


Comparative Genome Analysis of Old World and New World TYLCV Reveals a Biasness toward Highly Variable Amino Acids in Coat Protein
 ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Characteristics of BGVs Genomic Sequences
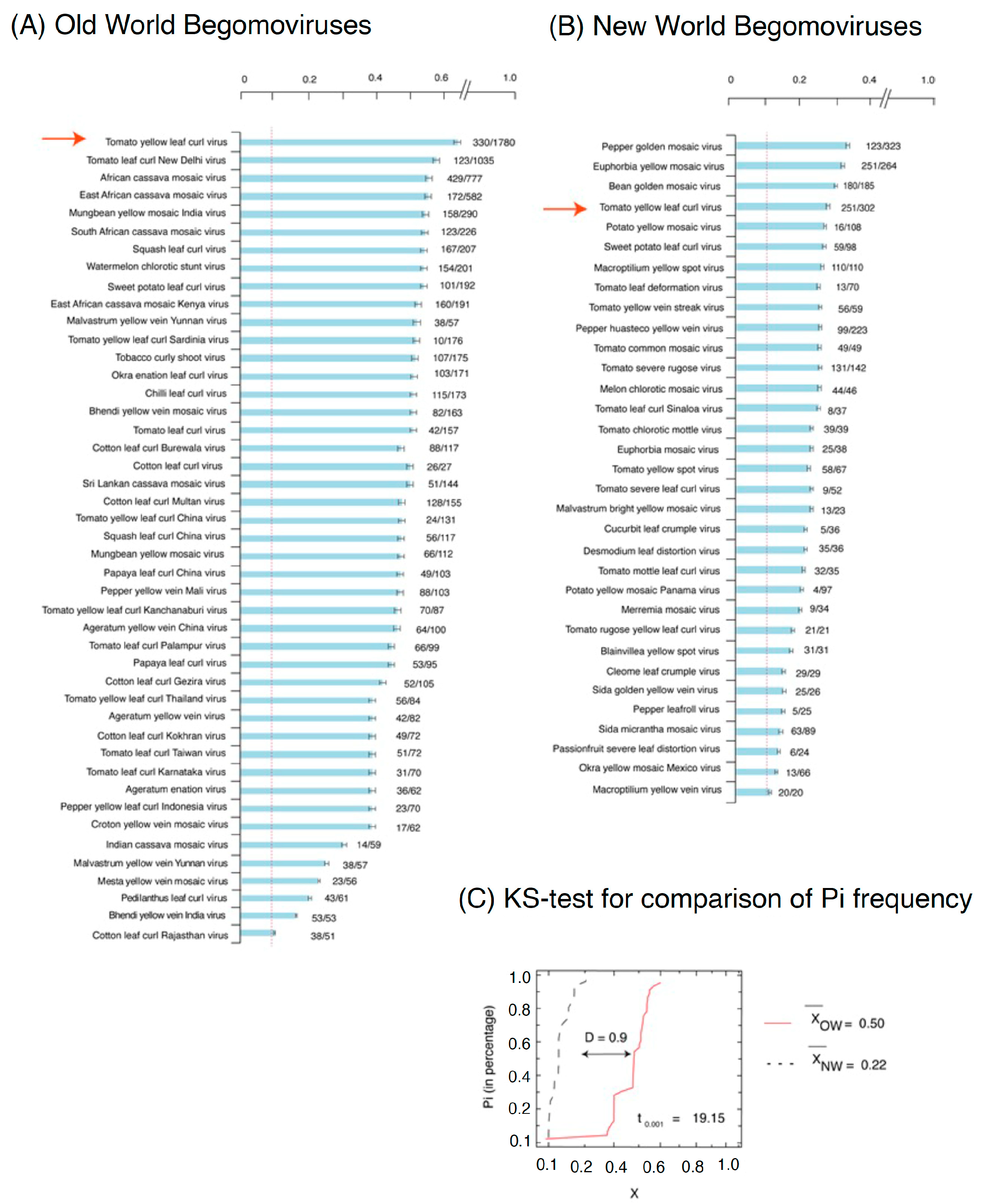
2.2. The Nucleotide Diversity Observed in BGVs from the Old World Differs from That Seen among New World BGVs
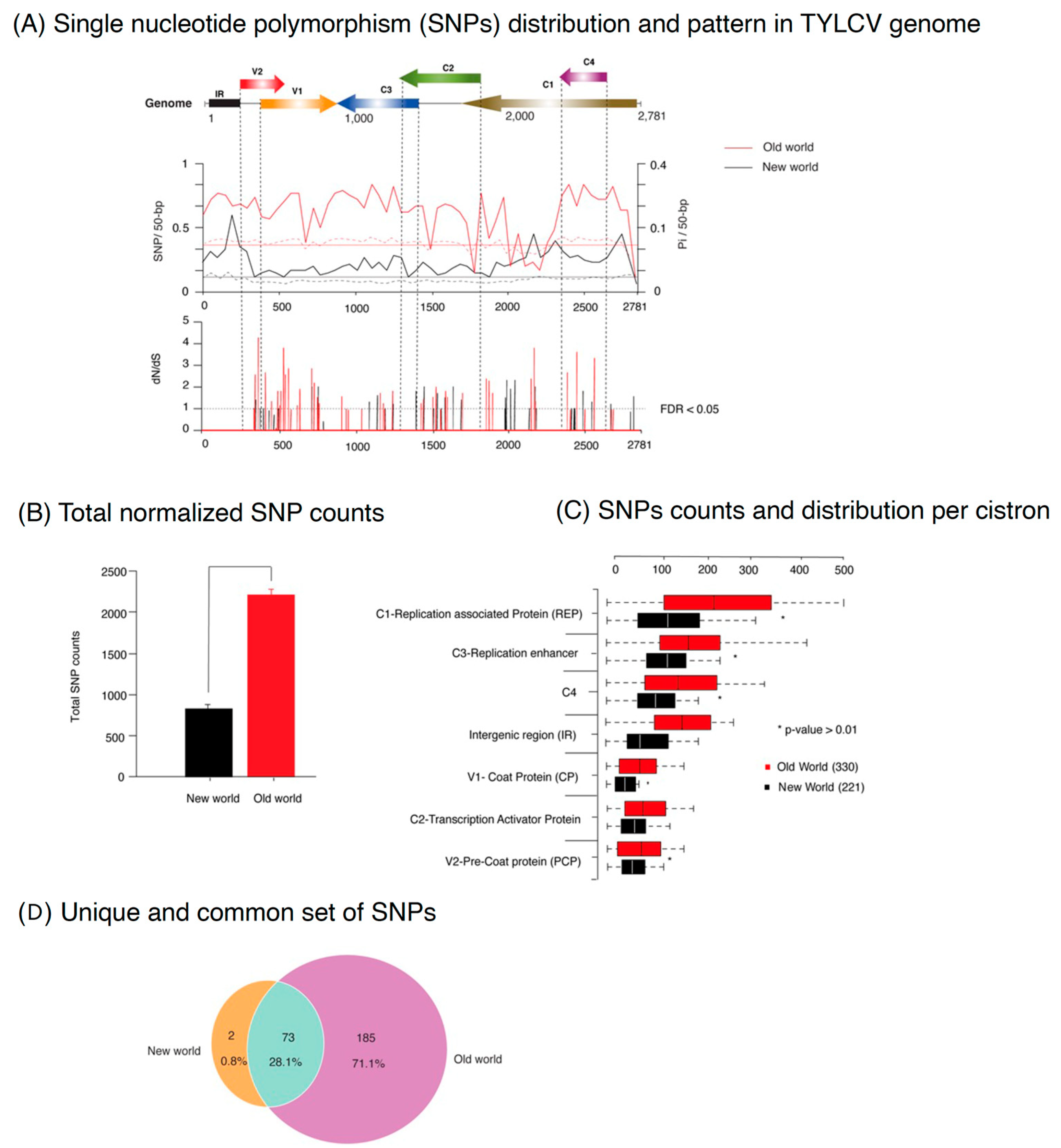
2.3. The Distribution of Nucleotide Diversity in Tomato Yellow Leaf Curl Virus Varies across the Genome and Differs among Old World versus New World Isolates
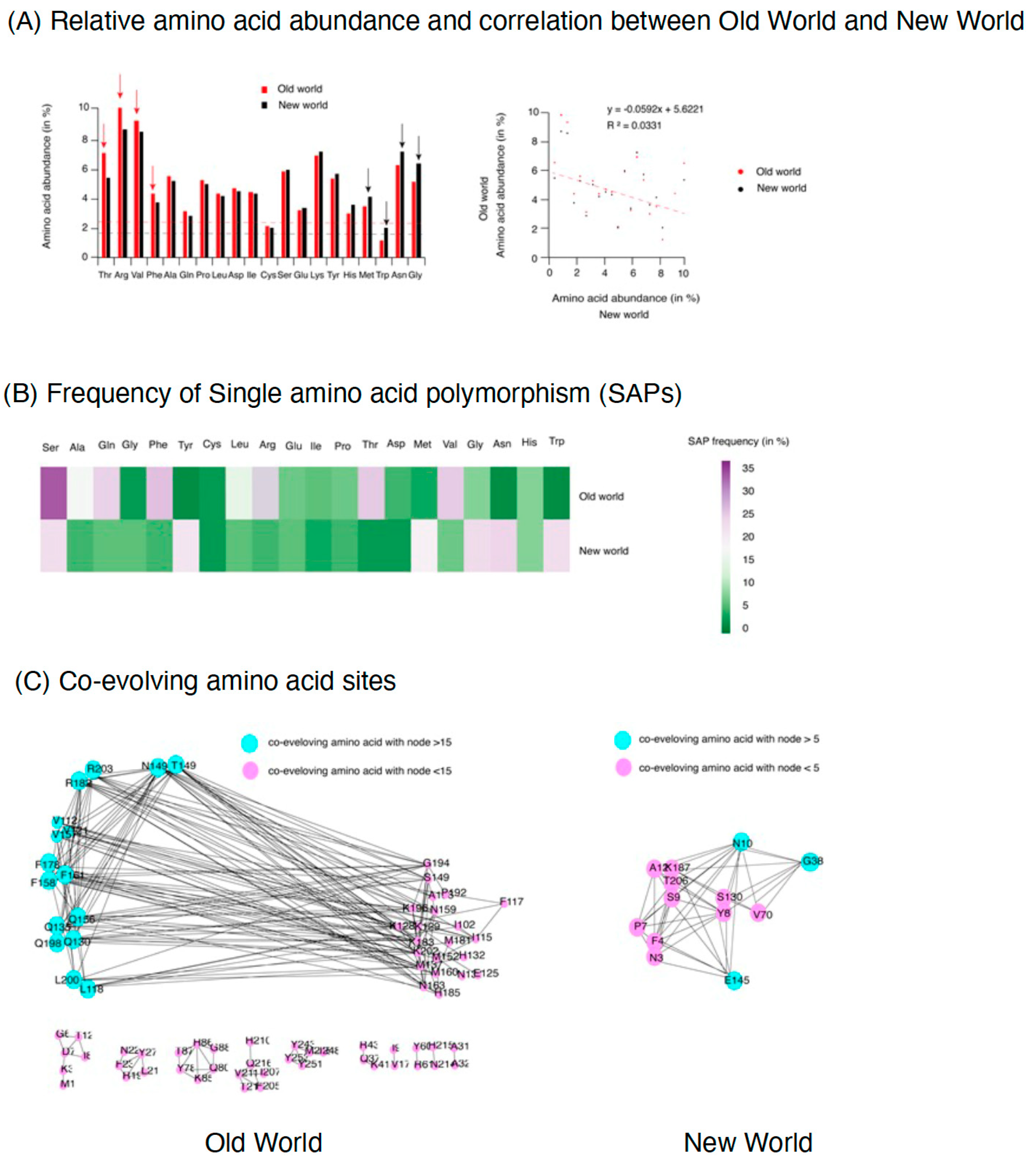
2.4. Amino Acid Substitution Profile in Coat Protein of TYLCV
2.5. Amino Acid Mutation in Coat Protein from Old World TYLCV Isolates Are Structure Changing
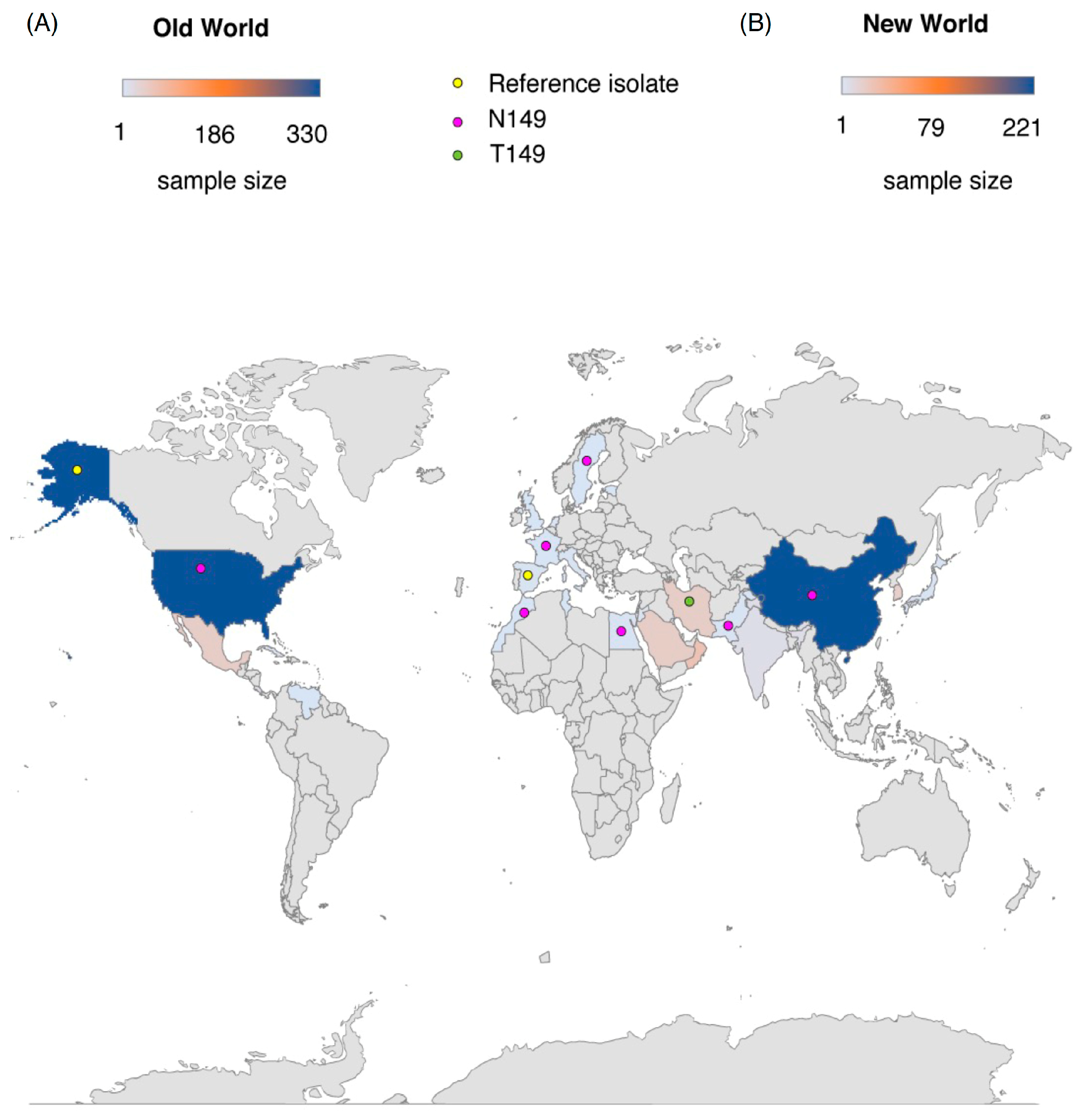
2.6. The S149 Mutation: Increasing Frequency and Worldwide Distribution
2.7. The S149 Mutation and TYLCV Evolution Are Linked to Host Geography
3. Discussion
3.1. Mutation Dynamics and Selection Constraint in Begomovirus Cistron; TYLCV as Model
3.2. Intergenic Region (IRs)
3.3. V2
3.4. Coat Protein/V1
3.5. C3 (Replication Enhancer Protein)
3.6. C2
3.7. C1
3.8. C4
4. Material and Methods
4.1. Genomic and Polyprotein Sequences
4.2. Single Nucleotide Polymorphism (SNP) Analysis
4.3. Kolmogorov-Smirnov Test
4.4. Discovery of Coevolving Groups in Coat Protein
4.5. Coat Protein 3-D Structure Prediction, Validation, Visualization, and Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fiallo-Olivé, E.; Pan, L.-L.; Liu, S.-S.; Navas-Castillo, J. Transmission of BGVs and Other Whitefly-Borne Viruses: Dependence on the Vector Species. Phytopathology 2020, 110, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Fiallo-Olivé, E.; Navas-Castillo, J. BGVs: What Is the Secret (S) of Their Success? Trends Plant Sci. 2023. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhao, P.; Wang, D.; Mubin, M.; Fang, R.; Ye, J. Diverse BGVs Evolutionarily Hijack Plant Terpenoid-Based Defense to Promote Whitefly Performance. Cells 2023, 12, 149. [Google Scholar] [CrossRef] [PubMed]
- Nigam, D. Genomic Variation and Diversification in Begomovirus Genome in Implication to Host and Vector Adaptation. Plants 2021, 10, 1706. [Google Scholar] [CrossRef] [PubMed]
- Lestari, S.M.; Khatun, M.F.; Acharya, R.; Sharma, S.R.; Shrestha, Y.K.; Jahan, S.M.H.; Aye, T.-T.; Lynn, O.M.; Win, N.K.K.; Hoat, T.X. Genetic Diversity of Cryptic Species of Bemisia Tabaci in Asia. Arch. Insect Biochem. Physiol. 2023, 112, e21981. [Google Scholar] [CrossRef]
- Venkataravanappa, V.; Kodandaram, M.H.; Prasanna, H.C.; Reddy, M.K.; Lakshminarayana Reddy, C.N. Unraveling Different BGVs, DNA Satellites and Cryptic Species of Bemisia Tabaci and Their Endosymbionts in Vegetable Ecosystem. Microb. Pathog. 2023, 174, 105892. [Google Scholar] [CrossRef]
- Liu, S.; Colvin, J.; De Barro, P.J. Species Concepts as Applied to the Whitefly Bemisia Tabaci Systematics: How Many Species Are There? J. Integr. Agric. 2012, 11, 176–186. [Google Scholar] [CrossRef]
