


Genomic Characteristics of Elite Maize Inbred Line 18-599 and Its Transcriptional Response to Drought and Low-Temperature Stresses

Abstract
:1. Introduction
2. Results
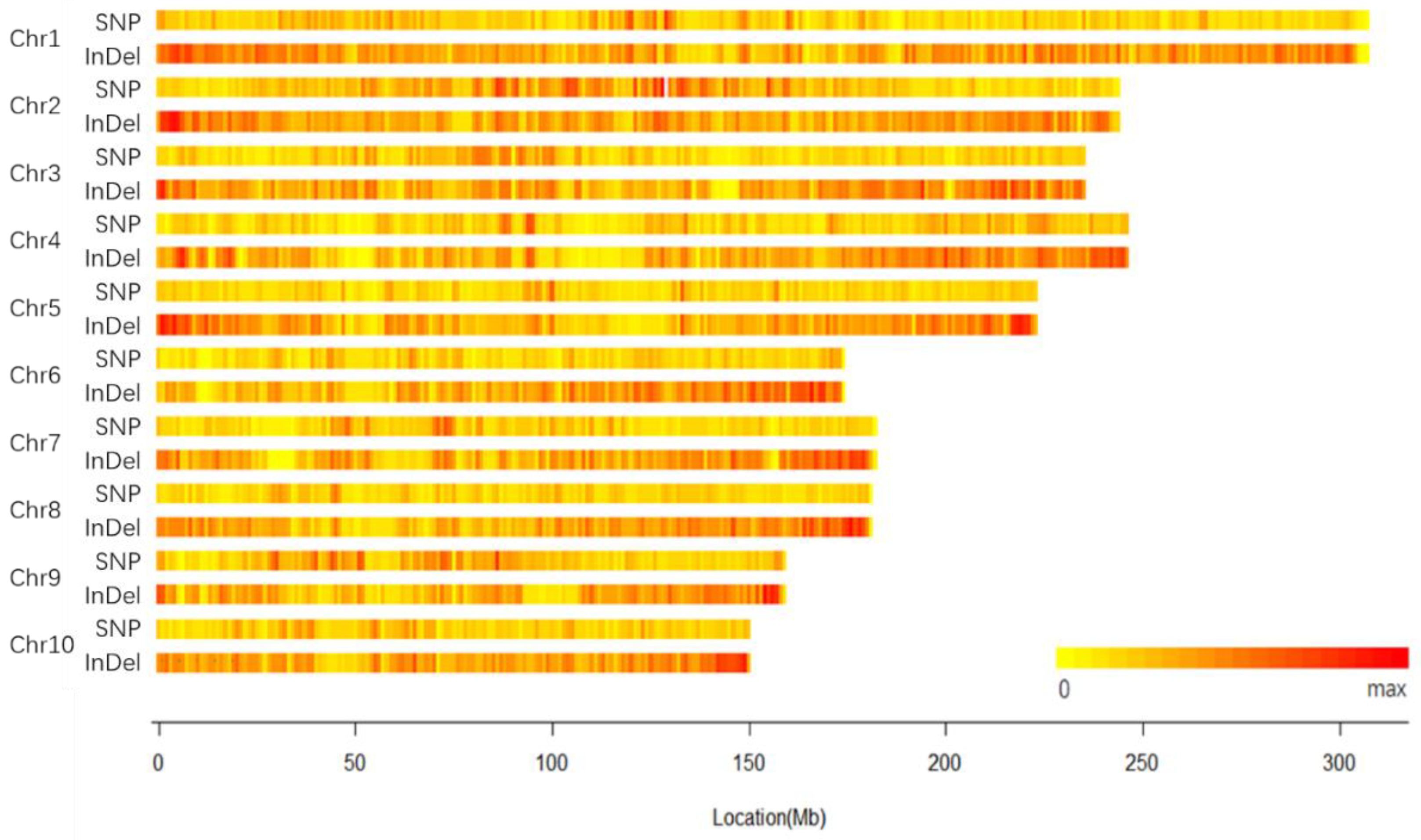
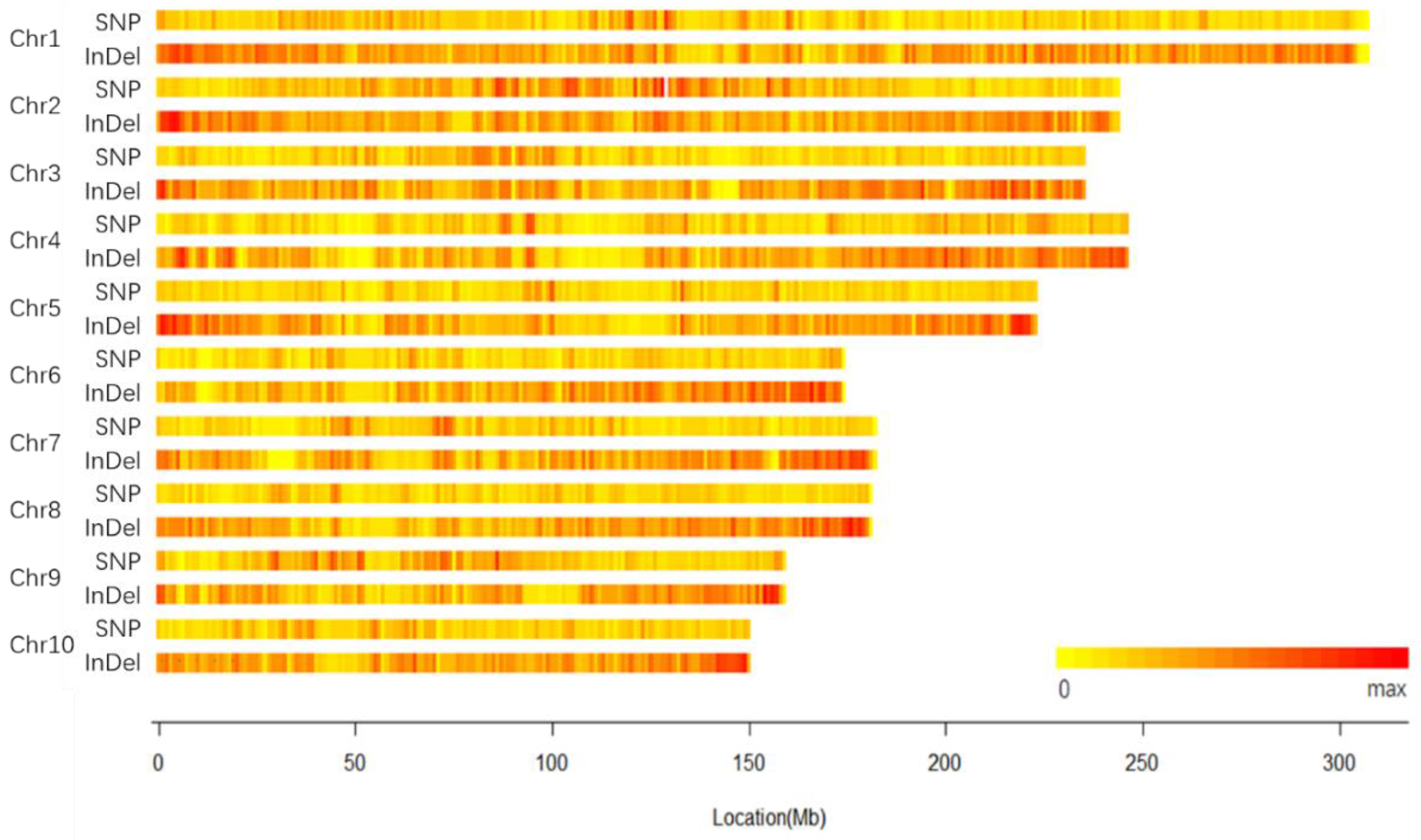
2.1. Genomic Variation of 18-599
2.2. Specific Variation Loci of 18-599
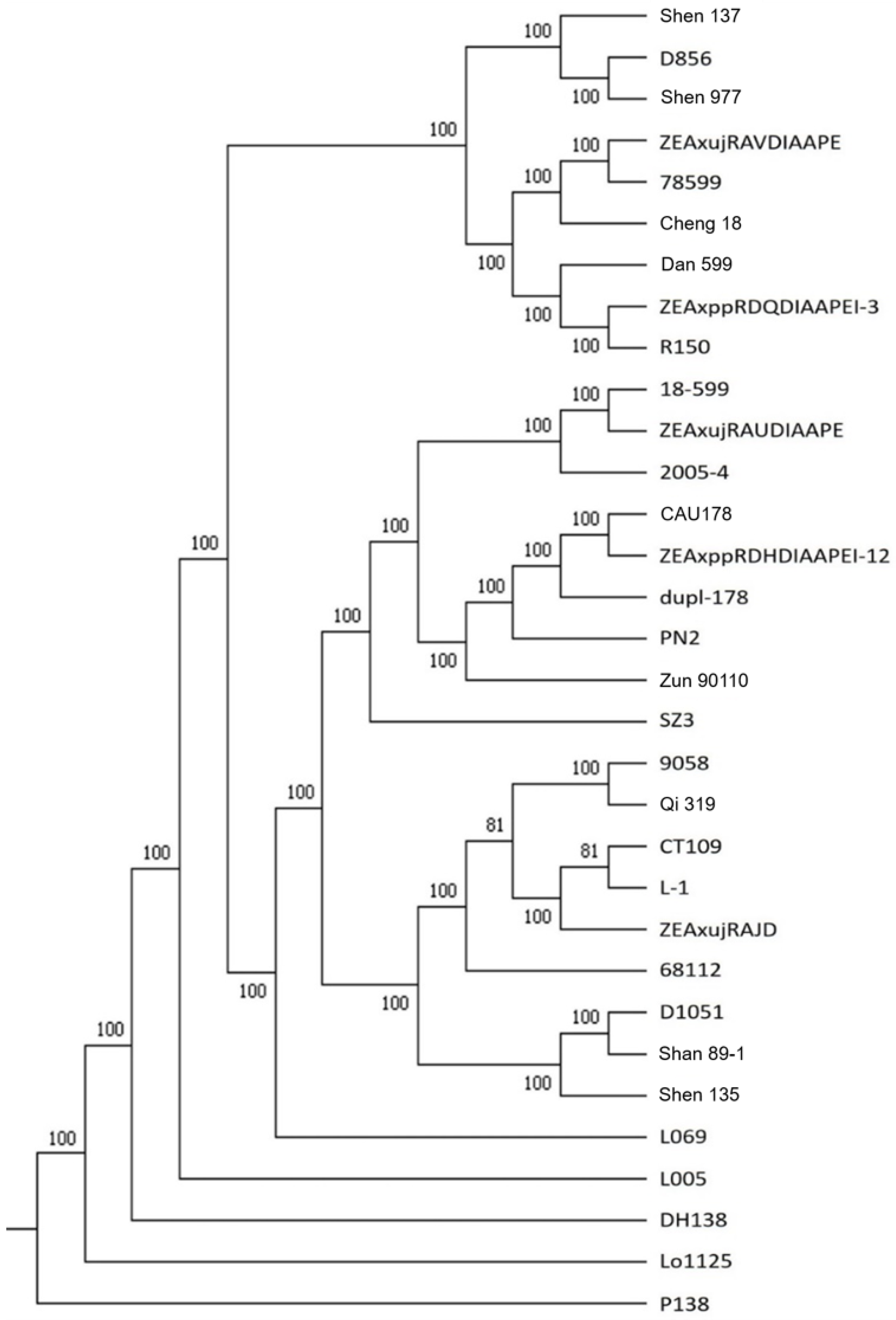
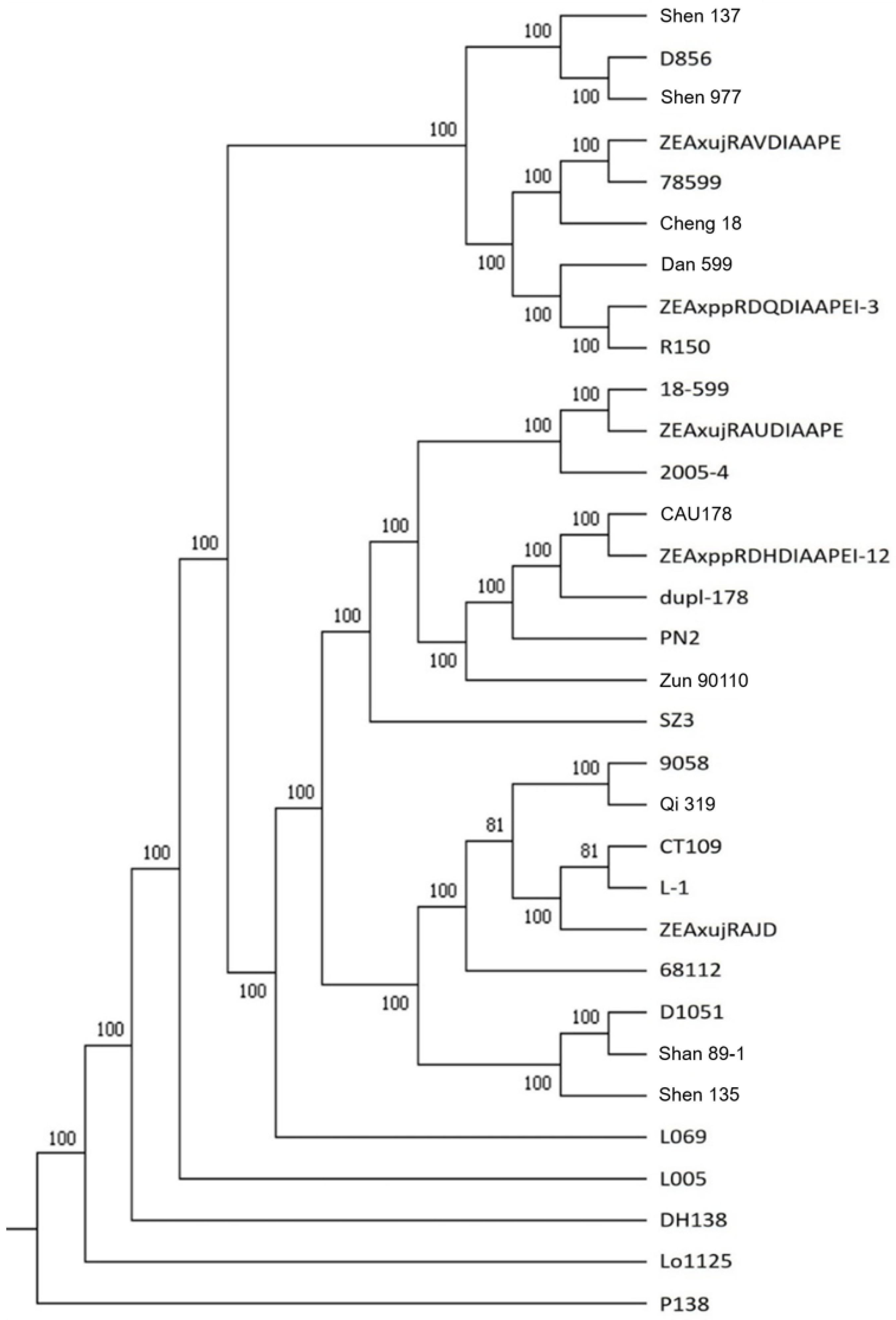
2.3. Identical Variation Loci and Phylogenetic Tree
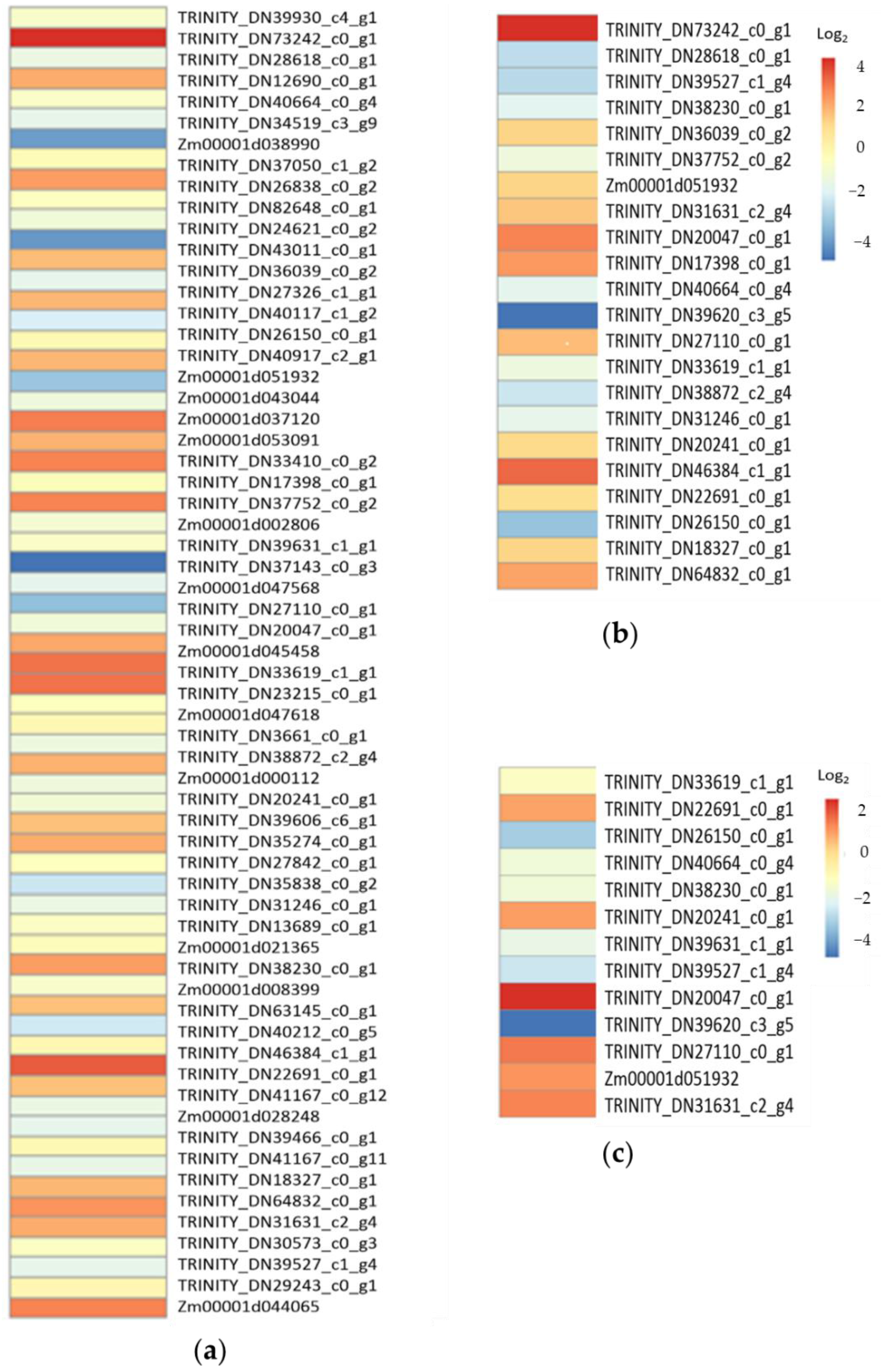
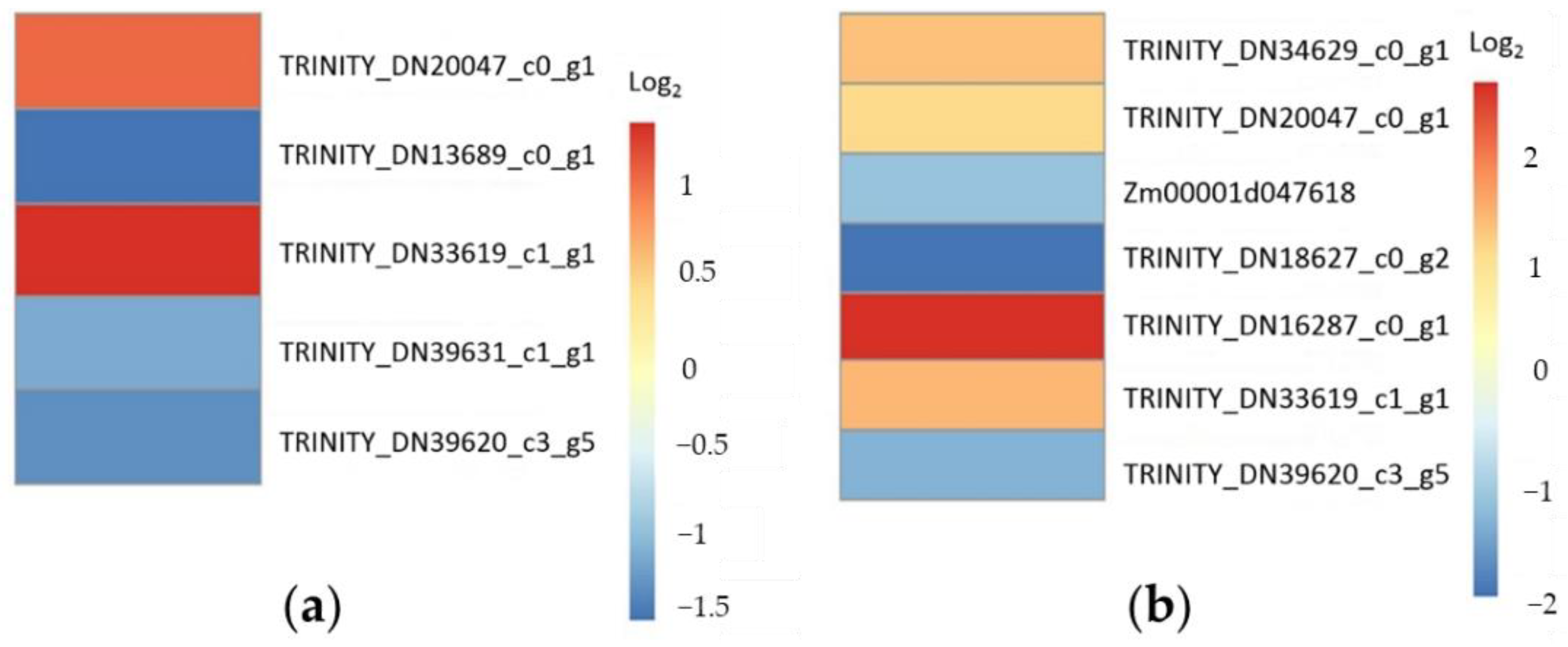
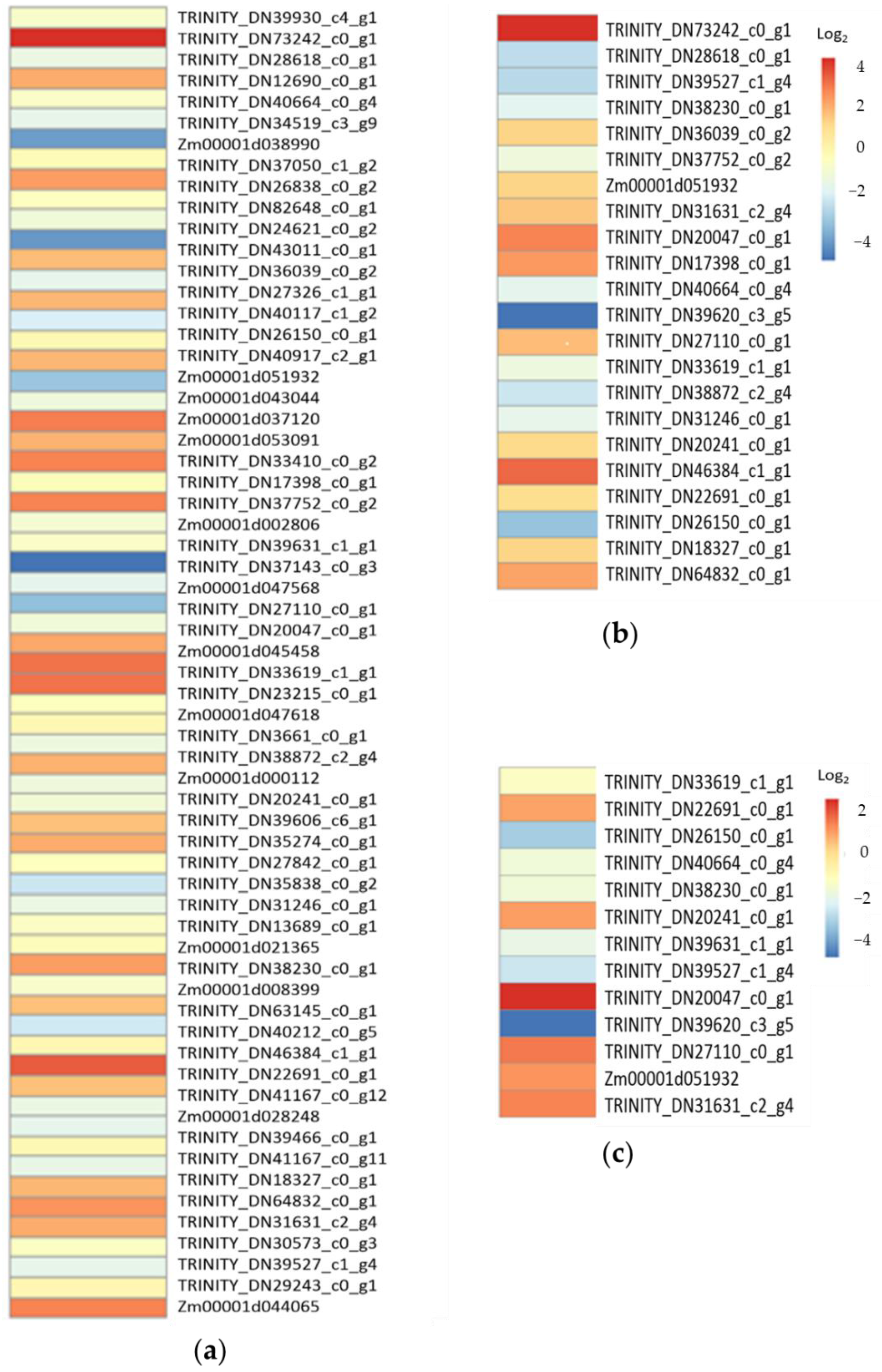
2.4. Transcriptional Response to Drought and Low-Temperature Stress
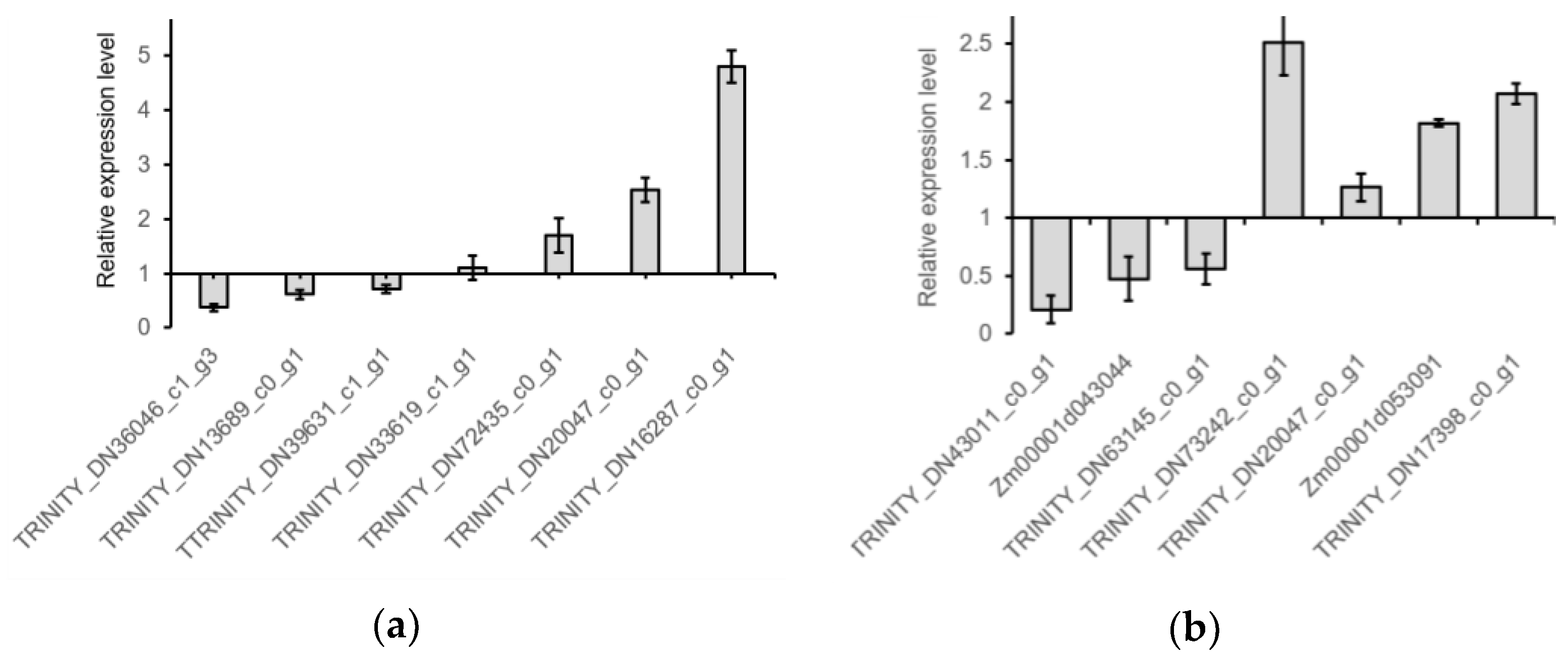
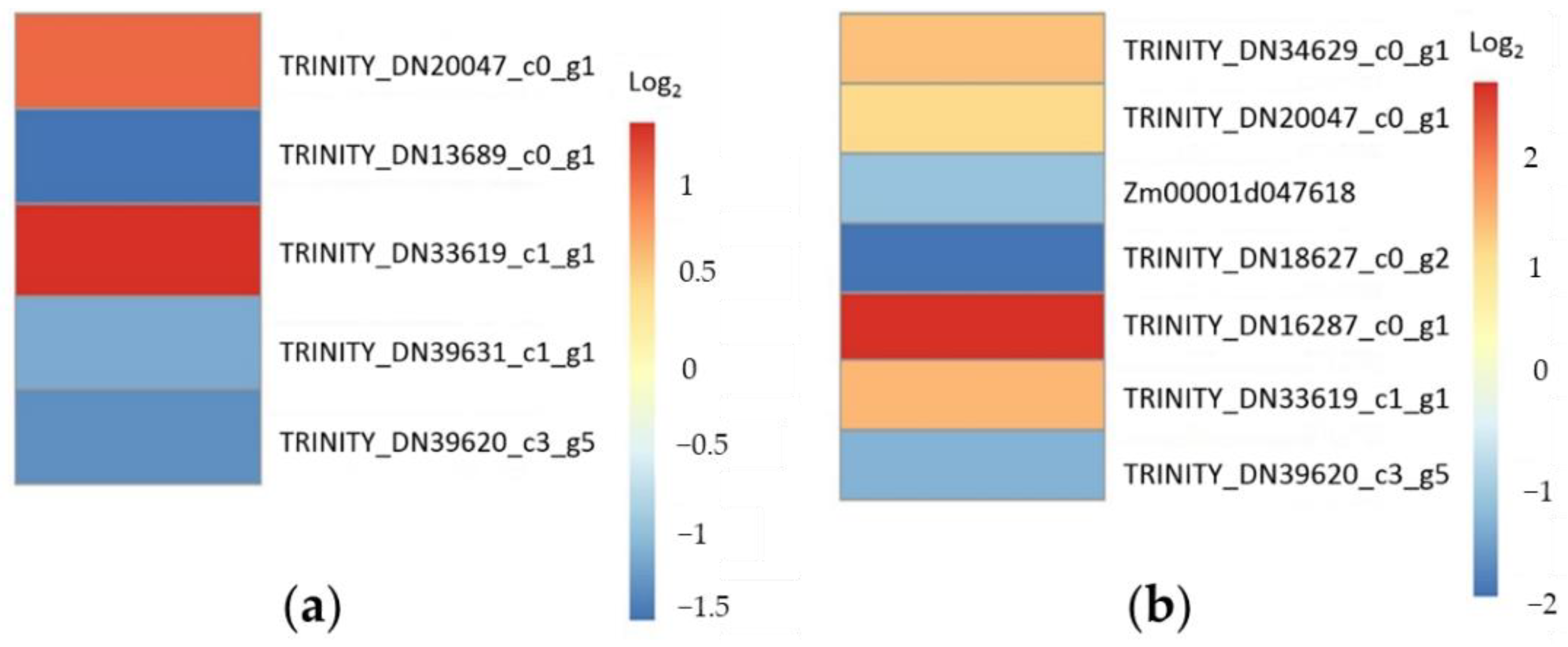
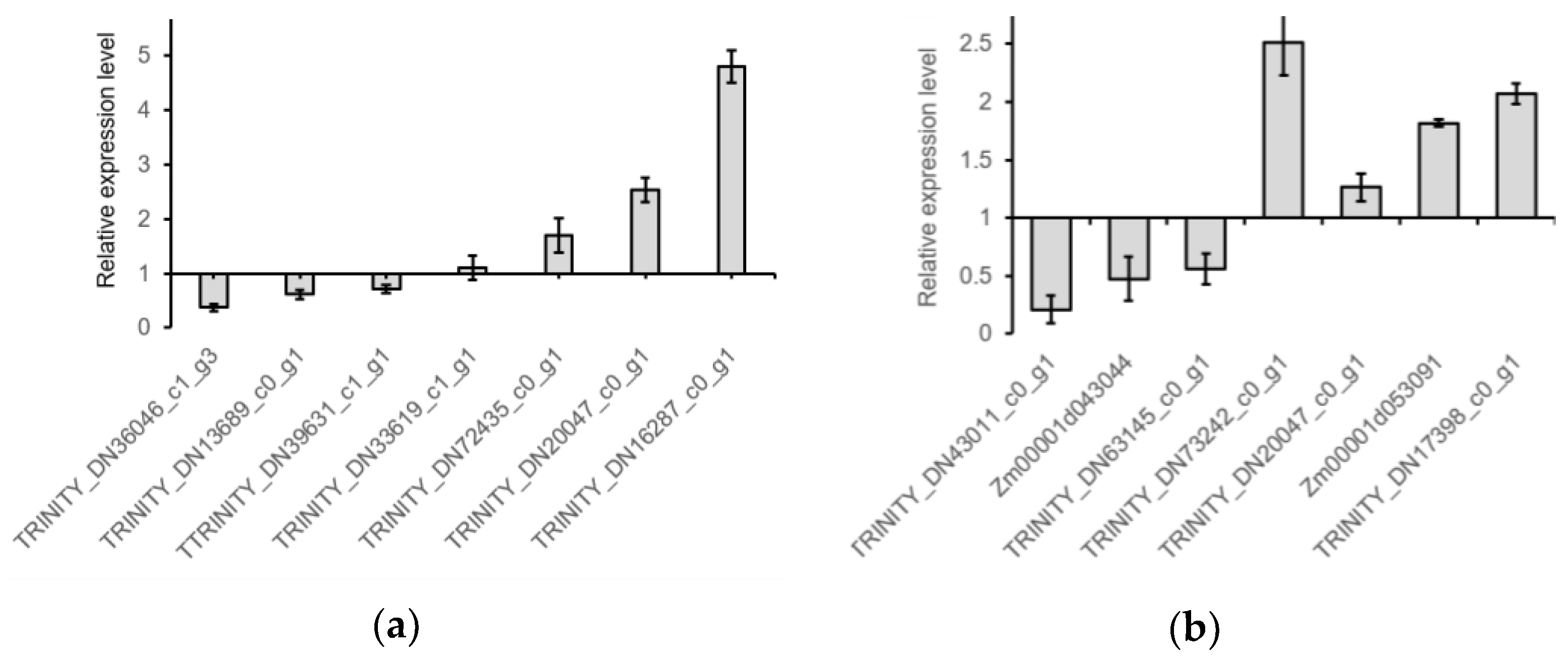
2.5. RT-qPCR Verification
2.6. Functional Annotation of Differentailly Expressed Genes
3. Discussion
4. Materials and Methods
4.1. Resequencing of Genomic DNA and Data Assembly
4.2. Identification of Specific Variation Loci and Phylogenetic Analysis
4.3. RNA Preparation and Sequencing
4.4. Identification and Functional Annotation of Differentially Expressed Genes
4.5. RT-qPCR Verification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sprague, G.F.; Dudley, J.W. Corn and Corn Improvement, 3rd ed.; American Society of Agronomy: Madison, WI, USA, 1988; pp. 1–774. [Google Scholar]
- Hallauer, A.R. Methods used in developing maize inbreds. Maydica 1990, 35, 1–16. [Google Scholar]
- Russell, W.A. Genetic improvement of maize yields. Adv. Agron. 1991, 46, 245–298. [Google Scholar]
- Liu, J.L. Maize Breeding, 2nd ed.; China Agriculture Press: Beijing, China, 2002; pp. 1–578. [Google Scholar]
- Wu, C. A review on the germplasm bases of the main corn hybrids in China. Sci. Agric. Sin. 1983, 16, 1–8. [Google Scholar]
