
An NmrA-Like Protein, Lws1, Is Important for Pathogenesis in the Woody Plant Pathogen Lasiodiplodia theobromae

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
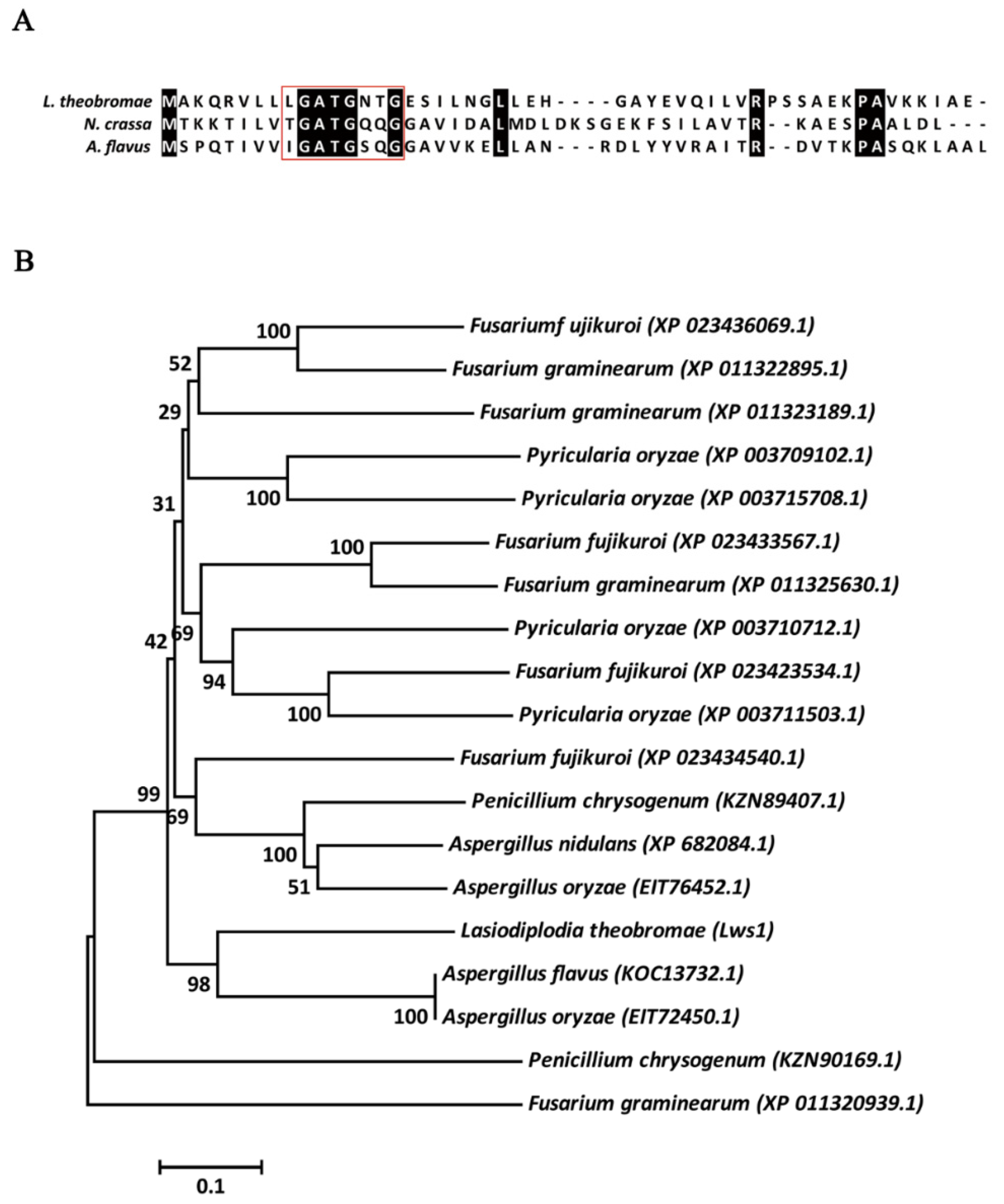
2.1. Structural Features of Lws1 Protein
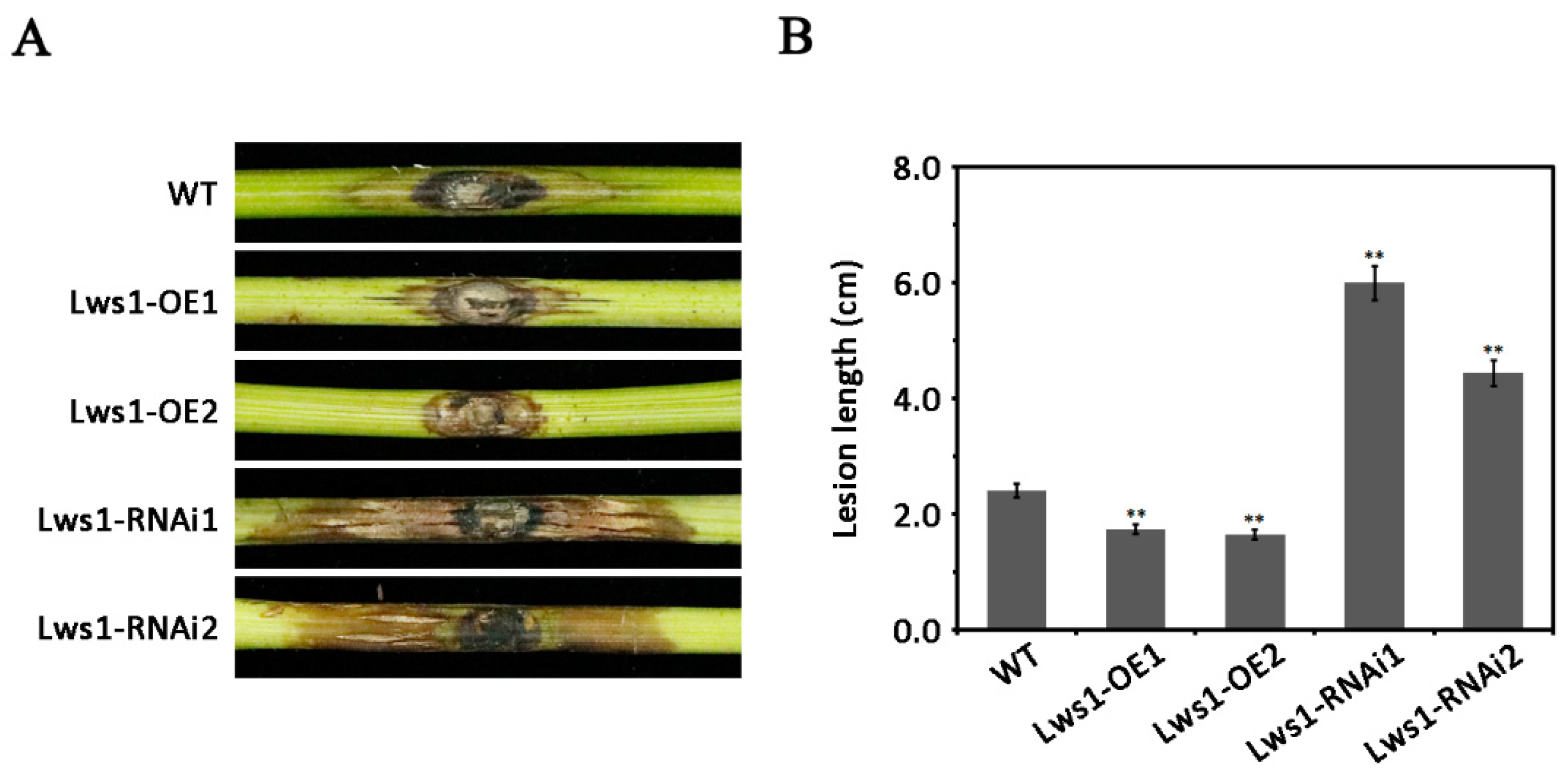
2.2. Regulation of the Pathogenicity of L. theobromae by Lws1
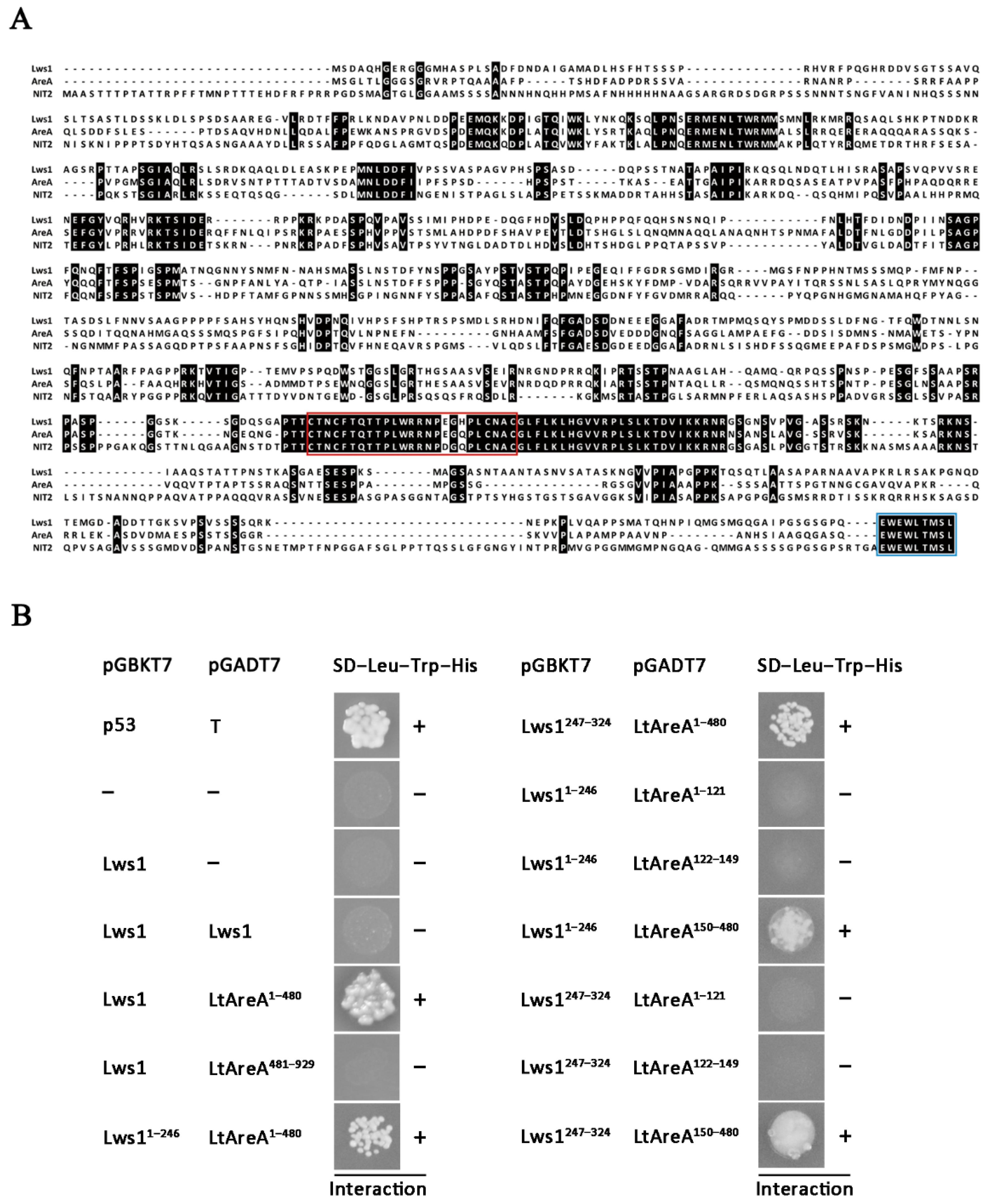
2.3. Interaction between Lws1 and LtAreA
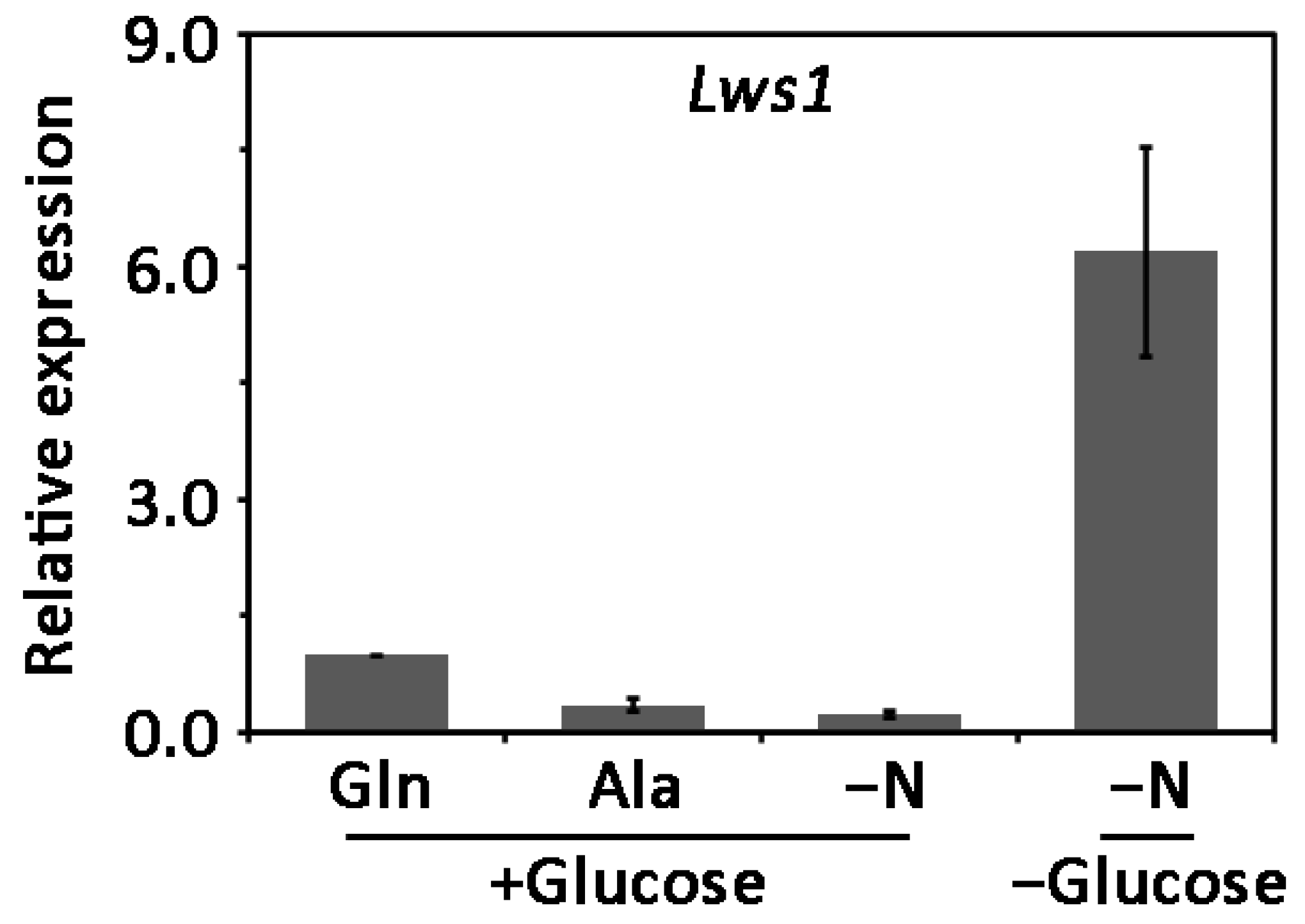
2.4. Expression of Lws1 under Different Nutrition Conditions in L. theobromae
3. Discussion
4. Materials and Methods
4.1. Fungal and Bacterial Strains, Growth Conditions, and Plant Material
4.2. RNA Extraction and Real-Time Quantitative PCR (qRT-PCR)
4.3. Pathogenicity Tests of Overexpressed and Silenced Transformants of the Lws1 Gene
4.4. Yeast Two-Hybrid Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marzluf, G.A. Genetic regulation of nitrogen metabolism in the fungi. Microbiol. Mol. Biol. Rev. 1997, 61, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.B.; Fraser, J.A.; Wong, K.H.; Davis, M.A.; Hynes, M.J. Nuclear accumulation of the GATA factor AreA in response to complete nitrogen starvation by regulation of nuclear export. Eukaryot Cell. 2005, 4, 1646–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.H.; Hynes, M.J.; Todd, R.B.; Davis, M.A. Transcriptional control of nmrA by the bZIP transcription factor MeaB reveals a new level of nitrogen regulation in Aspergillus nidulans. Mol. Microbiol. 2007, 66, 534–551. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Hume, S.L.; Johnson, C.; Thompson, P.; Huang, J.; Gray, J.; Lamb, H.K.; Hawkins, A.R. The transcription repressor NmrA is subject to proteolysis by three Aspergillus nidulans proteases. Protein sci. 2010, 19, 1405–1419. [Google Scholar] [CrossRef] [Green Version]