- Manivannan, K.; Renukadevi, P.; Malathi, V.G.; Karthikeyan, G.; Balakrishnan, N. A New Seed-Transmissible Begomovirus in Bitter Gourd (Momordica Charantia L.). Microb. Pathog. 2019, 128, 82–89. [Google Scholar] [CrossRef]
- Rocha, C.S.; Castillo-Urquiza, G.P.; Lima, A.T.M.; Silva, F.N.; Xavier, C.A.D.; Hora-Júnior, B.T.; Beserra-Júnior, J.E.A.; Malta, A.W.O.; Martin, D.P.; Varsani, A. Brazilian Begomovirus Populations Are Highly Recombinant, Rapidly Evolving, and Segregated Based on Geographical Location. J. Virol. 2013, 87, 5784–5799. [Google Scholar] [CrossRef]
- Ramesh, S.V.; Sahu, P.P.; Prasad, M.; Praveen, S.; Pappu, H.R. Geminiviruses and Plant Hosts: A Closer Examination of the Molecular Arms Race. Viruses 2017, 9, 256. [Google Scholar] [CrossRef]
- García-Andrés, S.; Accotto, G.P.; Navas-Castillo, J.; Moriones, E. Founder Effect, Plant Host, and Recombination Shape the Emergent Population of BGVs That Cause the Tomato Yellow Leaf Curl Disease in the Mediterranean Basin. Virology 2007, 359, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Monci, F.; Sánchez-Campos, S.; Navas-Castillo, J.; Moriones, E. A Natural Recombinant between the Geminiviruses Tomato Yellow Leaf Curl Sardinia Virus and Tomato Yellow Leaf Curl Virus Exhibits a Novel Pathogenic Phenotype and Is Becoming Prevalent in Spanish Populations. Virology 2002, 303, 317–326. [Google Scholar] [CrossRef]
- Lima, A.T.M.; Silva, J.C.F.; Silva, F.N.; Castillo-Urquiza, G.P.; Silva, F.F.; Seah, Y.M.; Mizubuti, E.S.G.; Duffy, S.; Zerbini, F.M. The Diversification of Begomovirus Populations Is Predominantly Driven by Mutational Dynamics. Virus Evol. 2017, 3, vex005. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Holmes, E.C. Phylogenetic Evidence for Rapid Rates of Molecular Evolution in the Single-Stranded DNA Begomovirus Tomato Yellow Leaf Curl Virus. J. Virol. 2008, 82, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Pita, J.S.; Fondong, V.N.; Sangare, A.; Otim-Nape, G.W.; Ogwal, S.; Fauquet, C.M. Recombination, Pseudorecombination and Synergism of Geminiviruses Are Determinant Keys to the Epidemic of Severe Cassava Mosaic Disease in Uganda. J. Gen. Virol. 2001, 82, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, Y.; Calvert, L.; Munoz, C.; Otim-Nape, G.W.; Robinson, D.J.; Harrison, B.D. Evidence That DNA-a of a Geminivirus Associated with Severe Cassava Mosaic Disease in Uganda Has Arisen by Interspecific Recombination. J. Gen. Virol. 1997, 78, 2101–2111. [Google Scholar] [CrossRef]
- Seal, S.E.; VandenBosch, F.; Jeger, M.J. Factors Influencing Begomovirus Evolution and Their Increasing Global Significance: Implications for Sustainable Control. Crit. Rev. Plant Sci. 2006, 25, 23–46. [Google Scholar] [CrossRef]
- Sharma, H.J.; Susheel, K.S.; Nongthombam, B.S. Genome Complexity of Begomovirus Disease and a Concern in Agro-Economic Loss. J. Appl. Biol. Biotechnol. 2019, 7, 78–83. [Google Scholar]
- Power, A.G. Insect Transmission of Plant Viruses: A Constraint on Virus Variability. Curr. Opin. Plant Biol. 2000, 3, 336–340. [Google Scholar] [CrossRef]
