
Investigation of Roles of TaTALE Genes during Development and Stress Response in Bread Wheat




Abstract
1. Introduction
2. Results
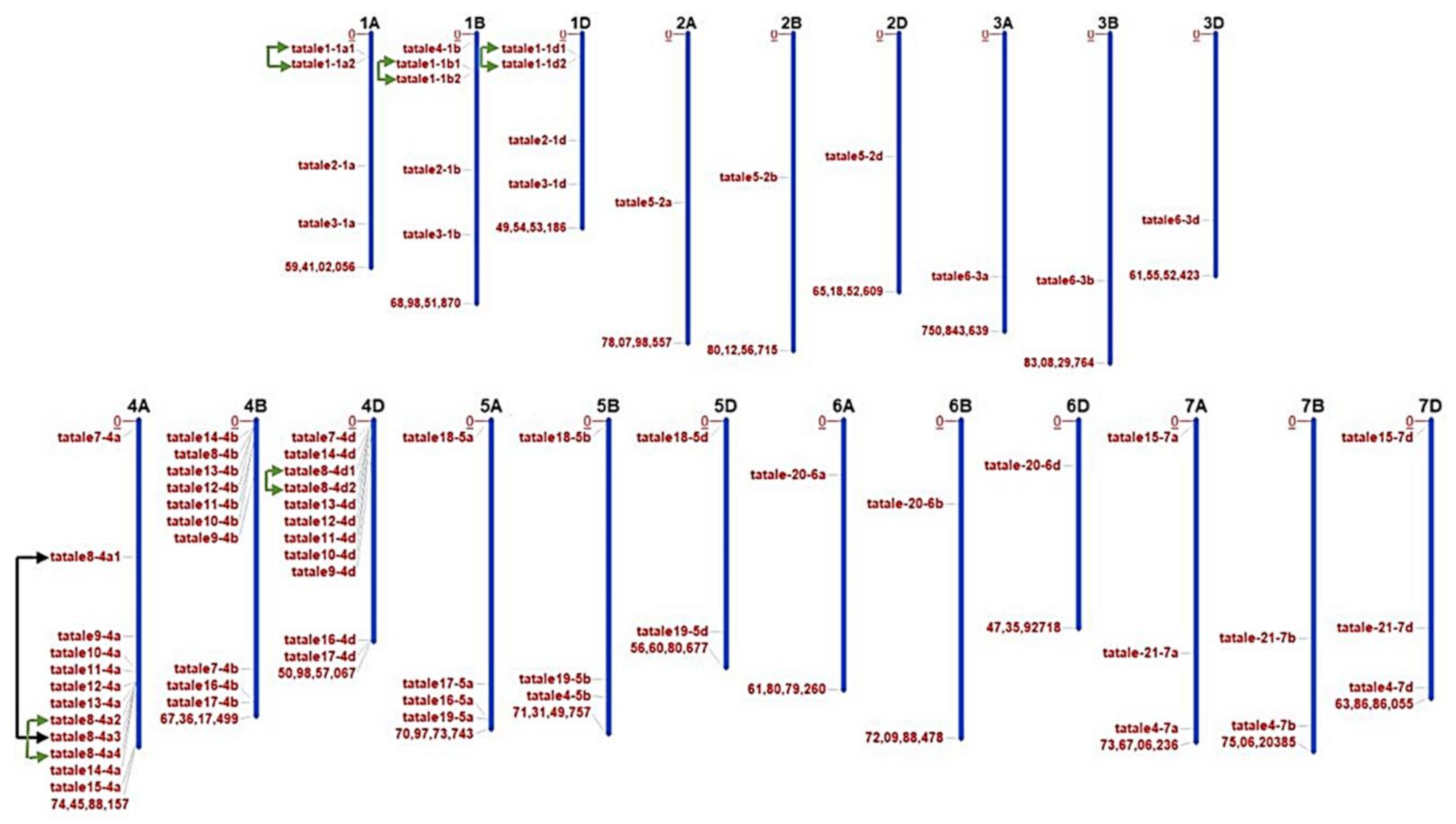
2.1. Identification, Chromosomal Localization, and Characterization of TaTALE Genes in Bread Wheat
2.2. Gene Duplication Analysis and Calculation of Non-Synonymous (Ka) and Synonymous (Ks) Substitution Rate
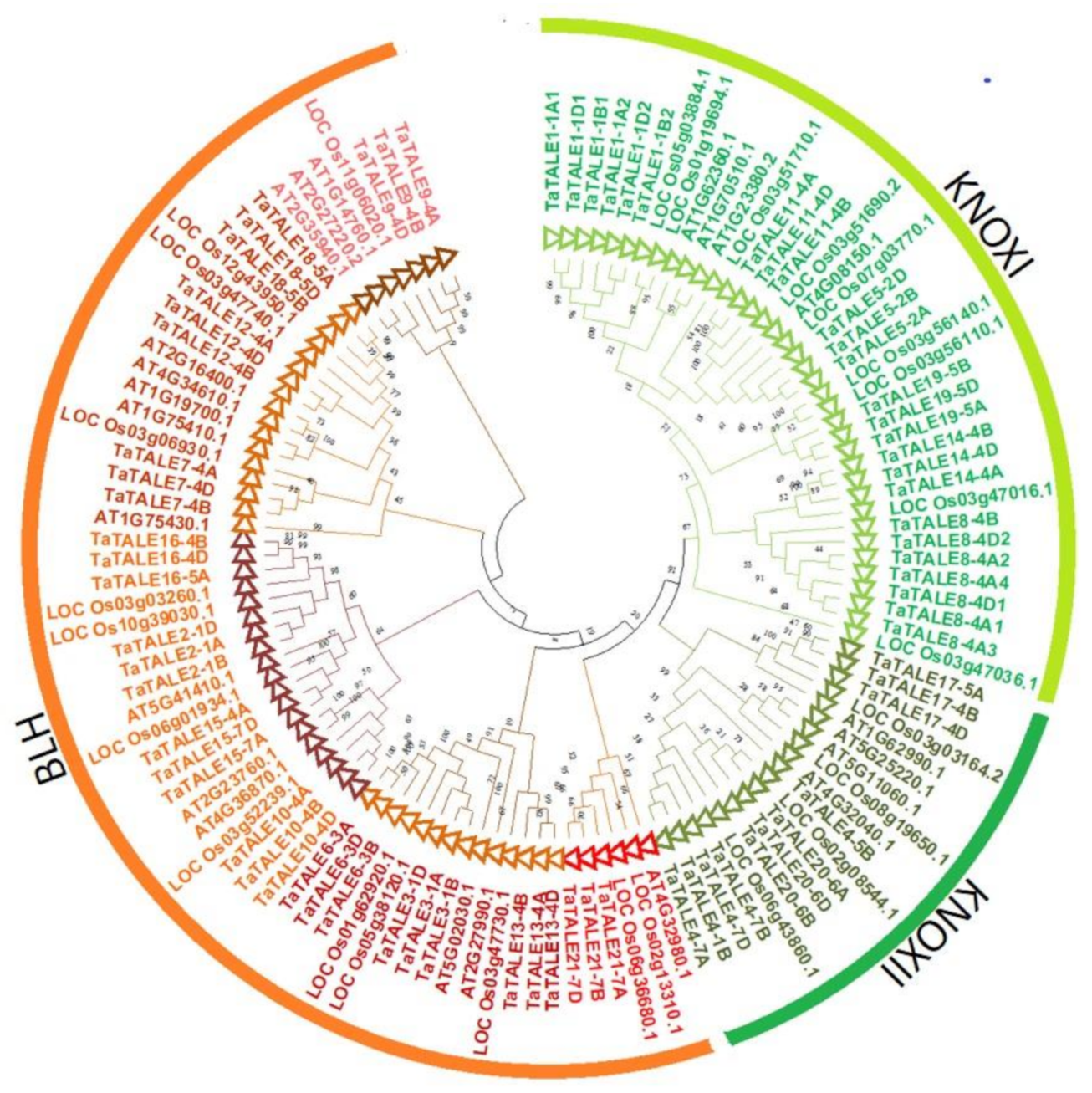
2.3. Phylogenetic Analysis
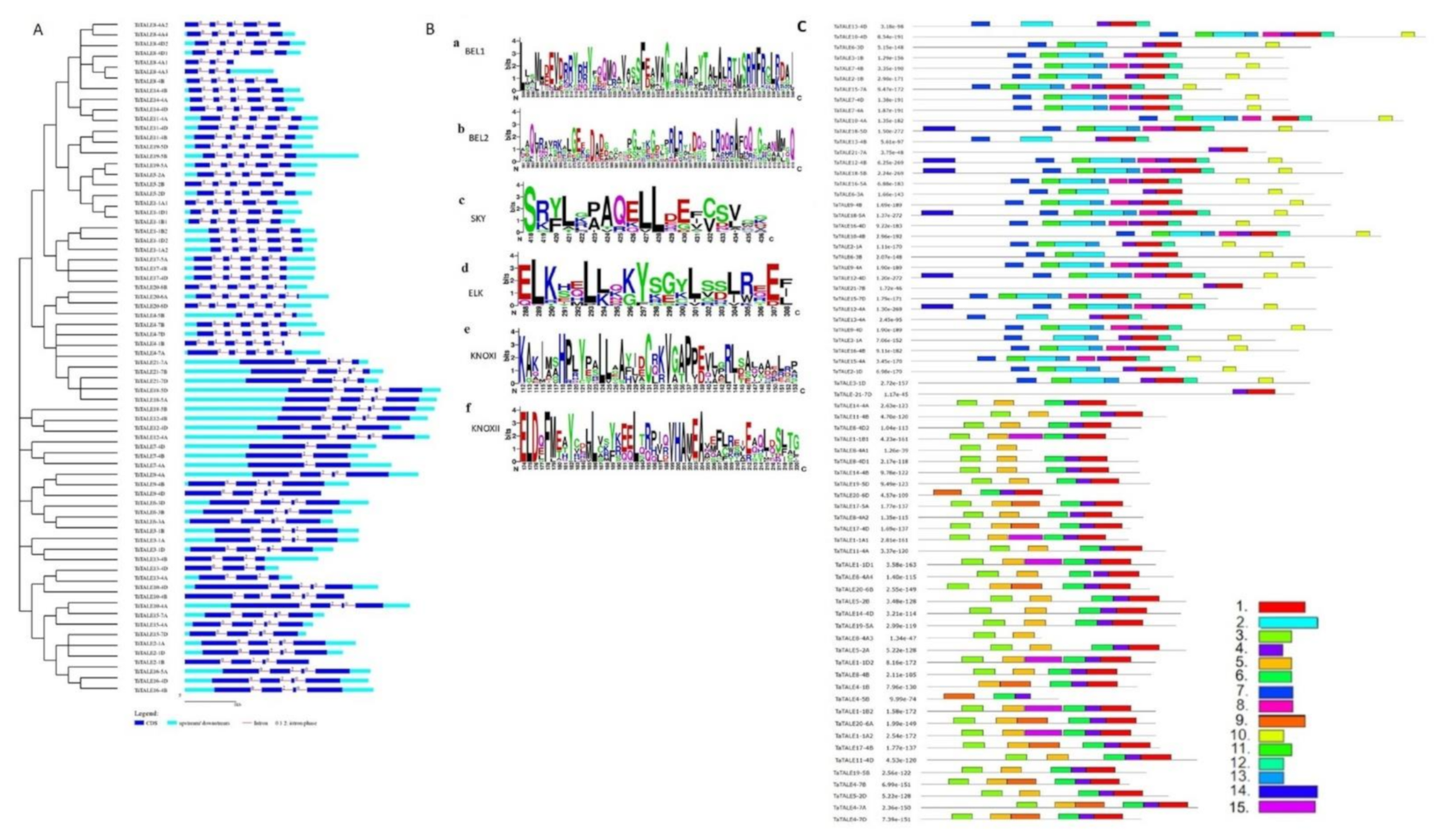
2.4. Exon-Intron, Domain, and Motif Analysis
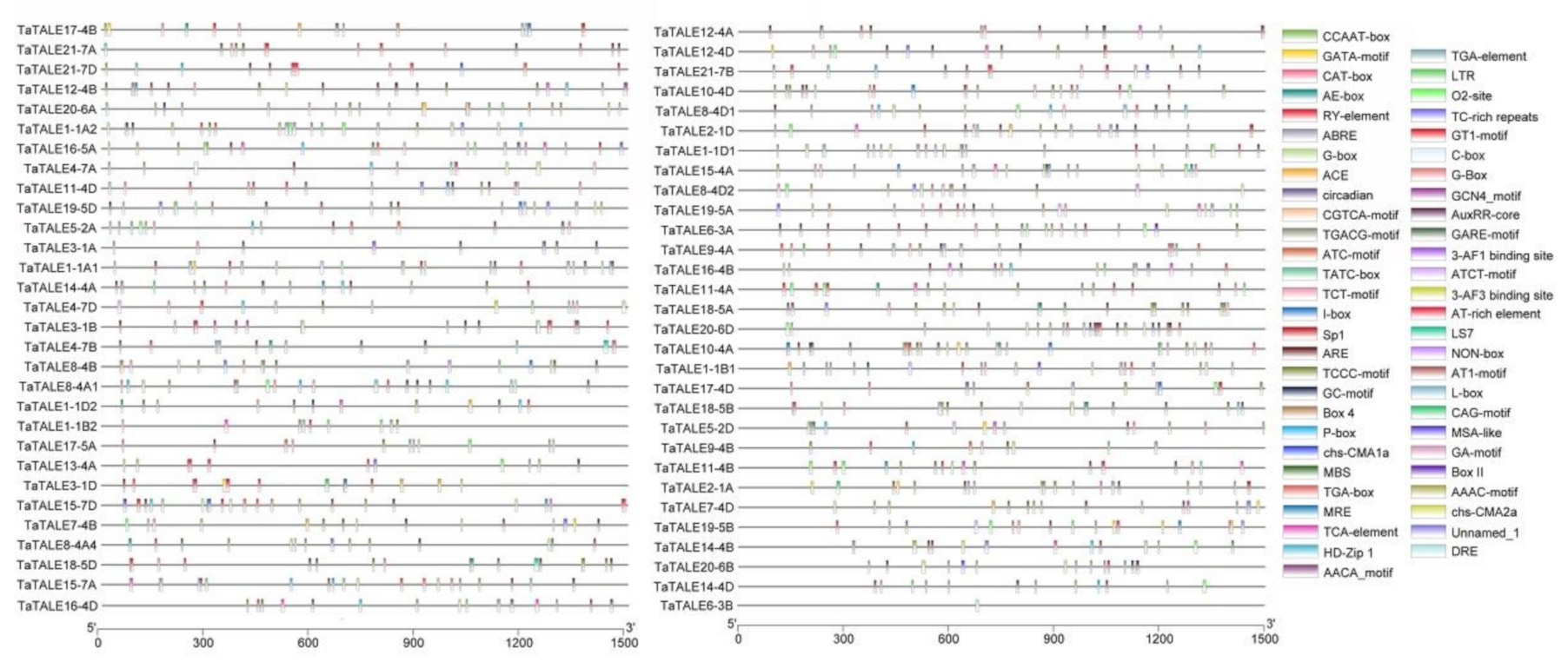
2.5. Promoter Analysis
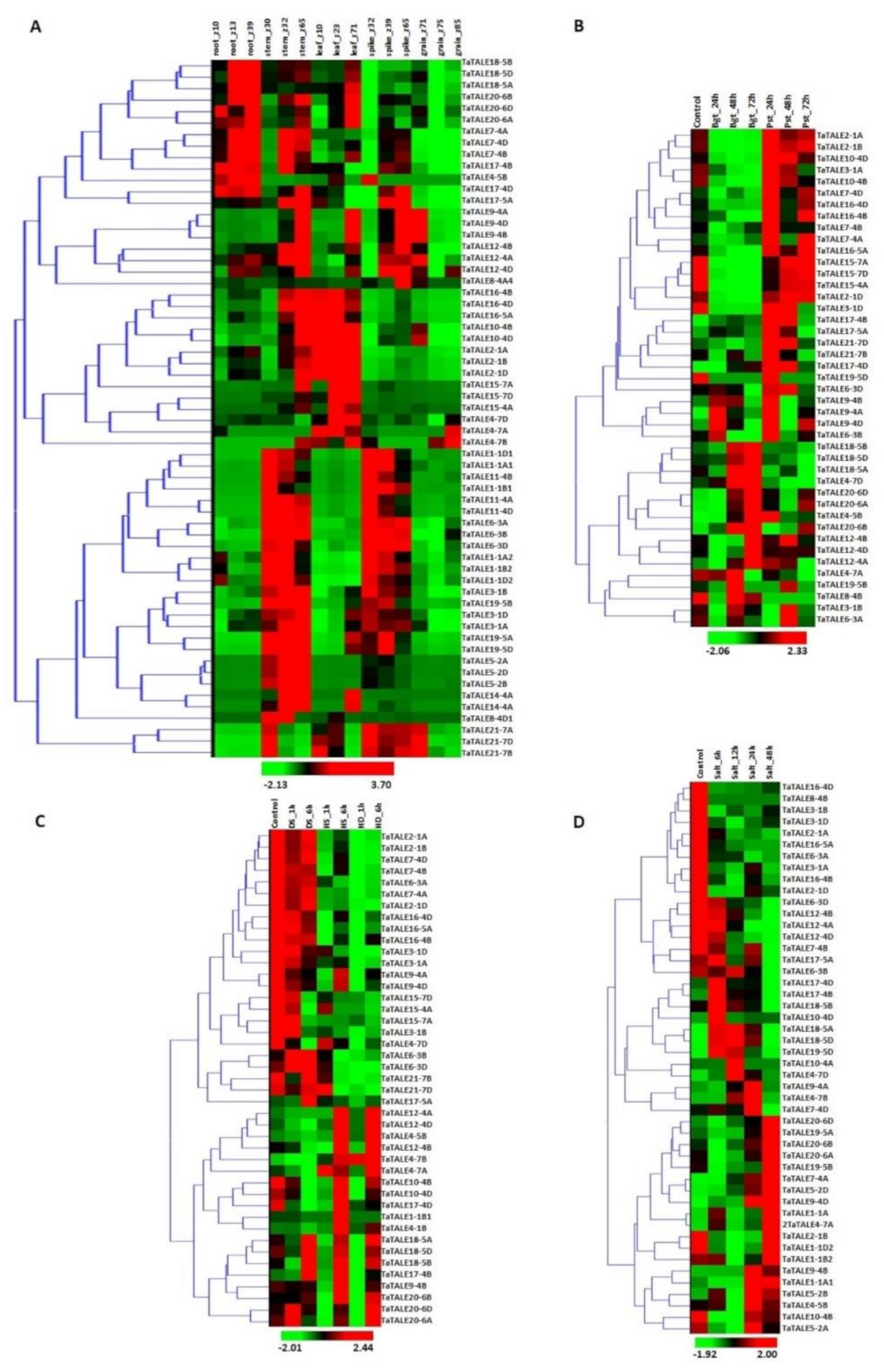
2.6. Expression Analysis
2.6.1. Expression Profile of TaTALE Genes under Tissue Developmental Stages
2.6.2. Expression Analysis under Fungal Pathogens Stress
2.6.3. Expression Analysis under Heat, Drought, and Combined Stress Conditions
2.6.4. Expression Analysis under Salt Stress
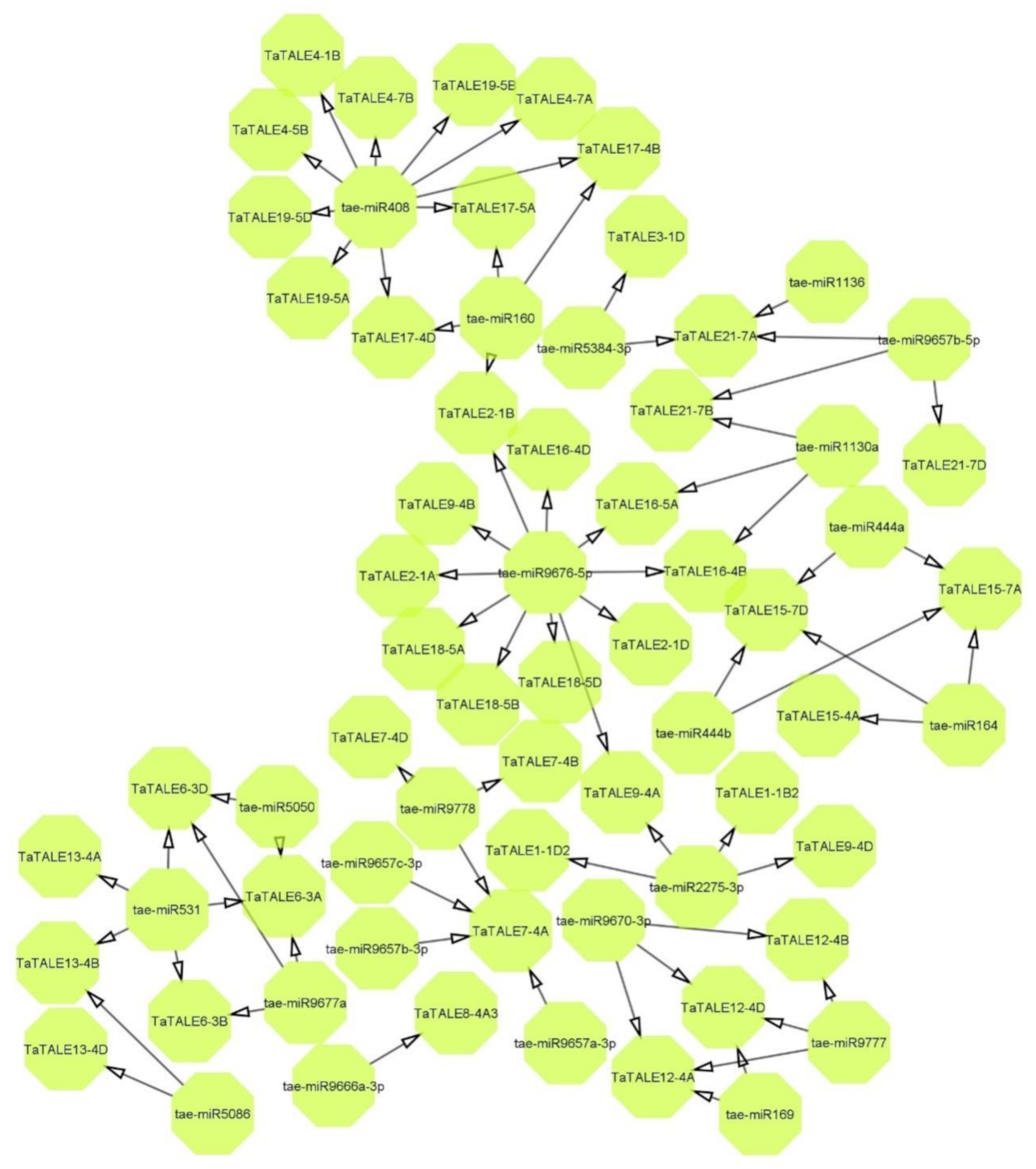
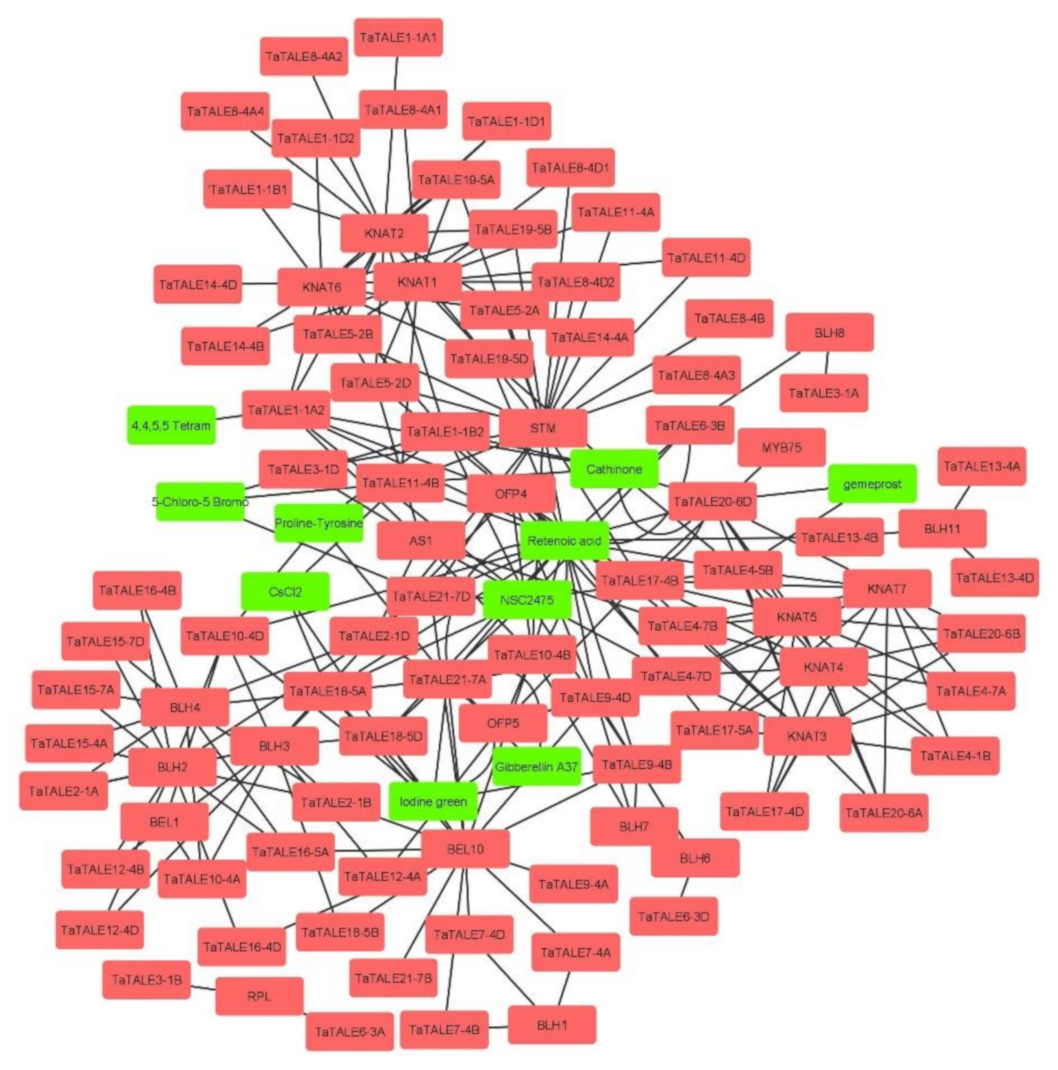
2.7. Interactome Analysis
3. Discussion
4. Materials and Methods
4.1. Identification and Nomenclature of TaTALE Genes
4.2. Chromosomal Localization, Homeologs Prediction, and Duplication Events
4.3. Phylogenetic Relationship and Multiple Sequence Alignment
4.4. Synonymous and Non-Synonymous Substitution Rates of TaTALEs
4.5. Gene Structure Analysis
4.6. Physicochemical Analysis of TaTALE Proteins
4.7. Expression Profiling under Tissue Developmental Stages and under Abiotic and Biotic Stress Conditions
4.8. miRNA-Targets and Interaction Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Desplan, C.; Theis, J.; O’Farrell, P.H. The sequence specificity of homeodomain-DNA interaction. Bone 2008, 23, 1–7. [Google Scholar] [CrossRef]
- Rathour, M.; Sharma, A.; Kaur, A.; Upadhyay, S.K. Genome-wide characterization and expression and co-expression analysis suggested diverse functions of WOX genes in bread wheat. Heliyon 2020, 6, e05762. [Google Scholar] [CrossRef]
- Chen, H.; Rosin, F.M.; Prat, S.; Hannapel, D.J. Interacting transcription factors from the three-amino acid loop extension superclass regulate tuber formation. Plant Physiol. 2003, 132, 1391–1404. [Google Scholar] [CrossRef] [PubMed]