- Macios, M.; Caddick, M.X.; Weglenski, P.; Scazzocchio, C.; Dzikowska, A. The GATA factors AREA and AREB together with the co-repressor NMRA, negatively regulate arginine catabolism in Aspergillus nidulans in response to nitrogen and carbon source. Fungal Genet. Biol. 2012, 49, 189–198. [Google Scholar] [CrossRef]
- Arst Jr, H.N.; Cove, D.J. Nitrogen metabolite repression in Aspergillus nidulans. Mol. gen. Genet. 1973, 126, 111–141. [Google Scholar] [CrossRef] [PubMed]
- Kudla, B.; Caddick, M.X.; Langdon, T.; Martinez-Rossi, N.M.; Bennett, C.F.; Sibley, S.; Davies, R.W.; Arst, H.N. The regulatory gene areA mediating nitrogen metabolite repression in Aspergillus nidulans. Mutations affecting specificity of gene activation alter a loop residue of a putative zinc finger. EMBO J. 1990, 9, 1355–1364. [Google Scholar] [CrossRef]
- Ravagnani, A.; Gorfinkiel, L.; Langdon, T.; Diallinas, G.; Adjadj, E.; Demais, S.; Gorton, D.; Arst, H.N.; Scazzocchio, C. Subtle hydrophobic interactions between the seventh residue of the zinc finger loop and the first base of an HGATAR sequence determine promoter-specific recognition by the Aspergillus nidulans GATA factor AreA. EMBO J. 1997, 16, 3974–3986. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.H.; Marzluf, G.A. nit-2, the major positive-acting nitrogen regulatory gene of Neurospora crassa, encodes a sequence-specific DNA-binding protein. Proc. Natl. Acad. Sci. USA 1990, 87, 5331–5335. [Google Scholar] [CrossRef] [Green Version]
- Haas, H.; Bauer, B.; Redl, B.; Stöffler, G.; Marzluf, G.A. Molecular cloning and analysis of nre, the major nitrogen regulatory gene of Penicillium chrysogenum. Curr. Genet. 1995, 27, 150–158. [Google Scholar] [CrossRef]
- Minehart, P.L.; Magasanik, B. Sequence and expression of GLN3, a positive nitrogen regulatory gene of Saccharomyces cerevisiae encoding a protein with a putative zinc finger DNA-binding domain. Mol. Cell Biol. 1991, 11, 6216–6228. [Google Scholar] [CrossRef] [PubMed]