- Guyader, S.; Ducray, D.G. Sequence Analysis of Potato Leafroll Virus Isolates Reveals Genetic Stability, Major Evolutionary Events and Differential Selection Pressure between Overlapping Reading Frame Products. J. Gen. Virol. 2002, 83, 1799–1807. [Google Scholar] [CrossRef]
- García-Arenal, F.; Fraile, A.; Malpica, J.M. Variation and Evolution of Plant Virus Populations. Int. Microbiol. 2003, 6, 225–232. [Google Scholar] [CrossRef]
- García-Arenal, F.; Fraile, A.; Malpica, J.M. Variability and Genetic Structure of Plant Virus Populations. Annu. Rev. Phytopathol. 2001, 39, 157–186. [Google Scholar] [CrossRef]
- Caracuel, Z.; Lozano-Durán, R.; Huguet, S.; Arroyo-Mateos, M.; Rodríguez-Negrete, E.A.; Bejarano, E.R. C2 from Beet Curly Top Virus Promotes a Cell Environment Suitable for Efficient Replication of Geminiviruses, Providing a Novel Mechanism of Viral Synergism. New Phytol. 2012, 194, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Rentería-Canett, I.; Xoconostle-Cázares, B.; Ruiz-Medrano, R.; Rivera-Bustamante, R.F. Geminivirus Mixed Infection on Pepper Plants: Synergistic Interaction between Phyvv and Pepgmv. Virol J. 2011, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.D.; Robinson, D.J. Natural Genomic and Antigenic Variation in Whitefly-Transmitted Geminiviruses (BGVs). Annu. Rev. Phytopathol. 1999, 37, 369–398. [Google Scholar] [CrossRef] [PubMed]
- Champeimont, R.; Laine, E.; Hu, S.-W.; Penin, F.; Carbone, A. Coevolution Analysis of Hepatitis C Virus Genome to Identify the Structural and Functional Dependency Network of Viral Proteins. Sci. Rep. 2016, 6, 26401. [Google Scholar] [CrossRef]
- Wu, B.; Shang, X.; Schubert, J.; Habekuß, A.; Elena, S.F.; Wang, X. Global-Scale Computational Analysis of Genomic Sequences Reveals the Recombination Pattern and Coevolution Dynamics of Cereal-Infecting Geminiviruses. Sci. Rep. 2015, 5, 8153. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Panchenko, A.R. Structural and Functional Roles of Coevolved Sites in Proteins. PLoS ONE 2010, 5, e8591. [Google Scholar] [CrossRef]
- Altschuh, D.; Lesk, A.M.; Bloomer, A.C.; Klug, A. Correlation of Co-Ordinated Amino Acid Substitutions with Function in Viruses Related to Tobacco Mosaic Virus. J. Mol. Biol. 1987, 193, 693–707. [Google Scholar] [CrossRef]
- Sruthi, C.K.; Prakash, M.K. Viral Complexity: Amino Acid Co-Evolution in Viral Genomes as a Possible Metric. BioRxiv 2017, BioRxiv:159541. [Google Scholar]
- Hasiów-Jaroszewska, B.; Fares, M.A.; Elena, S.F. Molecular Evolution of Viral Multifunctional Proteins: The Case of Potyvirus Hc-Pro. J. Mol. Evol. 2014, 78, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Mondal, D.; Mandal, S.; Shil, S.; Sahana, N.; Pandit, G.K.; Choudhury, A. Genome Wide Molecular Evolution Analysis of BGVs Reveals Unique Diversification Pattern in Coat Protein Gene of Old World and New World Viruses. Virusdisease 2019, 30, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Tatineni, S.; Hein, G.L. Plant Viruses of Agricultural Importance: Current and Future Perspectives of Virus Disease Management Strategies. Phytopathology 2023, 113, 117–141. [Google Scholar] [CrossRef]