- Bellaoui, M.; Pidkowich, M.S.; Samach, A.; Kushalappa, K.; Kohalmi, S.E.; Modrusan, Z.; Crosby, W.L.; Haughn, G.W. The Arabidopsis BELL1 and KNOX TALE Homeodomain Proteins Interact through a Domain Conserved between Plants and Animals. Plant Cell 2001, 13, 2455–2470. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Cho, Y.H.; Ryu, H.; Kim, Y.; Kim, T.H.; Hwang, I. BLH1 and KNAT3 modulate ABA responses during germination and early seedling development in Arabidopsis. Plant J. 2013, 75, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Wang, Y.; Franzen, R.; Santi, L.; Salamini, F.; Rohde, W. In vitro interactions between barley TALE proteins suggest a role for protein-protein associations in the regulation of Knox gene function. Plant J. 2001, 27, 13–23. [Google Scholar] [CrossRef]
- Vollbrecht, E.; Veit, B.; Sinha, N.; Hake, S. The developmental gene Knotted7 is a member of a maize homeobox gene family. Nature 1991, 350, 241–243. [Google Scholar] [CrossRef]
- Bürglin, T.R. Analysis of TALE superclass homeobox genes (MEIS, PBC, KNOX, Iroquois, TGIF) reveals a novel domain conserved between plants and animals. Nucleic Acids Res. 1991, 25, 4173–4180. [Google Scholar] [CrossRef]
- Magnani, E.; Hake, S. KNOX lost the OX: The Arabidopsis KNATM gene defines a novel class of KNOX transcriptional regulators missing the homeodomain. Plant Cell 2008, 20, 875–887. [Google Scholar] [CrossRef]
- Liu, Y.; You, S.; Taylor-Teeples, M.; Li, W.L.; Schuetz, M.; Brady, S.M.; Douglas, C.J. BEL1-LIKE HOMEODOMAIN6 and KNOTTED ARABIDOPSIS THALIANA7 Interact and Regulate Secondary Cell Wall Formation via Repression of REVOLUTA. Plant Cell 2015, 26, 4843–4861. [Google Scholar] [CrossRef]
- Sharma, P.; Lin, T.; Grandellis, C.; Yu, M.; Hannapel, D.J. The BEL1-like family of transcription factors in potato. J. Exp. Bot. 2014, 65, 709–723. [Google Scholar] [CrossRef] [PubMed]
- Kerstetter, R.A.; Laudencia-Chingcuanco, O.; Smith, L.G.; Hake, S. Loss of-function mutations in the maize homeobox gene, knotted1, are defective in shoot meristem maintenance. Development 1997, 124, 3045–3054. [Google Scholar] [CrossRef] [PubMed]
- Reiser, L.; Sanchez-Baracaldo, P.; Hake, S. Knots in the family tree: Evolutionary relationships and functions of KNOX homeobox genes. Plant Mol. Biol. 2000, 42, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Cho, L.H.; Kim, S.L.; Choi, H.; Koh, H.J.; An, G. The BEL1-type homeobox gene SH5 induces seed shattering by enhancing abscission-zone development and inhibiting lignin biosynthesis. Plant J. 2014, 79, 717–728. [Google Scholar] [CrossRef]
