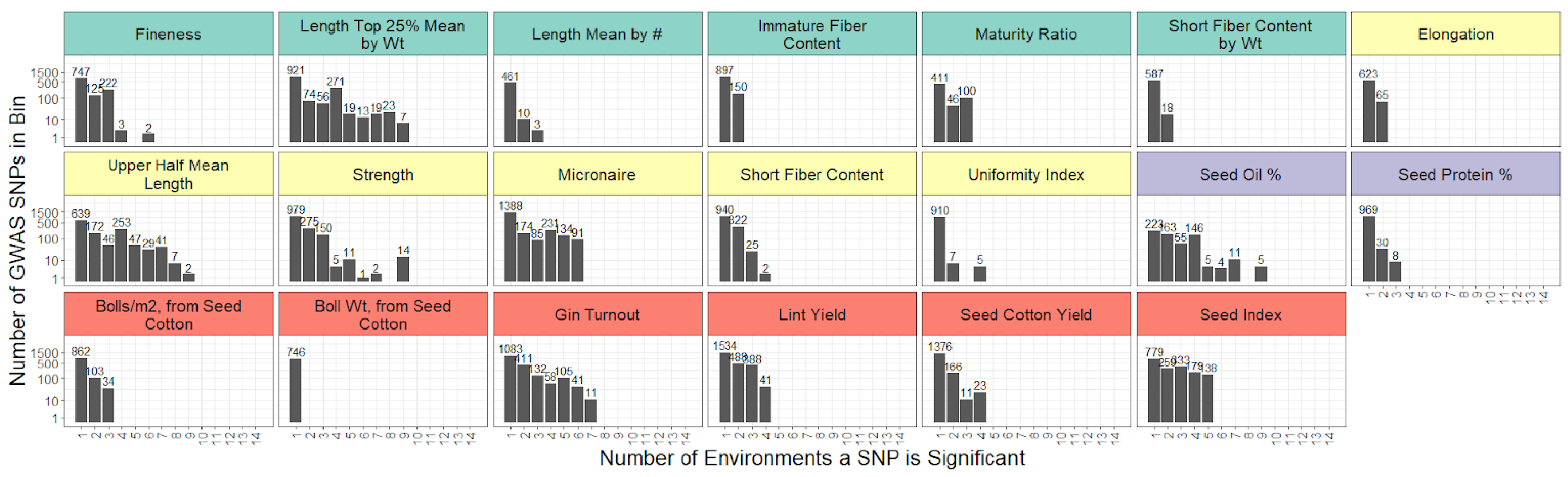
2.3. Genome-Wide Association Study
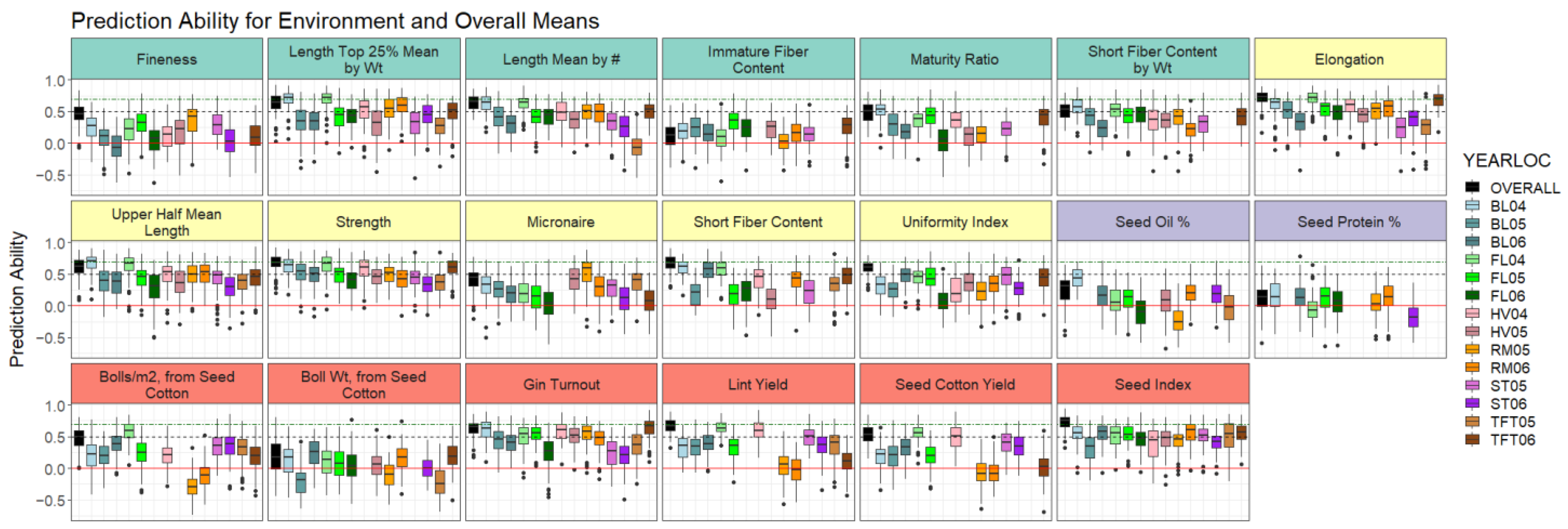
2.4. Genomic Prediction Evaluation through Cross Validation
2.4.1. Cross-Validation within Overall and Environment Means
2.4.2. Cross-Validation between Environments
2.4.3. Inclusion of GWAS Hits as Fixed Effects
2.5. Outlook for Genomic Prediction in Public Cotton Breeding
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campbell, B.T.; Chee, P.W.; Lubbers, E.; Bowman, D.T.; Meredith, W.R.; Johnson, J.; Fraser, D.; Bridges, W.; Jones, D.C. Dissecting Genotype × Environment Interactions and Trait Correlations Present in the Pee Dee Cotton Germplasm Collection following Seventy Years of Plant Breeding. Crop Sci. 2012, 52, 690. [Google Scholar] [CrossRef] [Green Version]
- Culp, T.W.; Harrell, D.C.; Kerr, T. Some Genetic Implications in the Transfer of High Fiber Strength Genes to Upland Cotton. Crop Sci. 1979, 19, 481–484. [Google Scholar] [CrossRef]
- May, O.L.; Green, C.C. Genetic Variation for Fiber Properties in Elite Pee Dee Cotton Populations. Crop Sci. 1994, 34, 684–690. [Google Scholar] [CrossRef]
- Meyer, L. Cotton Sector at a Glance. Available online: https://www.ers.usda.gov/topics/crops/cotton-wool/cotton-sector-at-a-glance/ (accessed on 28 March 2022).
- Dai, B.; Guo, H.; Huang, C.; Zhang, X.; Lin, Z. Genomic heterozygosity and hybrid breakdown in cotton (Gossypium): Different traits, different effects. BMC Genet. 2016, 17, 58. [Google Scholar] [CrossRef] [Green Version]
- Holladay, S.K.; Bridges, W.C.; Jones, M.A.; Campbell, B.T. Yield performance and fiber quality of Pima cotton grown in the southeast United States. Crop Sci. 2021, 61, 2423–2434. [Google Scholar] [CrossRef]
- Said, J.I.; Lin, Z.; Zhang, X.; Song, M.; Zhang, J. A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom. 2013, 14, 776. [Google Scholar] [CrossRef] [Green Version]
- Campbell, B.T.; Chee, P.W.; Lubbers, E.; Bowman, D.T.; Meredith, W.R.; Johnson, J.; Fraser, D.E. Genetic Improvement of the Pee Dee Cotton Germplasm Collection following Seventy Years of Plant Breeding. Crop Sci. 2011, 51, 955. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Sun, G.; Geng, X.; Gong, W.; Dai, P.; Jia, Y.; Shi, W.; Pan, Z.; Wang, J.; Wang, L.; et al. The genomic basis of geographic differentiation and fiber improvement in cultivated cotton. Nat. Genet. 2021, 53, 916–924. [Google Scholar] [CrossRef]
- Campbell, B.T.; Weaver, D.B.; Sharpe, R.; Wu, J.; Jones, D.C. Breeding Potential of Elite Pee Dee Germplasm in Upland Cotton Breeding Programs. Crop Sci. 2013, 53, 894–905. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Wu, J. Germplasm for genetic improvement of lint yield in Upland cotton: Genetic analysis of lint yield with yield components. Euphytica 2012, 187, 247–261. [Google Scholar] [CrossRef]