- Darrah, L.L.; Zuber, M.S. 1985 United States farm maize germplasm base and commercial breeding strategies. Crop Sci. 1986, 26, 287–291. [Google Scholar] [CrossRef]
- Zeng, S.X. The maize germplasm base of hybrids in China. Sci. Agri. Sin. 1990, 23, 1–9. [Google Scholar]
- Duvick, D.N. Genetic progress in yield of United States maize (Zea mays L.). Maydica 2005, 50, 193–202. [Google Scholar]
- Li, Y.; Wang, T.Y. Germplasm base of maize breeding in China and formation of foundation parents. J. Maize Sci. 2010, 18, 1–8. [Google Scholar]
- Hallauer, A.R.; Carena, M.J. Adaptation of tropical maize germplasm to temperate environments. Euphytica 2014, 196, 1–11. [Google Scholar] [CrossRef]
- Rong, T.Z.; Li, W.C.; Yang, K.C.; Zhang, B.; Zhang, S.K.; Tang, H.J.; Fan, X.M. Maize Breeding in the Southwest Ecological Region; China Agriculture Press: Beijing, China, 2003; pp. 1–309. [Google Scholar]
- Gao, X.; Wang, J.; Peng, Z.H.; Cao, S.S.; Shen, J.H.; Zhu, Y.F.; Chen, Z.H. Preliminary study on heterosis utilization model of foreign maize germplasm P78599. Crops 2004, 35, 46–50. [Google Scholar]
- Li, H.M.; Hu, R.F.; Zhang, S.H. The impacts of US and CGIAR’s germplasm on maize production in China. Sci. Agric. Sin. 2005, 38, 2189–2197. [Google Scholar]
- Guo, Q.; Zhang, Y.; Kang, H.; Liu, Z.; Liu, H.; Dou, B. Discussion on the introduction, selection and combination model of American maize germplasm. Mol. Plant Breed. 2016, 14, 3262–3272. [Google Scholar]
- He, D.Y.; Zhou, L.D.; Liu, J.W.; He, Q. The role of American maize germplasm in maize breeding in China. Seed Sci. Technol. 2009, 27, 20–21. [Google Scholar]
- Shi, L. The impact of US germplasm on maize breeding efforts in China. J. Maize Sci. 2007, 15, 1–4. [Google Scholar]
- Pan, G. Study on the application of TTC to population improvement. J. Sichuan Agric. Univ. 1986, 4, 129–138. [Google Scholar]