- Hake, S.; Smith, H.M.S.; Holtan, H.; Magnani, E.; Mele, G.; Ramirez, J. The role of Knox genes in plant development. Annu. Rev. Cell Dev. Biol. 2004, 20, 125–151. [Google Scholar] [CrossRef]
- Ma, Q.; Wang, N.; Hao, P.; Sun, H.; Wang, C.; Ma, L.; Wang, H.; Zhang, X.; Wei, H.; Yu, S. Genome-wide identification and characterization of TALE superfamily genes in cotton reveals their functions in regulating secondary cell wall biosynthesis. BMC Plant Biol. 2019, 19, 432. [Google Scholar] [CrossRef]
- Kim, J.S.; Mizoi, J.; Yoshida, T.; Fujita, Y.; Nakajima, J.; Ohori, T.; Todaka, D.; Nakashima, K.; Hirayama, T.; Shinozaki, K.; et al. An ABRE promoter sequence is involved in osmotic stress-responsive expression of the DREB2A gene, which encodes a transcription factor regulating drought-inducible genes in Arabidopsis. Plant Cell Physiol. 2011, 52, 2136–2146. [Google Scholar] [CrossRef]
- Zhao, K.; Zhang, X.; Cheng, Z.; Yao, W.; Li, R.; Jiang, T.; Zhou, B. Comprehensive analysis of the three-amino-acid-loop-extension gene family and its tissue-differential expression in response to salt stress in poplar. Plant Physiol. Biochem. 2018, 136, 1–12. [Google Scholar] [CrossRef]
- Razzaq, A.; Ashraf, J.; Malik, W.; Shaban, M.; Zhang, R.; Liang, C.; Hanif, M.; Abid, M.A.; Qayyum, A. In silico analyses of TALE transcription factors revealed its potential role for organ development and abiotic stress tolerance in Cotton. Int. J. Agric. Biol. 2020, 23, 1083–1094. [Google Scholar]
- Jia, P.; Zhang, C.; Xing, L.; Li, Y.; Shah, K.; Zuo, X.; Zhang, D.; An, N.; Han, M.; Ren, X. Genome-Wide Identification of the MdKNOX Gene Family and Characterization of Its Transcriptional Regulation in Malus domestica. Front. Plant Sci. 2020, 21, 128. [Google Scholar] [CrossRef]
- Wang, L.; Yang, X.; Gao, Y.; Yang, S. Genome-wide identification and characterization of tale superfamily genes in soybean (Glycine max L.). Int. J. Mol. Sci. 2021, 22, 4117. [Google Scholar] [CrossRef] [PubMed]
- Mitsis, T.; Efthimiadou, A.; Bacopoulou, F.; Vlachakis, D.; Chrousos, G.; Eliopoulos, E. Transcription factors and evolution: An integral part of gene expression (Review). World Acad. Sci. J. 2020, 2, 3–8. [Google Scholar] [CrossRef]