- Stanbrough, M.; Rowen, D.W.; Magasanik, B. Role of the GATA factors Gln3p and Nil1p of Saccharomyces cerevisiae in the expression of nitrogen-regulated genes. Proc. Natl. Acad. Sci. USA 1995, 92, 9450–9454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffman, J.A.; Rai, R.; Loprete, D.M.; Cunningham, T.; Svetlov, V.; Cooper, T.G. Cross regulation of four GATA factors that control nitrogen catabolic gene expression in Saccharomyces cerevisiae. J. Bacteriol. 1997, 179, 3416–3429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froeliger, E.H.; Carpenter, B.E. NUT1, a major nitrogen regulatory gene in Magnaporthe grisea, is dispensable for pathogenicity. Mol. Gen. Genet. 1996, 251, 647–656. [Google Scholar] [CrossRef]
- Christensen, T.; Hynes, M.J.; Davis, M.A. Role of the regulatory gene areA of Aspergillus oryzae in nitrogen metabolism. Appl Environ. Microbiol. 1998, 64, 3232–3237. [Google Scholar] [CrossRef] [Green Version]
- Tudzynski, B.; Homann, V.; Feng, B.; Marzluf, G.A. Isolation, characterization and disruption of the areA nitrogen regulatory gene of Gibberella fujikuroi. Mol. Gen. Genet. 1999, 261, 106–114. [Google Scholar] [CrossRef]
- Han, X.; Qiu, M.; Wang, B.; Yin, W.B.; Nie, X.; Qin, Q.; Ren, S.; Yang, K.; Zhang, F.; Zhuang, Z.; et al. Functional analysis of the nitrogen metabolite repression regulator gene nmrA in Aspergillus flavus. Front. Microbiol. 2016, 7, 1794. [Google Scholar] [CrossRef] [Green Version]
- Giese, H.; Sondergaard, T.E.; Sørensen, J.L. The AreA transcription factor in Fusarium graminearum regulates the use of some nonpreferred nitrogen sources and secondary metabolite production. Fungal Biol. 2013, 117, 814–821. [Google Scholar] [CrossRef]