- Sun, Q.Q.; Zhang, Y.; Rong, T.Z.; Dong, S.T.; Zhang, C.Q. Establishment of transgenic acceptor and transformation of barnase gene by particle gun in maize inbred line 18-599 (White). Acta Agron. Sin. 2007, 33, 738–743. [Google Scholar] [CrossRef]
- He, J.; Hu, Y.; Li, W.C.; Fu, F.L. Drought tolerant mutant induced by gamma-ray and sodium azide from maize calli. Maize Genet. Coop. News Lett. 2009, 83, 53–55. [Google Scholar]
- Popova, O.P.; Dietz, K.; Golldack, D. Salt-dependent expression of a nitrate transporter and two amino acid transporter genes in Mesembryanthemum crystallinum. Plant Mol. Biol. 2003, 52, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Barozai, M.Y.K.; Husnain, T. Identification of biotic and abiotic stress up-regulated ESTs in Gossypium arboreum. Mol. Biol. Rep. 2011, 39, 1011–1018. [Google Scholar] [CrossRef]
- Hou, J.Y.; Fu, J.Y.; Zhao, Y.; Qiu, Y.; Li, C.C.; Wang, Q. Expression analysis of terpene synthases in roots of maize inbred lines in response to drought and phytohormone treatments. J. Maize Sci. 2017, 25, 63–67. [Google Scholar]
- Ahmed, I.M.; Nadira, U.A.; Qiu, C.W.; Cao, F.; Zhang, G.; Holford, P.; Wu, F. Tolerance to drought, low pH and Al combined stress in Tibetan wild barley is associated with improvement of ATPase and modulation of antioxidant defense system. Int. J. Mol. Sci. 2018, 19, 3553. [Google Scholar] [CrossRef] [Green Version]
- Tegeder, M. Transporters involved in source to sink partitioning of amino acids and ureides: Opportunities for crop improvement. J. Exp. Bot. 2014, 65, 1865–1878. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Yang, T.; Zhang, Z.; Li, F.; Chen, Q.; Sun, J.; Shi, C.; Deng, W.; Tao, M.; Tai, Y.; et al. Identification and characterization of cationic amino acid transporters (CATs) in tea plant (Camellia sinensis). Plant Growth Regul. 2018, 84, 57–69. [Google Scholar] [CrossRef]