- Hernandez-Garcia, C.M.; Finer, J.J. Identification and validation of promoters and cis-acting regulatory elements. Plant Sci. 2014, 217–218, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Guo, Z.; Li, L. Evolutionary conservation of microRNA regulatory programs in plant flower development. Dev. Biol. 2013, 380, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Hamant, O.; Pautot, V. Plant development: A TALE story. Comptes Rendus Biol. 2010, 333, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; Poland, J.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar]
- Marcussen, T.; Sandve, S.R.; Heier, L.; Spannagl, M.; Pfeifer, M.; International Wheat Genome Sequencing Consortium; Jakobsen, K.S.; Wulff, B.B.H.; Steuernagel, B.; Mayer, K.F.X.; et al. Ancient hybridizations among the ancestral genomes of bread wheat. Science 2004, 345, 1250092. [Google Scholar] [CrossRef]
- Scofield, S.; Murray, J.A. KNOX gene function in plant stem cell niches. Plant Mol. Biol. 2006, 60, 929–946. [Google Scholar] [CrossRef]
- Lacerda, A.F.; Vasconcelos, É.A.; Pelegrini, P.B.; Grossi de Sa, M.F. Antifungal defensins and their role in plant defense. Front. Microbiol. 2014, 5, 116. [Google Scholar] [CrossRef]
- Kaur, A.; Pati, P.K.; Pati, A.M.; Nagpal, A.K. In-silico analysis of cis-acting regulatory elements of pathogenesis-related proteins of Arabidopsis thaliana and Oryza sativa. PLoS ONE 2017, 12, e0184523. [Google Scholar] [CrossRef]
- Kerstetter, R.; Vollbrecht, E.; Lowe, B.; Veit, B.; Yamaguchi, J.; Hake, S. Sequence analysis and expression patterns divide the maize knotted1-like homeobox genes into two classes. Plant Cell 1994, 6, 1877–1887. [Google Scholar] [PubMed]
- Sakamoto, T.; Nishimura, A.; Tamaoki, M.; Kuba, M.; Tanaka, H.; Iwahori, S.; Matsuoka, M. The conserved KNOX domain mediates specificity of tobacco KNOTTED1-type homeodomain proteins. Plant Cell 1999, 11, 1419–1431. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nagasaki, H.; Sakamoto, T.; Sato, Y.; Matsuoka, M. Functional analysis of the conserved domains of a rice KNOX homeodomain protein, OSH15. Plant Cell 2001, 13, 2085–2098. [Google Scholar] [CrossRef] [PubMed]
- He, M.; He, C.Q.; Ding, N.Z. Abiotic Stresses: General Defenses of Land Plants and Chances for Engineering Multistress Tolerance. Front Plant Sci. 2018, 9, 1771. [Google Scholar] [CrossRef]
- Bai, J.F.; Wang, Y.; Wang, P.; Duan, W.J.; Yuan, S.H.; Sun, H.; Yuan, G.L.; Ma, J.X.; Wang, N.; Zhang, F.T.; et al. Uncovering Male Fertility Transition Responsive miRNA in a Wheat Photo-Thermosensitive Genic Male Sterile Line by Deep Sequencing and Degradome Analysis. Front. Plant Sci. 2017, 8, 1370. [Google Scholar] [CrossRef]