- Nascimento, M.; Teodoro, P.E.; de Castro Sant’Anna, I.; Barroso, L.M.A.; Nascimento, A.C.C.; Azevedo, C.F.; Teodoro, L.P.R.; Farias, F.J.C.; Almeida, H.C.; de Carvalho, L.P. Influential Points in Adaptability and Stability Methods Based on Regression Models in Cotton Genotypes. Agronomy 2021, 11, 2179. [Google Scholar] [CrossRef]
- Paterson, A.H.; Saranga, Y.; Menz, M.; Jiang, C.-X.; Wright, R.J. QTL analysis of genotype x environment inter-actions affecting cotton fiber quality. Theor. Appl. Genet. 2003, 106, 384–396. [Google Scholar] [CrossRef]
- Lin, Z.; Hayes, B.J.; Daetwyler, H.D. Genomic selection in crops, trees and forages: A review. Crop Pasture Sci. 2014, 65, 1177. [Google Scholar] [CrossRef]
- Gapare, W.; Liu, S.; Conaty, W.; Zhu, Q.; Gillespie, V.; Llewellyn, D.; Stiller, W.; Wilson, I. Historical Datasets Support Genomic Selection Models for the Prediction of Cotton Fiber Quality Phenotypes Across Multiple Environments. G3 Genes|Genomes|Genetics 2018, 8, 1721–1732. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.S.; Fang, D.D.; Jenkins, J.N.; Guo, J.; McCarty, J.C.; Jones, D.C. Evaluation of genomic selection methods for predicting fiber quality traits in Upland cotton. Mol. Genet Genom. 2019, 295, 67–79. [Google Scholar] [CrossRef]
- Billings, G.T.; Jones, M.A.; Rustgi, S.; Hulse-Kemp, A.M.; Campbell, B.T. Population structure and genetic diversity of the Pee Dee cotton breeding program. G3 Genes|Genomes|Genetics 2021, 11, jkab145. [Google Scholar] [CrossRef]
- Campbell, B.T.; Myers, G.O. Quantitative Genetics. In Cotton; Crop Science Society of America: Madison, WI, USA, 2015; Volume 57, pp. 187–203. [Google Scholar]
- Campbell, B.T.; Chapman, K.D.; Sturtevant, D.; Kennedy, C.; Horn, P.; Chee, P.W.; Lubbers, E.; Meredith, W.R.; Johnson, J.; Fraser, D.; et al. Genetic Analysis of Cottonseed Protein and Oil in a Diverse Cotton Germplasm. Crop Sci. 2016, 56, 2457. [Google Scholar] [CrossRef]
- Shen, X.; Zhang, T.; Guo, W.; Zhu, X.; Zhang, X. Mapping Fiber and Yield QTLs with Main, Epistatic, and QTL × Environment Interaction Effects in Recombinant Inbred Lines of Upland Cotton. Crop Sci. 2006, 46, 61–66. [Google Scholar] [CrossRef]
- Kelly, C.M.; Hequet, E.F.; Dever, J.K.; Kelly, C.M.; Dever, J.K. Breeding and Genetics Interpretation of AFIS and HVI Fiber Property Measurements in Breeding for Cotton Fiber Quality Improvement. J. Cotton Sci. 2012, 16, 1. [Google Scholar]