- Kamthan, A.; Kamthan, M.; Azam, M.; Chakraborty, N.; Chakraborty, S.; Datta, A. Expression of a fungal sterol desaturase improves tomato drought tolerance, pathogen resistance and nutritional quality. Sci. Rep. 2012, 2, 951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.H.; Lee, K.; Lee, K.H.; Jung, S.; Jeon, Y.; Pai, H.S.; Kang, H. A nuclear-encoded chloroplast-targeted S1 RNA-binding domain protein affects chloroplast rRNA processing and is crucial for the normal growth of Arabidopsis thaliana. Plant J. 2015, 83, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Dinh, S.N.; Park, S.J.; Han, J.H.; Kang, H. A chloroplast-targeted S1 RNA-binding domain protein plays a role in Arabidopsis response to diverse abiotic stresses. J. Plant Biol. 2019, 62, 74–81. [Google Scholar] [CrossRef]
- Zhang, L.; Li, T.; Wang, Y.; Zhang, Y.; Dong, Y.S. FvC5SD overexpression enhances drought tolerance in soybean by reactive oxygen species scavenging and modulating stress-responsive gene expression. Plant Cell Rep. 2019, 38, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Ijaz, R.; Ejaz, J.; Gao, S.; Liu, T.; Imtiaz, M.; Ye, Z.; Wang, T. Overexpression of annexin gene AnnSp2, enhances drought and salt tolerance through modulation of ABA synthesis and scavenging ROS in tomato. Sci. Rep. 2017, 21, 12087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Wan, X.L.; Yu, J.Y.; Wang, K.L.; Zhang, J. Genome-wide identification, classification, and expression analysis of the Hsf gene family in carnation (Dianthus caryophyllus). Int. J. Mol. Sci. 2019, 20, 5233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.Y.; Yao, Y.; Hong, Y.H.; Hou, P.Y.; Li, C.X.; Xia, Z.Q.; Geng, M.T.; Chen, Y.H. Differential expression of the Hsf family in cassava under biotic and abiotic stresses. Genome 2019, 62, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Guo, Q.; Pang, X.; Zhang, P.; Kong, D.; Liu, J. New