- Mallory, A.C.; Reinhart, B.; Jones-Rhoades, M.W.; Tang, G.; Zamore, P.D.; Barton, M.K.; Bartel, D.P. MicroRNA control of PHABULOSA in leaf development: Importance of pairing to the microRNA 5’ region. EMBO J. 2004, 18, 3356–3364. [Google Scholar] [CrossRef]
- Feng, H.; Duan, X.Y.; Zhang, Q.; Li, X.R.; Wang, B.; Huang, L.L.; Wang, X.J.; Kang, Z. The target gene of tae-miR164, a novel NAC transcription factor from the NAM subfamily, negatively regulates resistance of wheat to stripe rust. Mol. Plant Pathol. 2014, 15, 284–296. [Google Scholar] [CrossRef]
- Anil, V.; Rao, S.K. Calcium-Mediated Signaling during Sandalwood Somatic Embryogenesis. Role for Exogenous Calcium as Second Messenger. Plant Physiol. 2000, 123, 1301–1312. [Google Scholar] [CrossRef]
- Kiselev, K.V.; Gorpenchenko, T.Y.; Tchernoded, G.K.; Dubrovina, A.S.; Grishchenko, O.V.; Bulgakov, V.P.; Zhuravlev, Y.N. Calcium-dependent mechanism of somatic embryogenesis in Panax ginseng cell cultures expressing the rolC oncogene. Mol. Biol. 2008, 42, 243–252. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, X. Regulation of somatic embryogenesis in higher plants. Crit. Rev. Plant Sci. 2010, 29, 36–57. [Google Scholar] [CrossRef]
- Kiselev, K.V.; Shumakova, O.A.; Manyakhin, A.Y.; Mazeika, A.N. Influence of calcium influx induced by the calcium ionophore, A23187, on resveratrol content and the expression of CDPK and STS genes in the cell cultures of Vitis amurensis. Plant Growth Regul. 2012, 68, 371–381. [Google Scholar] [CrossRef]
- Han, R.; Jian, C.; Lv, J.; Yan, Y.; Chi, Q.; Li, Z.; Wang, Q.; Zhang, J.; Liu, X.; Zhao, H. Identification and characterization of microRNAs in the flag leaf and developing seed of wheat (Triticum aestivum L.). BMC Genom. 2014, 15, 289. [Google Scholar] [CrossRef] [PubMed]
- Hay, A.; Tsiantis, M. KNOX genes: Versatile regulators of plant development and diversity. Development 2010, 137, 3153–3165. [Google Scholar] [CrossRef] [PubMed]
- Kanrar, S.; Onguka, O.; Smith, H.M.S. Arabidopsis inflorescence architecture requires the activities of KNOX-BELL homeodomain heterodimers. Planta 2006, 224, 1163–1173. [Google Scholar] [CrossRef]
- Sun, Y.; Zhou, Q.; Zhang, W.; Fu, Y.; Huang, H. ASYMMETRIC LEAVES1, an Arabidopsis gene that is involved in the control of cell differentiation in leaves. Planta 2002, 214, 694–702. [Google Scholar] [CrossRef]