- Platt, A.; Langdon, T.; Arst, H.N., Jr.; Kirk, D.; Tollervey, D.; Sanchez, J.M.; Caddick, M.X. Nitrogen metabolite signalling involves the C-terminus and the GATA domain of the Aspergillus transcription factor AREA and the 3′ untranslated region of its mRNA. EMBO J. 1996, 15, 2791–2801. [Google Scholar] [CrossRef]
- Morozov, I.Y.; Martinez, M.G.; Jones, M.G.; Caddick, M.X. A defined sequence within the 3’ UTR of the areA transcript is sufficient to mediate nitrogen metabolite signalling via accelerated deadenylation. Mol. Microbiol. 2000, 37, 1248–1257. [Google Scholar] [CrossRef]
- Morozov, I.Y.; Galbis-Martinez, M.; Jones, M.G.; Caddick, M.X. Characterization of nitrogen metabolite signalling in Aspergillus via the regulated degradation of areA mRNA. Mol. Microbiol. 2001, 42, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Langdon, T.; Sheerins, A.; Ravagnani, A.; Gielkens, M.; Caddick, M.X.; Arst, H.N., Jr. Mutational analysis reveals dispensability of the N-terminal region of the Aspergillus transcription factor mediating nitrogen metabolite repression. Mol. Microbiol. 1995, 17, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Small, A.J.; Hynes, M.J.; Davis, M.A. The TamA protein fused to a DNA-binding domain can recruit AreA, the major nitrogen regulatory protein, to activate gene expression in Aspergillus nidulans. Genetics 1999, 153, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Small, A.J.; Todd, R.B.; Zanker, M.C.; Delimitrou, S.; Hynes, M.J.; Davis, M.A. Functional analysis of TamA, a coactivator of nitrogen-regulated gene expression in Aspergillus nidulans. Mol. Genet. Genom. 2001, 265, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Andrianopoulos, A.; Kourambas, S.; Sharp, J.A.; Davis, M.A.; Hynes, M.J. Characterization of the Aspergillus nidulans nmrA gene involved in nitrogen metabolite repression. J. Bacterial. 1998, 180, 1973–1977. [Google Scholar] [CrossRef] [Green Version]