insights into evolution of plant heat shock factors (Hsfs) and expression analysis of tea genes in response to abiotic stresses. Plants 2020, 9, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ma, X.; Wang, H.; Li, B.; Clark, G.; Guo, Y.; Roux, S.; Sun, D.; Tang, W. Proteomic study of microsomal proteins reveals a key role for Arabidopsis annexin 1 in mediating heat stress-induced increase in intracellular calcium levels. Mol. Cell Proteomics 2015, 14, 686–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, R.B.; Romdhane, W.B.; Hsouna, A.B.; Mihoubi, W.; Harbaoui, M.; Brini, F. Insights into plant annexins function in abiotic and biotic stress tolerance. Plant Signal. Behav. 2020, 15, 1699264. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Liao, L.; Xie, S.; Yao, M.; Xie, P.; Liu, W.; Kang, Y.; Huang, L.; Wang, M.; Qian, L.; et al. Comprehensive analyses of the annexin (ANN) gene family in Brassica rapa, Brassica oleracea and Brassica napus reveals their roles in stress response. Sci. Rep. 2020, 10, 4295. [Google Scholar] [CrossRef] [Green Version]
- Talanova, V.V.; Titov, A.F.; Topchieva, L.V.; Malysheva, I.E.; Venzhik, Y.V.; Frolova, S.A. Expression of genes encoding the WRKY transcription factor and heat shock proteins in wheat plants during cold hardening. Dokl. Biol. Sci. 2008, 423, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhang, X.; Wang, W.; Wang, Y.; Ming, F. The suppression of WRKY44 by GIGANTEA-miR172 pathway is involved in drought response of Arabidopsis thaliana. PLoS ONE 2013, 8, e73541. [Google Scholar] [CrossRef]
- Mishra, A.; Heyer, A.G.; Mishra, K.B. Chlorophyll fluorescence emission can screen cold tolerance of cold acclimated Arabidopsis thaliana accessions. Plant Methods 2014, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.J.; Wang, B.; Dong, R.R.; Hou, B.K. AtUGT76C2, an Arabidopsis cytokinin glycosyltransferase is involved in drought stress adaptation. Plant Sci. 2015, 236, 157–167. [Google Scholar] [CrossRef]