- Verma, S.; Yusuf, A.; Sangeeta, S. A Novel Protocol to Identify the Sirna Hotspots for Creating Rnai-Based Begomovirus Resistance. Brief. Funct. Genom. 2023, 22, 49–60. [Google Scholar]
- Wang, Y.; Jiang, J.; Zhao, L.; Zhou, R.; Yu, W.; Zhao, T. Application of Whole Genome Resequencing in Mapping of a Tomato Yellow Leaf Curl Virus Resistance Gene. Sci. Rep. 2018, 8, 9592. [Google Scholar] [CrossRef] [PubMed]
- Mahfouz, M.M.; Tashkandi, M.; Ali, Z.; Aljedaani, F.R.; Shami, A. Engineering Resistance against Tomato Yellow Leaf Curl Virus Via the Crispr/Cas9 System in Tomato. bioRxiv 2017. [Google Scholar] [CrossRef]
- Mahmood, M.A.; Naqvi, R.Z.; Rahman, S.U.; Amin, I.; Mansoor, S. Plant Virus-Derived Vectors for Plant Genome Engineering. Viruses 2023, 15, 531. [Google Scholar] [CrossRef]
- Demirci, Y.; Zhang, B.; Unver, T. Crispr/Cas9: An Rna-Guided Highly Precise Synthetic Tool for Plant Genome Editing. J. Cell. Physiol. 2018, 233, 1844–1859. [Google Scholar] [CrossRef]
- Ali, Z.; Ali, S.; Tashkandi, M.; Zaidi, S.S.-E.; Mahfouz, M.M. Crispr/Cas9-Mediated Immunity to Geminiviruses: Differential Interference and Evasion. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Mehta, D.; Stürchler, A.; Anjanappa, R.B.; Zaidi, S.S.-e.-A.; Hirsch-Hoffmann, M.; Gruissem, W.; Vanderschuren, H. Linking Crispr-Cas9 Interference in Cassava to the Evolution of Editing-Resistant Geminiviruses. Genome Biol. 2019, 20, 1–10. [Google Scholar] [CrossRef]
- Tashkandi, M.; Ali, Z.; Aljedaani, F.; Shami, A.; Mahfouz, M.M. Engineering Resistance against Tomato Yellow Leaf Curl Virus Via the Crispr/Cas9 System in Tomato. Plant Signal. Behav. 2018, 13, e1525996. [Google Scholar] [CrossRef] [PubMed]
- Stokstad, E. Antibody-Based Defense May Protect Plants from Disease. Science 2023, 379, 867. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Tian, Y.; Xu, C.; Xing, Y.; Yang, L.; Qian, G.; Hua, X.; Gong, W.; Hu, B.; Wang, L. A Monoclonal Antibody-Based Immunochromatographic Test Strip and Its Application in the Rapid Detection of Cucumber Green Mottle Mosaic Virus. Biosensors 2023, 13, 199. [Google Scholar] [CrossRef]
- Naganur, P.; Shankarappa, K.S.; Mesta, R.K.; Rao, C.D.; Venkataravanappa, V.; Maruthi, M.N.; Reddy, L.R.C.N. Detecting Tomato Leaf Curl New Delhi Virus Causing Ridge Gourd Yellow Mosaic Disease, and Other Begomoviruses by Antibody-Based Methods. Plants 2023, 12, 490. [Google Scholar] [CrossRef]
- Bajpai, R.; Puyam, A.; Kashyap, P.L. Agro-Nanodiagnostics for Plant Diseases. In Nanotechnology in Agriculture and Agroecosystems; Elsevier: Amsterdam, The Netherlands, 2023; pp. 169–188. [Google Scholar]
- Wamaitha, M.J.; Nigam, D.; Maina, S.; Stomeo, F.; Wangai, A.; Njuguna, J.N.; Holton, T.A.; Wanjala, B.W.; Wamalwa, M.; Lucas, T. Metagenomic Analysis of Viruses Associated with Maize Lethal Necrosis in Kenya. Virol. J. 2018, 15, 90. [Google Scholar] [CrossRef]