- Lamb, H.K.; Ren, J.; Park, A.; Johnson, C.; Leslie, K.; Cocklin, S.; Thompson, P.; Mee, C.; Cooper, A.; Stammers, D.K.; et al. Modulation of the ligand binding properties of the transcription repressor NmrA by GATA-containing DNA and site-directed mutagenesis. Protein Sci. 2004, 13, 3127–3138. [Google Scholar] [CrossRef] [Green Version]
- Kotaka, M.; Johnson, C.; Lamb, H.K.; Hawkins, A.R.; Ren, J.; Stammers, D.K. Structural analysis of the recognition of the negative regulator NmrA and DNA by the zinc finger from the GATA-type transcription factor AreA. J. Mol. Biol. 2008, 381, 373–382. [Google Scholar] [CrossRef]
- Stammers, D.K.; Ren, J.; Leslie, K.; Nichols, C.E.; Lamb, H.K.; Cocklin, S.; Dodds, A.; Hawkins, A.R. The structure of the negative transcriptional regulator NmrA reveals a structural superfamily which includes the short-chain dehydrogenase/reductases. EMBO J. 2001, 20, 6619–6626. [Google Scholar] [CrossRef]
- Magomedova, Z.; Grecu, A.; Sensen, C.W.; Schwab, H.; Heidinger, P. Characterization of two novel alcohol short-chain dehydrogenases/reductases from Ralstonia eutropha H16 capable of stereoselective conversion of bulky substrates. J. Biotechnol. 2016, 221, 78–90. [Google Scholar] [CrossRef]
- Gräff, M.; Buchholz, P.; Stockinger, P.; Bommarius, B.; Bommarius, A.S.; Pleiss, J. The Short-chain Dehydrogenase/Reductase Engineering Database (SDRED): A classification and analysis system for a highly diverse enzyme family. Proteins. 2019, 87, 443–451. [Google Scholar] [CrossRef]
- Kristan, K.; Deluca, D.; Adamski, J.; Stojan, J.; Rizner, T.L. Dimerization and enzymatic activity of fungal 17beta-hydroxysteroid dehydrogenase from the short-chain dehydrogenase/reductase superfamily. BMC Biochem. 