- Li, P.; Li, Y.J.; Zhang, F.J.; Zhang, G.Z.; Jiang, X.Y.; Yu, H.M.; Hou, B.K. The Arabidopsis UDP-glycosyltransferases UGT79B2 and UGT79B3, contribute to cold, salt and drought stress tolerance via modulating anthocyanin accumulation. Plant J. 2017, 89, 85–103. [Google Scholar] [CrossRef] [Green Version]
- Desclos, M.; Dubousset, L.; Etienne, P.; Le Caherec, F.; Satoh, H.; Bonnefoy, J.; Ourry, A.; Avice, J.C. A proteomic profiling approach to reveal a novel role of Brassica napus drought 22 kD/water-soluble chlorophyll-binding protein in young leaves during nitrogen remobilization induced by stressful conditions. Plant Physiol. 2008, 147, 1830–1844. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Dai, X.; Zhang, W.H. A R2R3-type MYB gene, OsMYB2, is involved in salt, cold, and dehydration tolerance in rice. J. Exp. Bot. 2012, 63, 2541–2556. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Wang, S.; Xiong, B.; Cao, B.; Deng, X. Carbon/nitrogen imbalance associated with drought-induced leaf senescence in Sorghum bicolor. PLoS ONE 2015, 10, e0137026. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, J.; Hu, R.; Wu, P.; Hou, X.L.; Song, X.M.; Xiong, A.S. Genome-wide analysis of the R2R3-MYB transcription factor genes in Chinese cabbage (Brassica rapa ssp. pekinensis) reveals their stress and hormone responsive patterns. BMC Genom. 2015, 16, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.; Kim, Y.J.; Sukweenadhi, J.; Rahimi, S.; Kwon, W.S.; Yang, D.C. Molecular characterization of 5-chlorophyll a/b-binding protein genes from Panax ginseng Meyer and their expression analysis during abiotic stresses. Photosynthetica 2016, 54, 446–458. [Google Scholar] [CrossRef]