- Wang, S.; Chang, Y.; Guo, J.; Chen, J.G. Arabidopsis Ovate Family Protein 1 is a transcriptional repressor that suppresses cell elongation. Plant J. 2007, 50, 858–872. [Google Scholar] [CrossRef]
- Schmitz, A.J.; Begcy, K.; Sarath, G.; Walia, H. Rice ovate family protein 2 (OFP2) alters hormonal homeostasis and vasculature development. Plant Sci. 2015, 241, 177–188. [Google Scholar] [CrossRef]
- Xu, P.P.; Cai, W.M. Function of Brassica napus BnABI3 in Arabidopsis gs1, an allele of AtABI3, in seed development and stress response. Front. Plant Sci. 2019, 10, 67. [Google Scholar] [CrossRef]
- Yang, W.J.; Chen, Z.; Huang, Y.W.; Chang, G.X.; Li, P.; Wei, J.; Yuan, X.; Huang, J.; Hu, X. Powerdress as the novel regulator enhances Arabidopsis seeds germination tolerance to high temperature stress by histone modification of SOM locus. Plant Sci. 2019, 284, 91–98. [Google Scholar] [CrossRef]
- Haslekas, C.; Grini, P.E.; Nordgard, S.H.; Thorstensen, T.; Viken, M.K.; Nygaard, V.; Aalen, R.B. ABI3 mediates expression of the peroxiredoxin antioxidant AtPER1 gene and induction by oxidative stress. Plant Mol. Biol. 2003, 53, 313–326. [Google Scholar] [CrossRef]
- Chen, H.; Banerjee, A.K.; Hannapel, D.J. The tandem complex of BEL and KNOX partners is required for transcriptional repression of ga20ox1. Plant J. 2004, 38, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Barley, R.; Waites, R. Plant Meristems: The Interplay of KNOX and Gibberellins. Curr. Biol. 2002, 12, R696–R698. [Google Scholar] [CrossRef][Green Version]
- Bolduc, N.; Hake, S. The Maize Transcription Factor KNOTTED1 Directly Regulates the Gibberellin Catabolism Gene ga2ox1. Plant Cell 2009, 21, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, E.; Deflorian, G.; Bernardi, E.; Pauls, S.; Tiso, N.; Bortolussi, M.; Argenton, F. prep1.2 and aldh1a2 participate to a positive loop required for branchial arches development in zebrafish. Dev. Biol. 2010, 343, 94–103. [Google Scholar] [CrossRef]
- Letunic, T.; Doerks, P.; Bork, P. SMART: Recent updates, new developments and status in 2015. Nucleic Acids Res. 2015, 43, 257–260. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, 200–203. [Google Scholar] [CrossRef]
- Singh, K.; Upadhyay, S.K. LysM domain-containing proteins modulate stress response and signalling in Triticum aestivum L. Environ. Exp. Bot. 2021, 189, 104558. [Google Scholar]