- Gautam, S.; Mugerwa, H.; Buck, J.W.; Dutta, B.; Coolong, T.; Adkins, S.; Srinivasan, R. Differential Transmission of Old and New World Begomoviruses by Middle East-Asia Minor 1 (Meam1) and Mediterranean (Med) Cryptic Species of Bemisia Tabaci. Viruses 2022, 14, 1104. [Google Scholar] [CrossRef]
- Höhnle, M.; Höfer, P.; Bedford, I.D.; Briddon, R.W.; Markham, P.G.; Frischmuth, T. Exchange of Three Amino Acids in the Coat Protein Results in Efficient Whitefly Transmission of a Nontransmissible Abutilon Mosaic Virus Isolate. Virology. 2001, 290, 164–171. [Google Scholar] [CrossRef]
- Hajizadeh, M.; Sokhandan-Bashir, N. Population Genetic Analysis of Potato Virus X Based on the Cp Gene Sequence. Virusdisease. 2017, 28, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Sokhandan-Bashir, N.; Melcher, U. Population Genetic Analysis of Grapevine Fanleaf Virus. Arch. Virol. 2012, 157, 1919–1929. [Google Scholar] [CrossRef]
- Nigam, D.; LaTourrette, K.; Souza, P.F.N.; Garcia-Ruiz, H. Genome-Wide Variation in Potyviruses. Front. Plant Sci. 2019, 10, 1439. [Google Scholar] [CrossRef]
- Papayiannis, L.C.; Katis, N.I.; Idris, A.M.; Brown, J.K. Identification of Weed Hosts of Tomato Yellow Leaf Curl Virus in Cyprus. Plant Dis. 2011, 95, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, X.; Wang, Y.; Wu, X.; Wang, B.; Lu, Z.; Li, G. Genome-Wide Characterization and Expression Analysis of the Mlo Gene Family Sheds Light on Powdery Mildew Resistance in Lagenaria Siceraria. Heliyon 2023, 9, e14624. [Google Scholar] [CrossRef] [PubMed]
- Nigam, D.; Garcia-Ruiz, H. Variation Profile of the Orthotospovirus Genome. Pathogens 2020, 9, 521. [Google Scholar] [CrossRef]
- Sang, S.; Yue, Y.; Wang, Y.; Zhang, X. The Epidemiology and Evolutionary Dynamics of Massive Dengue Outbreak in China, 2019. Front. Microbiol. 2023, 14, 1156176. [Google Scholar] [CrossRef]
- Rabadán, M.P.; Juarez, M.; Gómez, P. Long-Term Monitoring of Aphid-Transmitted Viruses in Melon and Zucchini Crops: Genetic Diversity and Population Structure of Cucurbit Aphid-Borne Yellows Virus and Watermelon Mosaic Virus. Phytopathology 2023. [Google Scholar] [CrossRef]
- Dhobale, K.V.; Murugan, B.; Deb, R.; Kumar, S.; Sahoo, L. Molecular Epidemiology of Begomoviruses Infecting Mungbean from Yellow Mosaic Disease Hotspot Regions of India. Appl. Biochem. Biotechnol. 2023, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Neoh, Z.Y.; Lai, H.-C.; Lin, C.-C.; Suwor, P.; Tsai, W.-S. Genetic Diversity and Geographic Distribution of Cucurbit-Infecting Begomoviruses in the Philippines. Plants 2023, 12, 272. [Google Scholar] [CrossRef]
- Melero, I.; González, R.; Elena, S.F. Host Developmental Stages Shape the Evolution of a Plant Rna Virus. Philos. Trans. R. Soc. B Biol. Sci. 2023, 378, 20220005. [Google Scholar] [CrossRef]