- Jenkins, J.N.; McCarty, J.C.; Wubben, M.J.; Hayes, R.; Gutierrez, O.A.; Callahan, F.; Deng, D. SSR markers for marker assisted selection of root-knot nematode (Meloidogyne incognita) resistant plants in cotton (Gossypium hirsutum L). Euphytica 2011, 183, 49–54. [Google Scholar] [CrossRef]
- Bernardo, R. Molecular Markers and Selection for Complex Traits in Plants: Learning from the Last 20 Years. Crop Sci. 2008, 48, 1649–1664. [Google Scholar] [CrossRef] [Green Version]
- Cuevas, J.; Montesinos-López, O.A.; Martini, J.W.R.; Pérez-Rodríguez, P.; Lillemo, M.; Crossa, J.; Crossa, J. Approximate Genome-Based Kernel Models for Large Data Sets Including Main Effects and Interactions. Front. Genet. 2020, 11, 567757. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.A. 009: The Correlation between Relatives on the Supposition of Mendelian Inheritance. Trans. R. Soc. Edinb. 1918, 52, 399–433. [Google Scholar]
- de los Campos, G.; Gianola, D.; Rosa, G.J.M. Reproducing kernel Hilbert spaces regression: A general framework for genetic evaluation. J. Anim. Sci. 2009, 87, 1883–1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weselake, R.J.; Taylor, D.C.; Rahman, M.H.; Shah, S.; Laroche, A.; McVetty, P.B.E.; Harwood, J.L. Increasing the flow of carbon into seed oil. Biotechnol. Adv. 2009, 27, 866–878. [Google Scholar] [CrossRef]
- Liu, Y.H.; Xu, Y.; Zhang, M.; Cui, Y.; Sze, S.-H.; Smith, C.W.; Xu, S.; Zhang, H.-B. Accurate Prediction of a Quantitative Trait Using the Genes Controlling the Trait for Gene-Based Breeding in Cotton. Front. Plant Sci. 2020, 11, 583277. [Google Scholar] [CrossRef]
- Chen, Z.J.; Sreedasyam, A.; Ando, A.; Song, Q.; De Santiago, L.M.; Hulse-Kemp, A.M.; Ding, M.; Ye, W.; Kirkbride, R.C.; Jenkins, J.; et al. Genomic diversifications of five Gossypium allopolyploid species and their impact on cotton improvement. Nat. Genet. 2020, 52, 525. [Google Scholar] [CrossRef] [Green Version]
- Rogers, A.R.; Holland, J.B. Environment-specific genomic prediction ability in maize using environmental covariates depends on environmental similarity to training data. G3 Genes|Genomes|Genetics 2021, 12, jkab440. [Google Scholar] [CrossRef]
- Hulse-Kemp, A.M.; Lemm, J.; Plieske, J.; Ashrafi, H.; Buyyarapu, R.; Fang, D.D.; Frelichowski, J.; Giband, M.; Hague, S.; Hinze, L.L.; et al. Development of a 63K SNP Array for Cotton and High-Density Mapping of Intraspecific and Interspecific Populations of Gossypium spp. G3 Genes|Genomes|Genetics 2015, 5, 1187–1209. [Google Scholar] [CrossRef] [Green Version]
- Campbell, B.T.; Williams, V.E.; Park, W. Using molecular markers and field performance data to characterize the Pee Dee cotton germplasm resources. Euphytica 2009, 169, 285–301. [Google Scholar] [CrossRef]