- Tyagi, S.; Sharma, A.; Singh, K.; Upadhyay, S.K. Genomic dissection and transcriptional profiling of Cysteine-rich receptor-like kinases in five cereals and functional characterization of TaCRK68-A. Int. J. Biol. Macromol. 2019, 134, 316–329. [Google Scholar]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Gaut, B.S.; Morton, B.R.; McCaig, B.C.; Clegg, M. Substitution Rate comparisons Between Grasses and Palms: Synonymous Rate Differences at the Nuclear Gene Adh Parallel Rate Differences at the Plastid Gene rbcL. Proc. Natl. Acad. Sci. USA 1996, 93, 10274–10279. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene features visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, 585–587. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, 202–208. [Google Scholar] [CrossRef]
- Pingault, L.; Choulet, F.; Alberti, A.; Glover, N.; Wincker, P.; Feuillet, C.; Paux, E. Deep transcriptome sequencing provides new insights into the structural and functional organization of the wheat genome. Genome Biol. 2015, 16, 29. [Google Scholar] [CrossRef]
- Choulet, F.; Alberti, A.; Theil, S.; Glover, N.M.; Barbe, V.; Daron, J.; Pingault, L.; Sourdille, P.; Couloux, A.; Paux, E. Structural and Functional Partitioning of Bread Wheat Chromosome 3B. Science 2014, 345, 1249721. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Papatheodorou, I.; Fonseca, N.A.; Keays, M.; Tang, A.; Barrera, E.; Bazant, W.; Burke, M.; Füllgrabe, A.; Fuentes, A.M.-P.; George, N.; et al. Expression Atlas: Gene and protein expression across multiple studies and organisms. Nucleic Acids Res. 2018, 46, D246–D251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Z.; Khan, A.A.; Lin, Q.; Han, Y.; Mu, P.; Liu, Y.; Zhang, H.; Li, L.; Meng, X.; et al. Expression partitioning of homeologs and tandem duplications contribute to salt tolerance in wheat (Triticum aestivum L.). Sci. Rep. 2016, 6, 21476. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xin, M.; Qin, J.; Peng, H.; Ni, Z.; Yao, Y.; Sun, Q. Temporal transcriptome profiling reveals expression partitioning of homeologous genes contributing to heat and drought acclimation in wheat (Triticum aestivum L.). BMC Plant Biol. 2015, 15, 152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, Y.; Wang, C.; Liu, M.; Li, H.; Fu, Y.; Wang, Y.; Nie, Y.; Liu, X.; Ji, W. Large-scale transcriptome comparison reveals distinct gene activations in wheat responding to stripe rust and powdery mildew. BMC Genom. 2014, 15, 898. [Google Scholar] [CrossRef]