2005, 6, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Úrbez-Torres, J.R.; Leavitt, G.M.; Guerrero, J.C.; Guevara, J.; Gubler, W.D. Identification and pathogenicity of Lasiodiplodia theobromae and Diplodia seriata, the causal agents of bot canker disease of grapevines in Mexico. Plant. Dis. 2008, 92, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolinelli-Alfonso, M.; Villalobos-Escobedo, J.M.; Rolshausen, P.; Herrera-Estrella, A.; Galindo-Sánchez, C.; López-Hernández, J.F.; Hernandez-Martinez, R. Global transcriptional analysis suggests Lasiodiplodia theobromae pathogenicity factors involved in modulation of grapevine defensive response. BMC Genom. 2016, 17, 615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.S.; Asman, A.; Shao, J.; Balidion, J.F.; Strem, M.D.; Puig, A.S.; Meinhardt, L.W.; Bailey, B.A. Genome and transcriptome analysis of the latent pathogen Lasiodiplodia theobromae, an emerging threat to the cacao industry. Genome 2020, 63, 37–52. [Google Scholar] [CrossRef]
- Peng, J.; Wu, L.; Zhang, W.; Zhang, Q.; Xing, Q.; Wang, X.; Li, X.; Yan, J. Systemic identification and functional characterization of common in fungal extracellular membrane proteins in Lasiodiplodia theobromae. Front. Plant. Sci. 2021, 12, 804696. [Google Scholar] [CrossRef]
- Chethana, K.W.; Li, X.; Zhang, W.; Hyde, K.D.; Yan, J. Trail of decryption of molecular research on Botryosphaeriaceae in woody plants. Phytopathol. Mediterr. 2016, 55, 147–171. [Google Scholar] [CrossRef]
- Yan, J.Y.; Zhao, W.S.; Chen, Z.; Xing, Q.K.; Zhang, W.; Chethana, K.; Xue, M.F.; Xu, J.P.; Phillips, A.; Wang, Y.; et al. Comparative genome and transcriptome analyses reveal adaptations to opportunistic infections in woody plant degrading pathogens of Botryosphaeriaceae. DNA Res. 2018, 25, 87–102. [Google Scholar] [CrossRef] [Green Version]
- Songy, A.; Fernandez, O.; Clément, C.; Larignon, P.; Fontaine, F. Grapevine trunk diseases under thermal and water stresses. Planta 2019, 249, 1655–1679. [Google Scholar] [CrossRef]