- Holland, J.B.; Nyquist, W.E.; Cervantes-Martínez, C.T.; Janick, J. Estimating and interpreting heritability for plant breeding: An update. In Plant Breeding Reviews; Wiley: Hoboken, NJ, USA, 2003; Volume 22. [Google Scholar]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Chang, C. PLINK 1.9. Available online: www.cog-genomics.org/plink/1.9 (accessed on 1 February 2022).
- Bian, Y.; Holland, J.B. Enhancing genomic prediction with genome-wide association studies in multiparental maize popula-tions. Heredity 2017, 118, 585. [Google Scholar] [CrossRef] [PubMed]
- Pérez, P.; de los Campos, G. Genome-Wide Regression and Prediction with the BGLR Statistical Package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Overall | (Mean from up to 14 Tests in Separate Environments) Environment | Difference in H2 (Overall-Environment) | ||||
|---|---|---|---|---|---|---|
| Trait Type | Trait | Broad-Sense Heritability (H2) | Standard Error | Broad-Sense Heritability (H2) | Standard Error | |
| Advanced Fiber Information System (AFIS) | Fineness | 0.84 | 0.03 | 0.37 | 0.11 | 0.47 |
| Length Top 25% Mean by Wt | 0.96 | 0.01 | 0.79 | 0.05 | 0.17 | |
| Length Mean by Number | 0.92 | 0.01 | 0.47 | 0.11 | 0.45 | |
| Immature Fiber Content | 0.22 | 0.13 | 0.28 | 0.11 | −0.06 | |
| Maturity Ratio | 0.75 | 0.04 | 0.32 | 0.12 | 0.43 | |
| Short Fiber Content by Wt | 0.86 | 0.03 | 0.41 | 0.09 | 0.45 | |
| High Volume Instrument (HVI) | Elongation | 0.93 | 0.01 | 0.40 | 0.11 | 0.53 |
| Upper Half Mean Length | 0.96 | 0.01 | 0.64 | 0.08 | 0.32 | |
| Strength | 0.96 | 0.01 | 0.69 | 0.07 | 0.27 | |
| Micronaire | 0.88 | 0.02 | 0.68 | 0.07 | 0.20 | |
| Short Fiber Content | 0.80 | 0.03 | 0.47 | 0.10 | 0.33 | |
| Uniformity Index | 0.88 | 0.02 | 0.40 | 0.11 | 0.48 | |
| Seed Composition | Seed Oil % | 0.88 | 0.02 | 0.50 | 0.09 | 0.38 |
| Seed Protein % | 0.27 | 0.14 | 0.12 | 0.14 | 0.15 | |
| Yield | Bolls/m−2, from Seed Cotton | 0.65 | 0.06 | 0.35 | 0.15 | 0.30 |
| Boll Wt, from Seed Cotton | 0.71 | 0.05 | 0.27 | 0.13 | 0.44 | |
| Gin Turnout | 0.75 | 0.01 | 0.69 | 0.07 | 0.27 | |
| Lint Yield | 0.75 | 0.04 | 0.52 | 0.12 | 0.23 | |
| Seed Cotton Yield | 0.55 | 0.08 | 0.48 | 0.12 | 0.07 | |
| Seed Index | 0.93 | 0.01 | 0.60 | 0.08 | 0.33 | |
| Average | 0.78 | 0.04 | 0.47 | 0.10 | 0.31 | |
| Trait Type | Trait | Overall Means | Blackville | Florence | Hartsville | Rocky Mount | Stoneville | Tifton | Average per Trait | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2004 | 2005 | 2006 | 2004 | 2005 | 2006 | 2004 | 2005 | 2005 | 2006 | 2005 | 2006 | 2005 | 2006 | ||||
| Advanced Fiber Information System (AFIS) | Fineness | 657 | 292 | 8 | 30 | 34 | 238 | 96 | - | 2 | 240 | 1 | 35 | 18 | - | 36 | 112 |