- Pandey, V.; Srivastava, A.; Shahmohammadi, N.; Nehra, C.; Gaur, R.K.; Golnaraghi, A. Begomovirus: Exploiting the Host Machinery for Their Survival. J. Mod. Agric. Biotechnol. 2023, 2, 10. [Google Scholar] [CrossRef]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The Evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 361–379. [Google Scholar] [CrossRef]
- Wei, J.; He, Y.-Z.; Guo, Q.; Guo, T.; Liu, Y.-Q.; Zhou, X.-P.; Liu, S.-S.; Wang, X.-W. Vector Development and Vitellogenin Determine the Transovarial Transmission of Begomoviruses. Proc. Natl. Acad. Sci. USA 2017, 114, 6746–6751. [Google Scholar] [CrossRef]
- Gautam, S.; Buck, J.W.; Dutta, B.; Coolong, T.; Sanchez, T.; Smith, H.A.; Adkins, S.; Srinivasan, R. Sida Golden Mosaic Virus, an Emerging Pathogen of Snap Bean (Phaseolus Vulgaris L.) in the Southeastern United States. Viruses. 2023, 15, 357. [Google Scholar] [CrossRef]
- Borkosky, S.S.; Camporeale, G.; Chemes, L.B.; Risso, M.; Noval, M.G.; Sánchez, I.E.; Alonso, L.G.; de Prat Gay, G. Hidden Structural Codes in Protein Intrinsic Disorder. Biochemistry 2017, 56, 5560–5569. [Google Scholar] [CrossRef] [PubMed]
- Campen, A.; Williams, R.M.; Brown, C.J.; Meng, J.; Uversky, V.N.; Dunker, A.K. Top-Idp-Scale: A New Amino Acid Scale Measuring Propensity for Intrinsic Disorder. Protein Pept. Lett. 2008, 15, 956–963. [Google Scholar] [CrossRef]
- Kelley, L.; Jefferys, B. Phyre2: Protein Homology/Analogy Recognition Engine V 2.0; Structural Bioinformatics Group, Imperial College: London, UK, 2011. [Google Scholar]
- Pan, L.; Chen, Q.; Guo, T.; Wang, X.; Li, P.; Wang, X.; Liu, S. Differential Efficiency of a Begomovirus to Cross the Midgut of Different Species of Whiteflies Results in Variation of Virus Transmission by the Vectors. Sci. China Life Sci. 2018, 61, 1254–1265. [Google Scholar] [CrossRef]
- Pan, L.-L.; Cui, X.-Y.; Chen, Q.-F.; Wang, X.-W.; Liu, S.-S. Cotton Leaf Curl Disease: Which Whitefly Is the Vector? Phytopathology 2018, 108, 1172–1183. [Google Scholar] [CrossRef]
- Devendran, R.; Kumar, M.; Ghosh, D.; Yogindran, S.; Karim, M.J.; Chakraborty, S. Capsicum-Infecting Begomoviruses as Global Pathogens: Host–Virus Interplay, Pathogenesis, and Management. Trends Microbiol. 2022, 30, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Adkar, B.V. Computational and Experimental Studies on Protein Structure, Stability and Dynamics. 2014. Available online: https://etd.iisc.ac.in/handle/2005/2369 (accessed on 30 August 2021).
- Mondal, S.K.; Mukhoty, S.; Kundu, H.; Ghosh, S.; Sen, M.K.; Das, S.; Brogi, S. In Silico Analysis of Rna-Dependent Rna Polymerase of the Sars-Cov-2 and Therapeutic Potential of Existing Antiviral Drugs. Comput. Biol. Med. 2021, 135, 104591. [Google Scholar] [CrossRef] [PubMed]
- Sicard, A.; Michalakis, Y.; Gutiérrez, S.; Blanc, S. The Strange Lifestyle of Multipartite Viruses. PLoS Pathog. 2016, 12, e1005819. [Google Scholar] [CrossRef]
- Josefat, G.J. Evolución Forzada De Geminivirus: Inestabilidad De Mutaciones En La Hélice-4 Del Dominio De Unión a Retinoblastoma De La Proteína Rep. 2011. Available online: https://www.lareferencia.info/vufind/Record/MX_9ac268ee71658f5e99b9222d354b9cd4 (accessed on 30 August 2021).