- Cao, X.; Chen, X.; Liu, Y.; Xu, Z.; Li, L.; Zhou, Y.; Liu, J.; Zhao, Z.; Chen, M.; Ma, Y. An iNTT system for the large-scale screening of differentially expressed, nuclear-targeted proteins: Cold-treatment-induced nucleoproteins in rye (Secale cereale L.). BMC Genom. 2016, 17, 189. [Google Scholar] [CrossRef]
- Li, X.W.; Zhu, Y.L.; Chen, C.Y.; Geng, Z.J.; Li, X.Y.; Ye, T.T.; Mao, X.N.; Du, F. Cloning and characterization of two chlorophyll A/B binding protein genes and analysis of their gene family in Camellia sinensis. Sci. Rep. 2020, 10, 4602. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Bassham, D.C. COST1 balances plant growth and stress tolerance via attenuation of autophagy. Autophagy 2020, 16, 1157–1158. [Google Scholar] [CrossRef]
- Bao, Y.; Song, W.M.; Wang, P.; Yu, X.; Li, B.; Jiang, C.; Shiu, S.H.; Zhang, H.; Bassham, D.C. COST1 regulates autophagy to control plant drought tolerance. Proc. Natl. Acad. Sci. USA 2020, 117, 7482–7493. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. FASTP: An ultra-fast all-in-one FASTQ preprocessor FASTP: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Exploring single-sample SNP and INDEL calling with whole-genome de novo assembly. Bioinformatics 2012, 28, 1838–1844. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Bukowski, R.; Guo, X.; Lu, Y.; Zou, C.; He, B.; Rong, Z.; Wang, B.; Xu, D.; Yang, B.; Xie, C.; et al. Construction of the third-generation Zea mays haplotype map. Gigascience 2018, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 15, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.L.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variation Type | Number of Variation Loci | Number of Genes | Number of Variation Loci per Gene |
|---|---|---|---|
| Frameshift | 1023 | 10,201 | 1.67 |
| Change of splicing site | 2293 | 1994 | 1.15 |
| Stop gain | 1879 | 1600 | 1.17 |
| Change of start site | 550 | 532 | 1.03 |