| Length Top 25% Mean by Wt | 212 | 762 | 41 | - | 407 | 57 | 59 | 280 | 197 | 419 | 2 | 64 | 123 | 9 | 242 | 192 | |
| Length Mean by Number | - | 74 | - | 5 | 24 | 9 | - | - | 53 | 174 | 1 | - | - | 9 | 141 | 33 | |
| Immature Fiber Content | - | 88 | - | 39 | 17 | 187 | 199 | 164 | 10 | 52 | 58 | 78 | 9 | 184 | 112 | 80 | |
| Maturity Ratio | 3 | 4 | 58 | 18 | - | 300 | 32 | 41 | - | 110 | 9 | 65 | 13 | - | 150 | 54 | |
| Short Fiber Content by Wt | 1 | 1 | 5 | 72 | 21 | 19 | 112 | 168 | 44 | 125 | - | - | 9 | 9 | 37 | 42 | |
| High Volume Instrument (HVI) | Elongation | - | 18 | - | 201 | 434 | 8 | - | - | 3 | 19 | - | - | 15 | - | 55 | 50 |
| Upper Half Mean Length | 471 | 163 | 10 | 76 | 585 | 54 | - | 451 | 193 | 12 | 278 | 203 | 108 | 18 | 281 | 194 | |
| Strength | 284 | 41 | 15 | 30 | 112 | 15 | 570 | 35 | 41 | 47 | - | 364 | 535 | 31 | 80 | 147 | |
| Micronaire | 769 | 280 | 10 | 152 | 18 | - | 66 | - | 353 | 743 | 591 | 48 | 192 | 681 | 228 | 275 | |
| Short Fiber Content | 58 | 40 | 10 | 4 | 123 | 307 | 269 | 231 | - | - | 3 | 167 | 29 | 426 | - | 111 | |
| Uniformity Index | 17 | 30 | 3 | - | 26 | 726 | - | - | 3 | - | - | 1 | 26 | 112 | - | 63 | |
| Seed Composition | Seed Oil % | 180 | 89 | - | 155 | 161 | 33 | 151 | - | 89 | 129 | 156 | - | 230 | 96 | - | 98 |
| Seed Protein % | 104 | 21 | - | 16 | - | 299 | 119 | - | 13 | - | 454 | - | 3 | 24 | - | 70 | |
| Yield | Bolls/m2, from Seed Cotton | 91 | 38 | - | 2 | 150 | - | - | 9 | - | 6 | - | 6 | 381 | 47 | 440 | 78 |
| Boll Wt, from Seed Cotton | 9 | 105 | - | 10 | 29 | - | 3 | 33 | 7 | 22 | 34 | 30 | - | 4 | 460 | 50 | |
| Gin Turnout | 301 | 276 | 2 | - | 133 | 3 | 53 | 628 | 27 | 349 | 897 | 100 | 9 | - | 603 | 225 | |
| Lint Yield | 1007 | 9 | 38 | 201 | 192 | - | - | 1372 | - | - | - | 29 | 628 | 28 | 334 | 256 | |
| Seed Cotton Yield | 116 | 75 | 22 | 161 | 92 | - | - | 785 | - | 3 | - | 5 | 469 | 38 | 67 | 122 | |
| Seed Index | 605 | 831 | 11 | 15 | 1 | - | 278 | - | 168 | - | 732 | 237 | 8 | 1 | 815 | 247 | |
| Average per Environment | 244 | 162 | 12 | 59 | 128 | 113 | 100 | 210 | 60 | 123 | 161 | 72 | 140 | 86 | 204 | ||
| Prediction Ability Compared to within Year and Location (% of within Prediction Ability) | |||||
|---|---|---|---|---|---|
| Within Location, Different Year | Within Year, Different Location | Different Year and Location | Interpretation | ||
| Advanced Fiber Information System (AFIS) | Fineness | 95% | 84% | 94% | Within location is easier |
| Length Mean by Number | 100% | 88% | 96% | Within location is easier | |
| Length Top 25% Mean by Wt | 100% | 90% | 98% | Within location is easier | |