- Félix, C.; Meneses, R.; Gonçalves, M.; Tilleman, L.; Duarte, A.S.; Jorrín-Novo, J.V.; Van de Peer, Y.; Deforce, D.; Van Nieuwerburgh, F.; Esteves, A.C.; et al. A multi-omics analysis of the grapevine pathogen Lasiodiplodia theobromae reveals that temperature affects the expression of virulence- and pathogenicity-related genes. Sci. Rep. 2019, 9, 13144. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, M.; Nunes, R.B.; Tilleman, L.; Van de Peer, Y.; Deforce, D.; Van Nieuwerburgh, F.; Esteves, A.C.; Alves, A. Dual RNA sequencing of Vitis vinifera during Lasiodiplodia theobromae infection unveils host-pathogen interactions. Int. J. Mol. Sci. 2019, 20, 6083. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Yan, J.; Li, X.; Xing, Q.; Chethana, K.; Zhao, W. Transcriptional response of grapevine to infection with the fungal pathogen Lasiodiplodia theobromae. Sci. Rep. 2019, 9, 5387. [Google Scholar] [CrossRef] [PubMed]
- Harishchandra, D.L.; Zhang, W.; Li, X.; Chethana, K.; Hyde, K.D.; Brooks, S.; Yan, J.; Peng, J. A LysM domain-vontaining protein LtLysM1 is important for vegetative growth and pathogenesis in woody plant pathogen Lasiodiplodia theobromae. Plant. Pathol. J. 2020, 36, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Chethana, K.W.; Peng, J.; Li, X.; Xing, Q.; Liu, M.; Zhang, W.; Hyde, K.D.; Zhao, W.; Yan, J. LtEPG1, a secretory endopolygalacturonase protein, regulates the virulence of Lasiodiplodia theobromae in Vitis vinifera and is recognized as a microbe-associated molecular patterns. Phytopathology 2020, 110, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Aluthmuhandiram, J.V.S.; Chethana, K.W.T.; Zhang, W.; Peng, J.; Zhao, E.; Li, X.H.; N, S.; Yan, J.Y. Impact of temperature variation on the phytotoxic secondary metabolite production by Lasiodiplodia theobromae. J. Phytopathol. 2021, 169, 716–723. [Google Scholar] [CrossRef]