- Seo, J.; Gordish-Dressmanm, H.; Hoffman, E.P. An interactive power analysis tool for microarray hypothesis testing and generation. Bioinformatics 2006, 22, 808–814. [Google Scholar] [CrossRef]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2018, 46, 49–54. [Google Scholar] [CrossRef]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, 808–815. [Google Scholar] [CrossRef]
- Kuhn, M.; Szklarczyk, D.; Franceschini, A.; von Mering, C.; Jensen, L.J.; Bork, P. STITCH 3: Zooming in on protein-chemical interactions. Nucleic Acids Res. 2012, 40, 876–880. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Paralogous Genes | Ka | Ks | Ka_Ks | Duplication Event | T(MYA) | Selection Pressure | |
|---|---|---|---|---|---|---|---|
| TaTALE8-4A3 | TaTALE8-4A1 | 0.0244421 | 0.028216 | 0.8662338 | SD | 2.150647397 | Purifying |
| TaTALE8-4A2 | TaTALE8-4A4 | 0.0013357 | 0.004329 | 0.3085478 | TD | 0.329955514 | Purifying |
| TaTALE8-4D2 | TaTALE8-4D1 | 0.0515728 | 0.102674 | 0.5022977 | TD | 7.825746904 | Purifying |
| TaTALE1-1D2 | TaTALE1-1D1 | 0.0517904 | 0.25453 | 0.2034744 | TD | 19.40018629 | Purifying |
| TaTALE1-1A2 | TaTALE1-1A1 | 0.0572889 | 0.239382 | 0.2393203 | TD | 18.24556926 | Purifying |
| TaTALE1-1B2 | TaTALE1-1B1 | 0.0595657 | 0.235113 | 0.2533489 | TD | 17.92021991 | Purifying |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rathour, M.; Shumayla; Alok, A.; Upadhyay, S.K. Investigation of Roles of TaTALE Genes during Development and Stress Response in Bread Wheat. Plants 2022, 11, 587. https://doi.org/10.3390/plants11050587
Rathour M, Shumayla, Alok A, Upadhyay SK. Investigation of Roles of TaTALE Genes during Development and Stress Response in Bread Wheat. Plants. 2022; 11(5):587. https://doi.org/10.3390/plants11050587
Chicago/Turabian StyleRathour, Meenakshi, Shumayla, Anshu Alok, and Santosh Kumar Upadhyay. 2022. "Investigation of Roles of TaTALE Genes during Development and Stress Response in Bread Wheat" Plants 11, no. 5: 587. https://doi.org/10.3390/plants11050587
APA StyleRathour, M., Shumayla, Alok, A., & Upadhyay, S. K. (2022). Investigation of Roles of TaTALE Genes during Development and Stress Response in Bread Wheat. Plants, 11(5), 587. https://doi.org/10.3390/plants11050587