| Stop loss | 424 | 411 | 1.03 |
| Total | 21,961 | 12,297 | 1.79 |
| Inbred Line | Identity | Inbred Line | Identity |
|---|---|---|---|
| ZEAxujRAUDIAAPE | 73.65% | L005 | 54.40% |
| ZEAxppRDHDIAAPEI-12 | 60.11% | Shen 135 | 54.13% |
| ZEAxujRAVDIAAPE | 59.99% | DH138 | 54.01% |
| CAU 178 | 59.82% | CT109 | 53.92% |
| dupl-178 | 59.82% | 9058 | 53.83% |
| 78599 | 59.78% | L-1 | 53.51% |
| D856 | 58.64% | Cheng 18 | 53.22% |
| 2005-4 | 58.12% | P138 | 53.00% |
| Shen 137 | 58.01% | L069 | 52.53% |
| Shen 977 | 56.66% | Lo1125 | 52.12% |
| ZEAxujRAJDIBAPE | 55.48% | Shan 89-1 | 52.08% |
| Dan 599 | 55.33% | D1051 | 51.80% |
| Zun 90110 | 55.28% | PN2 | 51.16% |
| 68122 | 55.14% | R150 | 50.95% |
| ZEAxppRDQDIAAPEI-3 | 54.69% | SZ3 | 50.50% |
| Qi 319 | 54.41% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Y.; Qu, J.; Yu, H.; Yang, Q.; Li, W.; Fu, F. Genomic Characteristics of Elite Maize Inbred Line 18-599 and Its Transcriptional Response to Drought and Low-Temperature Stresses. Plants 2022, 11, 3242. https://doi.org/10.3390/plants11233242
Cao Y, Qu J, Yu H, Yang Q, Li W, Fu F. Genomic Characteristics of Elite Maize Inbred Line 18-599 and Its Transcriptional Response to Drought and Low-Temperature Stresses. Plants. 2022; 11(23):3242. https://doi.org/10.3390/plants11233242
Chicago/Turabian StyleCao, Yang, Jingtao Qu, Haoqiang Yu, Qingqing Yang, Wanchen Li, and Fengling Fu. 2022. "Genomic Characteristics of Elite Maize Inbred Line 18-599 and Its Transcriptional Response to Drought and Low-Temperature Stresses" Plants 11, no. 23: 3242. https://doi.org/10.3390/plants11233242
APA StyleCao, Y., Qu, J., Yu, H., Yang, Q., Li, W., & Fu, F. (2022). Genomic Characteristics of Elite Maize Inbred Line 18-599 and Its Transcriptional Response to Drought and Low-Temperature Stresses. Plants, 11(23), 3242. https://doi.org/10.3390/plants11233242