| Immature Fiber Content | 95% | 63% | 92% | Within location is easier | |
| Maturity Ratio | 93% | 67% | 91% | Within location is easier | |
| Short Fiber Content by Wt | 84% | 72% | 82% | Within location is easier | |
| High Volume Instrument (HVI) | Elongation | 95% | 92% | 94% | Within location is easier |
| Upper Half Mean Length | 100% | 90% | 98% | Within location is easier | |
| Strength | 100% | 95% | 97% | Within location is easier | |
| Micronaire | 67% | 65% | 62% | Within location is easier | |
| Short Fiber Content | 36% | 71% | 47% | Within year is easier | |
| Uniformity Index | 96% | 88% | 93% | Within location is easier | |
| Seed Composition | Seed Oil % | 100% | 76% | 100% | Within location is easier |
| Seed Protein % | 56% | 30% | 5% | Within location is easier | |
| Yield | Bolls/m2, from Seed Cotton | 1% | 48% | 29% | Within year is easier |
| Boll Wt, from Seed Cotton | 18% | 100% | 81% | Within year is easier | |
| Gin Turnout | 98% | 96% | 95% | Within location is easier | |
| Lint Yield | 49% | 68% | 59% | Within year is easier | |
| Seed Cotton Yield | 13% | 44% | 34% | Within year is easier | |
| Seed Index | 96% | 99% | 99% | Within year is easier | |
| Location | Year | Comments |
|---|---|---|
| Blackville, SC | 2004 | ✔ |
| 2005 | No seed oil % or seed protein % | |
| 2006 | ✔ | |
| Florence, SC | 2004 | ✔ |
| 2005 | ✔ | |
| 2006 | No bolls m−2, boll weight, lint, or seed cotton yield | |
| Hartsville, SC | 2004 | No seed oil % or seed protein % |
| 2005 | No bolls m−2, boll weight, lint, or seed cotton yield | |
| Rocky Mount, NC | 2005 | No high-volume instrument short fiber content |
| 2006 | ✔ | |
| Stoneville, MS | 2005 | No seed oil % or seed protein % |
| 2006 | ✔ | |
| Tifton, GA | 2005 | ✔ |
| 2006 | ✔ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Billings, G.T.; Jones, M.A.; Rustgi, S.; Bridges, W.C., Jr.; Holland, J.B.; Hulse-Kemp, A.M.; Campbell, B.T. Outlook for Implementation of Genomics-Based Selection in Public Cotton Breeding Programs. Plants 2022, 11, 1446. https://doi.org/10.3390/plants11111446
Billings GT, Jones MA, Rustgi S, Bridges WC Jr., Holland JB, Hulse-Kemp AM, Campbell BT. Outlook for Implementation of Genomics-Based Selection in Public Cotton Breeding Programs. Plants. 2022; 11(11):1446. https://doi.org/10.3390/plants11111446
Chicago/Turabian StyleBillings, Grant T., Michael A. Jones, Sachin Rustgi, William C. Bridges, Jr., James B. Holland, Amanda M. Hulse-Kemp, and B. Todd Campbell. 2022. "Outlook for Implementation of Genomics-Based Selection in Public Cotton Breeding Programs" Plants 11, no. 11: 1446. https://doi.org/10.3390/plants11111446