- Bonnet, J.; Fraile, A.; Sacristán, S.; Malpica, J.M.; García-Arenal, F. Role of Recombination in the Evolution of Natural Populations of Cucumber Mosaic Virus, a Tripartite Rna Plant Virus. Virology 2005, 332, 359–368. [Google Scholar] [CrossRef]
- Lucía-Sanz, A.; Manrubia, S. Multipartite Viruses: Adaptive Trick or Evolutionary Treat? NPJ Syst. Biol. Appl. 2017, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Moriones, E.; Navas-Castillo, J. Tomato Yellow Leaf Curl Virus, an Emerging Virus Complex Causing Epidemics Worldwide. Virus Res. 2000, 71, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Sharma, N.; Hari-Gowthem, G.; Muthamilarasan, M.; Prasad, M. Tomato Yellow Leaf Curl Virus: Impact, Challenges, and Management. Trends Plant Sci. 2020, 25, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of Evolutionary Change in Viruses: Patterns and Determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Sánchez-Campos, S.; Domínguez-Huerta, G.; Díaz-Martínez, L.; Tomás, D.M.; Navas-Castillo, J.; Moriones, E.; Grande-Pérez, A. Differential Shape of Geminivirus Mutant Spectra across Cultivated and Wild Hosts with Invariant Viral Consensus Sequences. Front. Plant Sci. 2018, 9, 932. [Google Scholar] [CrossRef]
- Wang, B.; Li, F.; Huang, C.; Yang, X.; Qian, Y.; Xie, Y.; Zhou, X. V2 of Tomato Yellow Leaf Curl Virus Can Suppress Methylation-Mediated Transcriptional Gene Silencing in Plants. J. Gen. Virol. 2014, 95, 225–230. [Google Scholar] [CrossRef]
- Csorba, T.; Kontra, L.; Burgyán, J. Viral Silencing Suppressors: Tools Forged to Fine-Tune Host-Pathogen Coexistence. Virology 2015, 479, 85–103. [Google Scholar] [CrossRef]
- Bar-Ziv, A.; Levy, Y.; Hak, H.; Mett, A.; Belausov, E.; Citovsky, V.; Gafni, Y. The Tomato Yellow Leaf Curl Virus (Tylcv) V2 Protein Interacts with the Host Papain-Like Cysteine Protease Cyp1. Plant Signal. Behav. 2012, 7, 983–989. [Google Scholar] [CrossRef]
- Hussain, M.; Mansoor, S.; Iram, S.; Zafar, Y.; Briddon, R.W. The Hypersensitive Response to Tomato Leaf Curl New Delhi Virus Nuclear Shuttle Protein Is Inhibited by Transcriptional Activator Protein. Mol. Plant-Microbe Interact. 2007, 20, 1581–1588. [Google Scholar] [CrossRef]
- Melgarejo, T.A.; Kon, T.; Rojas, M.R.; Paz-Carrasco, L.; Zerbini, F.M.; Gilbertson, R.L. Leaf, Monopartite Begomovirus Causing. “Characterization of a New World. J. Virol. 2013, 87, 5397. [Google Scholar] [CrossRef]
- Chowda-Reddy, R.V.; Achenjang, F.; Felton, C.; Etarock, M.T.; Anangfac, M.-T.; Nugent, P.; Fondong, V.N. Role of a Geminivirus Av2 Protein Putative Protein Kinase C Motif on Subcellular Localization and Pathogenicity. Virus Res. 2008, 135, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Rouhibakhsh, A.; Haq, Q.M.; Malathi, V.G. Mutagenesis in Orf Av2 Affects Viral Replication in Mungbean Yellow Mosaic India Virus. J. Biosci. 2011, 36, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Mubin, M.; Amin, I.; Amrao, L.; Briddon, R.W.; Mansoor, S. The Hypersensitive Response Induced by the V2 Protein of a Monopartite Begomovirus Is Countered by the C2 Protein. Mol. Plant Pathol. 2010, 11, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, F.; Pallas, V.; Sanchez-Navarro, J. Implication of the C Terminus of the Prunus Necrotic Ringspot Virus Movement Protein in Cell-to-Cell Transport and in Its Interaction with the Coat Protein. J. Gen. Virol. 2010, 91, 1865–1870. [Google Scholar] [CrossRef]
- Rojas, M.R.; Hagen, C.; Lucas, W.J.; Gilbertson, R.L. Exploiting Chinks in the Plant’s Armor: Evolution and Emergence of Geminiviruses. Annu. Rev. Phytopathol. 2005, 43, 361–394. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.K.; Idris, A.M.; Torres-Jerez, I.; Banks, G.K.; Wyatt, S.D. The Core Region of the Coat Protein Gene Is Highly Useful for Establishing the Provisional Identification and Classification of BGVs. Arch. Virol. 2001, 146, 1581–1598. [Google Scholar] [CrossRef]
- Carbonell, A.; Maliogka, V.I.; de Jesús Pérez, J.; Salvador, B.; León, D.S.; García, J.A.; Simón-Mateo, C. Diverse Amino Acid Changes at Specific Positions in the N-Terminal Region of the Coat Protein Allow Plum Pox Virus to Adapt to New Hosts. Mol. Plant-Microbe Interact. 2013, 26, 1211–1224. [Google Scholar] [CrossRef]
- Dennehy, J.J. Evolutionary Ecology of Virus Emergence. Ann. N. Y. Acad. Sci. 2017, 1389, 124–146. [Google Scholar] [CrossRef]
- Sun, M.; Jiang, K.; Li, C.; Du, J.; Li, M.; Ghanem, H.; Wu, G.; Qing, L. Tobacco Curly Shoot Virus C3 Protein Enhances Viral Replication and Gene Expression in Nicotiana Benthamiana Plants. Virus Res. 2020, 281, 197939. [Google Scholar] [CrossRef]
- Settlage, S.B.; See, R.G.; Hanley-Bowdoin, L. Geminivirus C3 Protein: Replication Enhancement and Protein Interactions. J. Virol. 2005, 79, 9885–9895. [Google Scholar] [CrossRef]
- Cantú-Iris, M. Estudio Del Promotor Ac2 Y Secuencias Que Responden Al Transactivador Trap En Begomovirus. 2019. Available online: https://www.lareferencia.info/vufind/Record/MX_c8a9ce95787b9ef0395035002538cf8f (accessed on 30 August 2021).