- Pham, J.; Stam, R.; Heredia, V.M.; Csukai, M.; Huitema, E. An NMRA-Like protein regulates gene expression in Phytophthora capsici to drive the infection cycle on tomato. Mol. Plant. Microbe Interact. 2018, 31, 665–677. [Google Scholar] [CrossRef]
- Xiao, X.; Fu, Y.H.; Marzluf, G.A. The negative-acting NMR regulatory protein of Neurospora crassa binds to and inhibits the DNA-binding activity of the positive-acting nitrogen regulatory protein NIT2. Biochemistry 1995, 34, 8861–8868. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Becker, D.M.; Lundblad, V. Introduction of DNA into yeast cells. Curr. Protoc. Mol. Biol. 1994, 27, 13–17. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, J.; Aluthmuhandiram, J.V.S.; Chethana, K.W.T.; Zhang, Q.; Xing, Q.; Wang, H.; Liu, M.; Zhang, W.; Li, X.; Yan, J. An NmrA-Like Protein, Lws1, Is Important for Pathogenesis in the Woody Plant Pathogen Lasiodiplodia theobromae. Plants 2022, 11, 2197. https://doi.org/10.3390/plants11172197
Peng J, Aluthmuhandiram JVS, Chethana KWT, Zhang Q, Xing Q, Wang H, Liu M, Zhang W, Li X, Yan J. An NmrA-Like Protein, Lws1, Is Important for Pathogenesis in the Woody Plant Pathogen Lasiodiplodia theobromae. Plants. 2022; 11(17):2197. https://doi.org/10.3390/plants11172197
Chicago/Turabian StylePeng, Junbo, Janith V. S. Aluthmuhandiram, K. W. Thilini Chethana, Qi Zhang, Qikai Xing, Hui Wang, Mei Liu, Wei Zhang, Xinghong Li, and Jiye Yan. 2022. "An NmrA-Like Protein, Lws1, Is Important for Pathogenesis in the Woody Plant Pathogen Lasiodiplodia theobromae" Plants 11, no. 17: 2197. https://doi.org/10.3390/plants11172197
APA StylePeng, J., Aluthmuhandiram, J. V. S., Chethana, K. W. T., Zhang, Q., Xing, Q., Wang, H., Liu, M., Zhang, W., Li, X., & Yan, J. (2022). An NmrA-Like Protein, Lws1, Is Important for Pathogenesis in the Woody Plant Pathogen Lasiodiplodia theobromae. Plants, 11(17), 2197. https://doi.org/10.3390/plants11172197