- Etessami, P.; Saunders, K.; Watts, J.; Stanley, J. Mutational Analysis of Complementary-Sense Genes of African Cassava Mosaic Virus DNA A. J. Gen. Virol. 1991, 72, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, C. Geminivirus DNA Replication. Cell Mol. Life Sci. 1999, 56, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Medina-Puche, L.; Orílio, A.F.; Zerbini, F.M.; Lozano-Durán, R. Small but Mighty: Functional Landscape of the Versatile Geminivirus-Encoded C4 Protein. PLoS Pathog. 2021, 17, e1009915. [Google Scholar] [CrossRef]
- Dai, K.-W.; Tsai, Y.-T.; Wu, C.-Y.; Lai, Y.-C.; Lin, N.-S.; Hu, C.-C. Identification of Crucial Amino Acids in Begomovirus C4 Proteins Involved in the Modulation of the Severity of Leaf Curling Symptoms. Viruses 2022, 14, 499. [Google Scholar] [CrossRef]
- Deom, C.M.; Alabady, M.S.; Yang, L. Early Transcriptome Changes Induced by the Geminivirus C4 Oncoprotein: Setting the Stage for Oncogenesis. BMC Genom. 2021, 22, 1–19. [Google Scholar] [CrossRef]
- Rishishwar, R.; Dasgupta, I. Suppressors of Rna Silencing Encoded by Geminiviruses and Associated DNA Satellites. Virusdisease 2019, 30, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J. DNA Sequence Polymorphism Analysis Using Dnasp. Methods Mol. Biol. 2009, 537, 337–350. [Google Scholar] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The Variant Call Format and Vcftools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Lopes, R.H.C.; Reid, I.D.; Hobson, P.R. The Two-Dimensional Kolmogorov-Smirnov Test. In Proceedings of the XI International Workshop on Advanced Computing and Analysis Techniques in Physics Research, Amsterdam, The Netherlands, 23–27 April 2007. [Google Scholar]
- Zhang, G.; Wang, X.; Liang, Y.-C.; Liu, J. Fast and Robust Spectrum Sensing Via Kolmogorov-Smirnov Test. IEEE Trans. Commun. 2010, 58, 3410–3416. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. Mafft: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of Phyml 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Oteri, F.; Nadalin, F.; Champeimont, R.; Carbone, A. Bis2analyzer: A Server for Co-Evolution Analysis of Conserved Protein Families. Nucleic Acids Res. 2017, 45, W307–W314. [Google Scholar] [CrossRef]
- Fares, M.A.; McNally, D. Caps: Coevolution Analysis Using Protein Sequences. Bioinformatics 2006, 22, 2821–2822. [Google Scholar] [CrossRef] [PubMed]
- Grantham, R. Amino Acid Difference Formula to Help Explain Protein Evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Go, M.; Miyazawa, S. Relationship between Mutability, Polarity and Exteriority of Amino Acid Residues in Protein Evolution. Int. J. Pept. Protein Res. 1980, 15, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Banerjee, A.K.; Tripathi, P.P.; Srivastava, A.K.; Ray, U. A Virus That Has Gone Viral: Amino Acid Mutation in S Protein of Indian Isolate of Coronavirus Covid-19 Might Impact Receptor Binding, and Thus, Infectivity. Biosci. Rep. 2020, 40, 5. [Google Scholar] [CrossRef]
- Dougherty, W.G.; Carrington, J.C.; Cary, S.M.; Parks, T.D. Biochemical and Mutational Analysis of a Plant Virus Polyprotein Cleavage Site. EMBO J. 1988, 7, 1281–1287. [Google Scholar] [CrossRef]
- Storey, J.D. False Discovery Rate. Ann. Statist. 2003, 31, 2013–2035. [Google Scholar]
- Kohl, M.; Wiese, S.; Warscheid, B. Cytoscape: Software for Visualization and Analysis of Biological Networks. Methods Mol. Biol. 2011, 696, 291–303. [Google Scholar]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, J.E.M. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. Ucsf Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. Tm-Align: A Protein Structure Alignment Algorithm Based on the Tm-Score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure Modeling Features | Old World -CP (ALN12561.1) | New World -CP (QCG74731.1) | |
|---|---|---|---|
| Top 10 templates predicted by I-TASSER | 1i3yA, 1i4yA, 1i6zA, 6cmlA, 6cmnA, 6i9yA, 8cmlA, 8cmnA, 1i5yA, 1i7zA | 1i2yA, 1i2yA, 1i6zA, 8cmlA, 7cmnA, 4i9yA, 4cmlA, 4cmnA, 1i4yA, 9i7zA | |
| Model Evaluation data of predicted structures | C- score | −2.86 | −2.23 |
| Expected TM score | 0.29 ± 0.10 | 0.61 ± 0.12 | |
| Expected RMSD | 15.5 ± 2.4 | 14.8 ± 2.9 | |
| Number of Decoys | 322 | 412 | |
| Cluster Identity | 0.0143 | 0.02 | |
| Energy value (KJ/mole) of predicted protein models | Before energy minimization | −1048.821 | 140.521 |
| After energy minimization | −14,412.217 | −12,125.214 | |
| Ramachandran Plot statistics (% residues) | Favored regions | 89 | 88.9 |
| Additional region | 8 | 7 | |
| Allowed regions | 2 | 3 | |
| Disallowed regions | 1 | 1.1 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nigam, D.; Muthukrishnan, E.; Flores-López, L.F.; Nigam, M.; Wamaitha, M.J. Comparative Genome Analysis of Old World and New World TYLCV Reveals a Biasness toward Highly Variable Amino Acids in Coat Protein. Plants 2023, 12, 1995. https://doi.org/10.3390/plants12101995
Nigam D, Muthukrishnan E, Flores-López LF, Nigam M, Wamaitha MJ. Comparative Genome Analysis of Old World and New World TYLCV Reveals a Biasness toward Highly Variable Amino Acids in Coat Protein. Plants. 2023; 12(10):1995. https://doi.org/10.3390/plants12101995
Chicago/Turabian StyleNigam, Deepti, Ezhumalai Muthukrishnan, Luis Fernando Flores-López, Manisha Nigam, and Mwathi Jane Wamaitha. 2023. "Comparative Genome Analysis of Old World and New World TYLCV Reveals a Biasness toward Highly Variable Amino Acids in Coat Protein" Plants 12, no. 10: 1995. https://doi.org/10.3390/plants12101995
APA StyleNigam, D., Muthukrishnan, E., Flores-López, L. F., Nigam, M., & Wamaitha, M. J. (2023). Comparative Genome Analysis of Old World and New World TYLCV Reveals a Biasness toward Highly Variable Amino Acids in Coat Protein. Plants, 12(10), 1995. https://doi.org/10.3390/plants12101995